- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Isolation of Myofibres and Culture of Muscle Stem Cells from Adult Zebrafish
Published: Vol 11, Iss 17, Sep 5, 2021 DOI: 10.21769/BioProtoc.4149 Views: 7053
Reviewed by: Chiara AmbrogioJohn W PetersonValerian DORMOY
Original research article
The authors used this protocol in:

Oct 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Skeletal muscles generate force throughout life and require maintenance and repair to ensure efficiency. The population of resident muscle stem cells (MuSCs), termed satellite cells, dwells beneath the basal lamina of adult myofibres and contributes to both muscle growth and regeneration. Upon exposure to activating signals, MuSCs proliferate to generate myoblasts that differentiate and fuse to grow or regenerate myofibres. This myogenic progression resembles aspects of muscle formation and development during embryogenesis. Therefore, the study of MuSCs and their associated myofibres permits the exploration of muscle stem cell biology, including the cellular and molecular mechanisms underlying muscle formation, maintenance and repair. As most aspects of MuSC biology have been described in rodents, their relevance to other species, including humans, is unclear and would benefit from comparison to an alternative vertebrate system. Here, we describe a procedure for the isolation and immunolabelling or culture of adult zebrafish myofibres that allows examination of both myofibre characteristics and MuSC biology ex vivo. Isolated myofibres can be analysed for morphometric characteristics such as the myofibre volume and myonuclear domain to assess the dynamics of muscle growth. Immunolabelling for canonical stemness markers or reporter transgenes identifies MuSCs on isolated myofibres for cellular/molecular studies. Furthermore, viable myofibres can be plated, allowing MuSC myogenesis and analysis of proliferative and differentiative dynamics in primary progenitor cells. In conclusion, we provide a comparative system to amniote models for the study of vertebrate myogenesis, which will reveal fundamental genetic and cellular mechanisms of MuSC biology and inform aquaculture.
Graphic abstract:
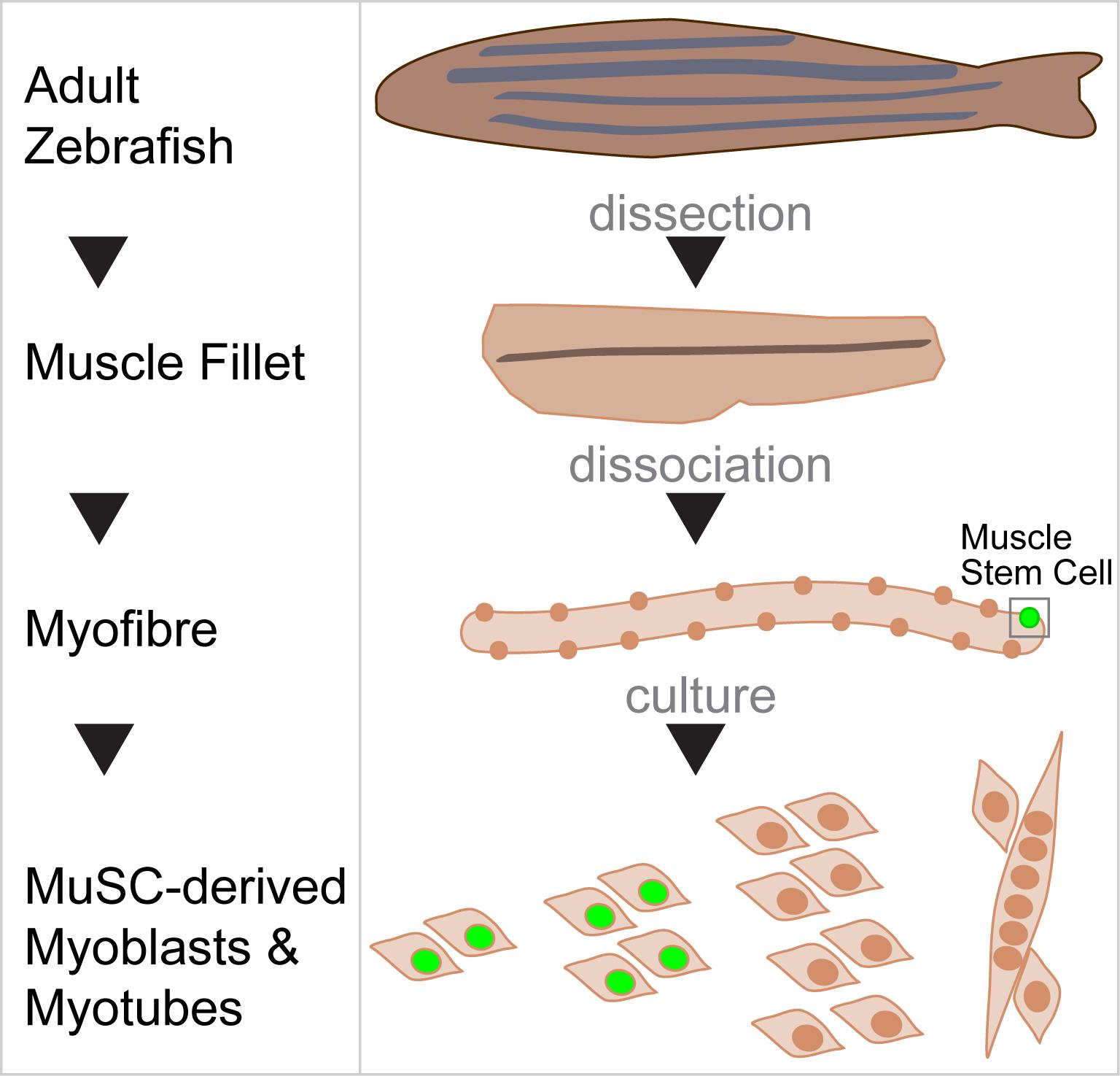
Schematic of Myofibre Isolation and Culture of Muscle Stem Cells from Adult Zebrafish.
Background
Skeletal musculature provides lifelong body support and movement through coordinated contraction of myofibres, highly specialised syncytial cells containing hundreds of post-mitotic myonuclei. Myofibres constantly adapt to both exogenous and endogenous stimuli in part thanks to resident muscle stem cells (MuSCs), also known as satellite cells, located underneath the basal lamina of most myofibres (Katz, 1961; Mauro, 1961; Relaix and Zammit, 2012; Purohit and Dhawan, 2019). In response to exercise or damage, quiescent MuSCs quickly activate and become muscle progenitor cells (MPCs) called myoblasts, which proliferate, differentiate and fuse either to pre-existing multinucleated myofibres or to one another to form new myofibres (Fukada et al., 2020). Isolation of rodent myofibres and culture of associated MuSCs is a well-established tool to explore muscle stem cell biology, providing not only an understanding of MuSC behaviour and regulation of quiescence, activation, proliferation, self-renewal and differentiation (Zammit et al., 2004), but also yielding insights into both embryonic muscle development and adult myofibre growth and maintenance (Buckingham and Relaix, 2015). However, mechanisms underlying murine MuSC biology may not fully resemble those found in human myogenesis. Therefore, alternative vertebrate models to study adult/MuSC myogenesis are desirable to consolidate findings of ancestral mechanisms in vertebrate muscle. Notably, despite substantial overlap of molecular pathways shared in myogenesis between zebrafish and amniotes (Hammond et al., 2007; Hinits et al., 2009 and 2011; Ganassi et al., 2018; Osborn et al., 2020), the complementary study of adult muscle homeostasis in zebrafish is limited to mechanical trituration of bulk muscle, hindering the purity of myogenic yield (Alexander et al., 2011; Froehlich et al., 2014). Adult myofibre isolation in fish has permitted the study of their physical or contractile properties (Johnston and Altringham 1988; Davies 1995; Johnston et al., 2004), and some studies have more recently exploited it to investigate zebrafish MuSC biology (Anderson et al., 2012; Zhang and Anderson, 2014; Ganassi et al., 2018 and 2020).
Here, we describe how to isolate single viable myofibres and associated MuSCs through enzymatic digestion and fine trituration of the trunk musculature of adult zebrafish. This method is adapted from the standard mouse protocol (Bischoff, 1975; Cardasis and Cooper, 1975a and 1975b; Bekoff and Betz, 1977a and 1977b; Rosenblatt et al., 1995; Moyle and Zammit, 2014). We explain how to plate myofibres to study MuSC activation and progression through myogenesis in vitro, ensuring a virtually pure myogenic population (Ganassi et al., 2018 and 2020). Our protocol provides an appropriate toolbox for comparative analysis of adult myogenesis across vertebrates and has been recently developed and used to explore the function of the transcription factor Myogenin in adult MuSC activation, proliferation and differentiation (Ganassi et al., 2018 and 2020). Myofibre isolation, culture, and analysis from adult fish exploits the advantages of the zebrafish model, such as the spatial segregation of slow and fast myofibres that facilitates fibre-type specific studies (Blagden et al., 1997; Pipalia et al., 2016; Hromowyk et al., 2020), and provides insight useful to aquaculture. As an alternative to the classical rodent procedure, the analysis of fish MuSC also offers an independent benchmark to verify genetic and cellular mechanisms identified using rodent models. Therefore, application of our techniques to adult zebrafish muscle has the potential to contribute to understanding genetic, molecular, and cellular mechanisms maintaining and adapting human musculature.
The method is simple, efficient, and cost-effective and permits the study of 1) myofibre characteristics ex vivo, 2) MuSC-derived myoblasts/myotubes ex vivo and 3) mechanisms of adult muscle formation, development, and maintenance.
Materials and Reagents
Materials required for dissection and dissociation of adult muscle
Deep Petri dishes (150 mm and 100 mm) sterile, cell culture grade (Corning, catalog numbers: 430599 and 430167)
Glass Pasteur pipettes (22 cm), sterile (Volac, catalog number: D812)
0.45 μm and 0.2 μm sterile syringe filters (ThermoFisher, catalog numbers: 15216869 and 15206869)
Sterile syringe, 50 ml (Terumo, catalog number: SS+50ES1)
Aluminium foil
Bijou tubes, 7 ml
Tricaine methanesulfonate (MS-222) solution (Sigma-Aldrich, catalog number: E10521)
70% Ethanol solution (in deionised water) (70% EtOH) (Ethanol absolute; Sigma-Aldrich, catalog number: 1024282500)
5% Bovine serum albumin (BSA) (powder, Sigma-Aldrich, catalog number: A7906)
Collagenase from Clostridium histolyticum (Sigma-Aldrich, catalog number: C0130)
Dulbecco’s modified Eagle’s medium (DMEM), high glucose, GlutaMAX, Pyruvate (ThermoFisher, catalog number: 31966)
Phosphate-buffered saline Ca2+ and Mg2+ free (PBS), sterile (Oxoid, catalog number: BR0014G)
Penicillin and Streptomycin solution (Sigma-Aldrich, catalog number: P0781)
1% Virkon solution (in deionised water) (powder, 3S Healthcare) (see Recipes)
P/S-PBS (see Recipes)
BSA-PBS (see Recipes)
cDMEM (complete DMEM) (see Recipes)
Collagenase-cDMEM (see Recipes)
Materials required for myofibre and MuSC-derived cell immunolabelling
Cover glasses 50 mm × 22 mm (Academy, catalog number: 400-04-17)
Glass Slides (Fisher, catalog number: 1157-2203)
Crystal-clear plastic microcentrifuge tubes, 2 ml (Starlab, catalog number: S-1620-2700)
Transparent nail varnish
Paraformaldehyde (PFA) solution, 4% in PBS (PFA-PBS) (Alfa Aesar, catalog number: J61899)
Liquid blocker super pap pen (Pyramid Innovation, catalog number: R62002-E)
Triton X-100 detergent solution (Sigma-Aldrich, catalog number: X100)
Chicken anti-GFP (RRID:AB_300798; Abcam, catalog number: 13970; use 1:400)
Goat anti-chicken IgY (H+L), Alexa Fluor® 488 (RRID:AB_2534091; Thermo Fisher Scientific, catalog number: A11032, use 1:1000)
Hoechst 33342 solution (ThermoFisher, catalog number: H3570, use 1:1000)
Normal goat serum (NGS) (Agilent, catalog number: x0907)
Glycerol-based mounting medium (Agilent, catalog number: 50001)
PBSTx (see Recipes)
Materials required for myofibre and MuSC-derived cell culture
24-well plates cell culture grade (ThermoFisher, catalog number: 142475)
Fetal bovine serum (FBS), heat inactivated (ThermoFisher, catalog number: 10500-064)
Horse serum (HS) (ThermoFisher, catalog number: 26050088)
Matrigel (Corning, catalog number: 354263)
5-ethynyl-2’-deoxyuridine (EdU) solution (From Click-iT EdU kit; ThermoFisher, catalog number: C10646)
Gentamicin (Gibco, catalog number: 15750-060)
Matrigel solution (see Recipes)
Proliferation Medium (PM) (see Recipes)
Differentiation Medium (DM) (see Recipes)
Equipment
Dissection and dissociation of adult muscle
Tissue culture hood or lamina flow cabinet
Tissue culture incubator (humidified, 28.5°C, 5% CO2)
Cork dissection board (IKEA, catalog number: 870.777.00)
Dissection metal pins
Fine forceps, one pair (Idealtek, No. 5A.s)
Sterile disposable scalpels No. 10 (Swann-Morton, catalog number: 0501)
Bunsen burner
Diamond-tipped pen (VWR, catalog number: 201-0392)
Dissection microscope with transmission illumination (Zeiss Stemi SV6 and Leica M50)
Software
Image Analysis: Fiji; NIH (www.Fiji.sc)
Data presentation: GraphPad Prism 8 (https://www.graphpad.com/scientific-software/prism/)
Procedure
Muscle Dissection
Where possible, perform steps under sterile conditions in a tissue culture hood or laminar flow cabinet.
Euthanise the fish by immersion in ice-cold 0.3 mg/ml tricaine solution. Immerse fish in chilled tricaine solution aliquoted into a 50 ml tube for the required amount of time. To minimise animal distress, keep the tube on ice during incubation. Please note that the duration of tricaine incubation must be determined empirically, depending on fish size and age as described before (Westerfield, 2000; Harper and Lawrence, 2011) (see Note 1).
Remove fish carcass from tricaine solution and immerse it in 25 ml of 1% Virkon (see Recipe 1) solution in a 100 mm dish. Incubate for 5 min to kill bacteria and fungi.
Use clean forceps to transfer the fish carcass to a new 100 mm dish containing 25 ml P/S-PBS (see Recipe 2) and incubate for 5 min.
Transfer fish carcass into a new empty 100 mm Petri dish. Use a disposable scalpel to remove scales. To increase scaling efficiency, position the blade perpendicular to the antero-posterior axis of the fish body and gently scrub the skin surface from tail to head (see Note 2).
Wash the descaled carcass into a new 100 mm dish containing 25 ml of fresh P/S-PBS for 5 min. Meanwhile, carefully wipe dissection metal pins, corkboard and fine forceps with 70% EtOH to reduce chances of contamination.
Move fish carcass to a new 100 mm dish, gently dry residual P/S-PBS with cloth and spray with 70% EtOH on both sides.
Move fish to the dissecting corkboard and place one pin passing through tissue just behind the gill operculum and a second pin penetrating the tissue just anterior to the base of the caudal fin (Figure 1A).
Use the scalpel to cut fins as close as possible to the fish body. Removed fins can be processed to extract genomic DNA for fish genotyping (Figures 1A and 1B) (see Note 3).
Make a curved incision along the ventral side of the carcass to facilitate evisceration using blade and fine forceps (Figure 1A).
At this point, different portions of the carcass can be collected for required analyses. As indicated in Figure 1B: i) fins are useful for retrospective genotyping, ii) the muscle region near the tail tip is usually damaged by the dissecting pin but can be used for whole muscle RNA/Protein analysis, iii) the adjacent 5 mm section of muscle can be cryopreserved for histological analysis and iv) most of the trunk musculature is processed for myofibre isolation.
Use the scalpel to make a light incision on the skin just behind the gill operculum and perpendicular to the antero-posterior axis, carefully avoiding incision of the muscle beneath. Use the fine forceps to gently pinch and lift the skin along the incision edge. Carefully grab and pull the skin toward the fish tail to expose the underlying muscle (Figure 1C).
Continue to pull gently until reaching the pin positioned close to the tail (Figure 1C). Slow muscle is strongly attached to the overlying skin, so pull very gently to avoid damaging the slow myofibres. Most of the trunk musculature should now be exposed.
Use the same procedure to remove skin from the contralateral side.
When skinning is completed, unpin the fish and rotate it 90° onto its back so that the ventral side (belly) points upward towards the operator (Figures 1D-1F).
Re-pin the fish to the corkboard in the new position, using one pin passing through the lower jaw and head and the second at the base of the tail. The vertebral column should be visible and accessible through the opening in the belly (Figures 1E and 1F).
Use the blade to cut on the right of the vertebral column along the entire antero-posterior axis to create two muscle fillets, one bearing the associated vertebral column and spinal cord and the other without. It is important to angle the scalpel so that its tip points toward the dorsal midline, penetrating the anterior-most part of the muscle tissue close to the vertebral column (Figure 1E). Draw the blade posteriorly until the tail pin is reached, leaving the ribs in the fillet (Figure 1E’). Stopping or hesitating whilst cutting along the column can lead to varying fillet thickness and damage the medial-most muscle.
Use scalpel to remove the fish head and fully release the two muscle fillets. The fish fillets display slow and fast muscle compartments (Figure 1G). The spinal cord should be visible in the right fillet and can be removed with the scalpel, but this is not essential. We usually do not remove it to reduce possible damage to the surrounding muscle tissue.
Muscle dissection should require 30 min and can be performed on multiple fish in parallel.
Myofibre Dissociation and Isolation
Rinse one 150 mm and two 100 mm new sterile Petri dishes per fish with BSA-PBS solution (see Recipe 3) to prevent myofibre adhesion to the dish. Remove excess BSA-PBS solution and add 25 ml and 10 ml of complete DMEM (cDMEM, see Recipe 4) to the 150 mm and 100 mm dishes, respectively. Place dishes in a 28.5°C 5% CO2 incubator for at least 30 min to allow the cDMEM to warm.
Place the freshly dissected fillets in the bijou tube with Collagenase-cDMEM solution (see Recipe 5 and Note 4), apply cap loosely and incubate at 28.5°C in 5% CO2 incubator for 120 min with occasional (every 30 min) very gentle swirling of the tube (Figure 1H).
Meanwhile, use a diamond pen to score two glass Pasteur pipettes per fish and create openings with diameters of approximately 1 and 3-4 mm, respectively (Figure 1I). Use a Bunsen burner to melt the glass around the opening to smoothly polish any sharp edges (Figure 1I and Video 1). Test the polishing by circling the pipette edge on aluminium foil. No cut/tear should be produced. Quickly flame the prepared glass pipettes to sterilise, wrap in aluminium foil and store in the tissue culture hood until use.
 Video 1. Glass Pasteur pipette cut and heat-polish process
Video 1. Glass Pasteur pipette cut and heat-polish processWhen incubation is complete, place the bijou tube in the tissue culture hood. A well-digested muscle looks slightly swollen and, under the microscope, hair-like myofibres appear dislodged around the edge of the muscle mass (Figures 1J and J’ and see Note 5). Also collect the 150 mm dish with warm cDMEM from the incubator and place it in the culture hood.
Gently decant and discard most of Collagenase-cDMEM solution from the bijou tube. Rapidly invert the bijou tube to pour the muscle fillets into the 150 mm Petri containing cDMEM. Return the Petri dish with fillets to the incubator for 20-30 min. This allows the muscle to rest and dilute the Collagenase, promoting inactivation of the enzyme.
Place the dissecting microscope in the culture hood, if possible; otherwise, use a clean area away from doors, windows and draughts or other contamination sources. Collect the 150 mm dish with fillets and place under the lens of the microscope.
Rinse the heat-polished glass pipettes with BSA-PBS solution to prevent myofibre adhesion.
Using the pipette with the larger diameter (~3-4 mm), direct cDMEM onto the fillets repeatedly for at least 10 min, pipetting up and down to expel a continuous stream of liquid (Figure 1K). Tissue dissociation can be enhanced by carefully passing the fillets once or twice in and out of the glass pipette, but not continuously as this will damage the myofibres and reduce the final yield. Myofibres are visible hair-like structures that will be released from the muscle bulk (Figures 1K’ and 1L).
Continue the trituration process until most myofibres have been released. The procedure will also result in the release of debris, including fat droplets and hypercontracted myofibres (Figure 1L), which will increase the turbidity of the medium. If trituration is prolonged, allow a further 5-10 min incubation at 28.5°C, 5% CO2 to re-equilibrate the temperature and pH of the medium. If medium reaches below the range of physiological temperatures (22-29°C) for an extended time, myofibers will hypercontract and die.
Place the 150 mm plate back in the incubator for 10-15 min to allow released myofibres to rest and sink to the bottom.
Using the glass pipette with the smaller diameter (~1 mm), carefully collect intact myofibres and transfer them onto a 100 mm dish with fresh cDMEM (Figure 1K’). If needed, the remaining muscle bulk can be further processed to enhance the release of residual myofibres, but not longer than 30-60 min as this will reduce the viability of residual myofibres.
Place the 100 mm dish containing cleaned myofibres back in the incubator for another 10-15 min to allow them to rest and sink to the bottom.
Viable myofibres appear translucent and with a smooth surface (Figure 1M). If needed, myofibres can be transferred into a new cDMEM-containing 100 mm dish to clean further. If significant debris is still present in the dish, repeat Steps B11 and B12.
The entire muscle dissection procedure should require 180 min.
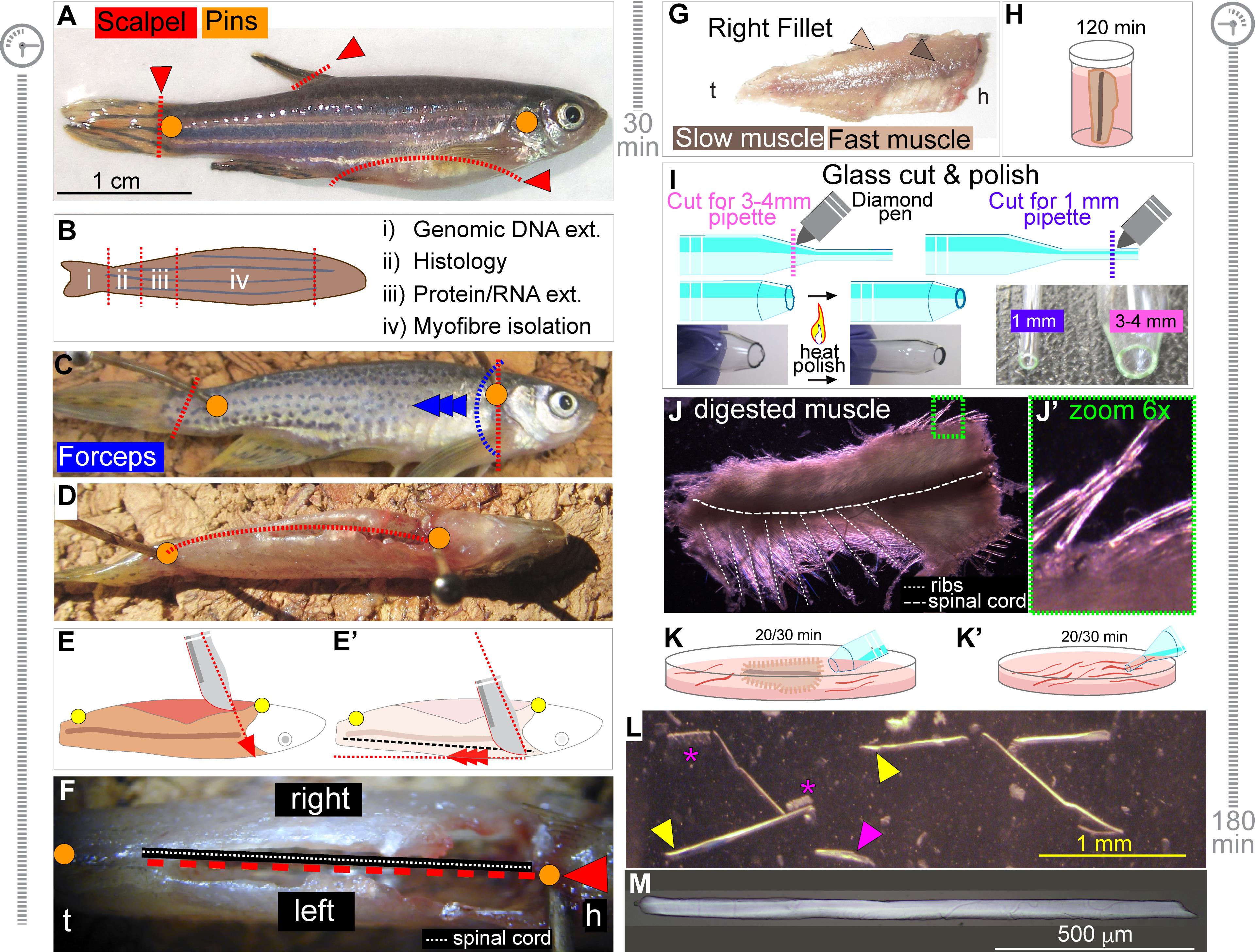
Figure 1. Dissection and Isolation of Myofibres from Adult Zebrafish. A. Representative picture of an 8-month-old adult zebrafish depicts pin positioning (orange dots) to anchor fish carcass to the dissecting board. Red dashed lines indicate cuts to remove fins and to perform ventral incision for evisceration. B. Summary of analysis performable from different portions of the fish (ext; extraction). C. Blue dashed line and arrowheads indicate position of the skin incision, pinch and pulling direction for skinning with forceps. D. After skinning, 90° rotation of the carcass on its dorsal side exposes the ventral incision upward. Red dashed line and orange dots indicate cut direction and pin positioning, respectively. E, E’. Diagram of scalpel angle and cut direction during fish filleting. Red dashed arrows indicate inclination toward the dorsal midline (E) and cut direction towards tail pin (E’). Dashed black line shows position of spinal cord (E’). F. View of the ventral incision upward. Dashed red line and arrow indicate cut direction for filleting, with the spinal cord used as a guide (dashed white line). Antero-posterior orientation is indicated (h; head, t; tail). G. Slow (dark arrowhead) and fast (light arrowhead) muscle portions are visible in the dissected fillets (h; head, t; tail). H. Fillet is incubated with Collagenase-cDMEM solution for 120 min in the bijou tube at 28.5°C. I. Diagram of Pasteur glass pipettes cut and heat-polished to obtain two pipettes with wide (3-4 mm, pink) and small diameter (1 mm, purple) apertures, for muscle trituration and single muscle fibre handling, respectively. Purple (cut for 1 mm opening) or pink (cut for 3-4 mm opening) dashed lines indicate cut position on each pipette with diamond pen and heat polishing with a flame. Desired result for cut and edge heat-polish is shown in bottom pictures (see Video 1). J, J’. Representative images of muscle fillet after 120 min incubation in Collagenase-cDMEM solution (J). Note the hair-like myofibres dislodged around the edge of the muscle mass (J’, zoomed area in green). Position of ribs and spinal cord is also indicated. K, K’. Schematic of fillet trituration (K) and single myofibre isolation and wash (K’) with estimated duration in minutes. L. Representative images of single myofibres during washes. Yellow arrowheads denote intact viable myofibres, while the magenta arrowhead and asterisks indicate damaged or hypercontracted myofibres, respectively. Estimated time for fish preparation/muscle dissection (C-F) and myofibre dissociation/isolation (G-J) are reported beside panels. M. Representative single viable myofibre following muscle dissection and isolation.
Analysis of Isolated Myofibres
Isolated myofibres are now ready for analysis, such as morphometrical measurements. Myofibres can be photographed prior to fixation using a brightfield microscope, which allows exclusion of those that are hypercontracted or damaged. Depending on the microscope used, damaged myofibres that are not yet fully hypercontracted appear shorter and more opaque, with a rough and irregular surface (Figure 1L).
Nuclear counting and subsequent analyses may require myofibre fixation. Under the microscope, use the BSA-PBS pre-rinsed glass pipette with smaller diameter (Figures 1I and 1K’) to collect isolated myofibres and place them in a 2 ml clear round-bottomed microcentrifuge tube that has been rinsed with BSA-PBS to prevent myofibre adhesion. Gently swirl the dish to gather all myofibres at the centre of the plate to reduce the volume of cDMEM medium collected with myofibres. We suggest limiting the number of myofibres to 40 per tube to avoid clumping and possible damage.
Leave the collection microcentrifuge tube standing upright for 5 min at room temperature to allow the myofibres to sink to the bottom of the tube.
Carefully remove the medium above the myofibres with a pipette and replenish the tube with 1 ml of 4% PFA in PBS solution (PFA-PBS) by gentle trickling down the side of the tilted tube. Incubate for 10-15 min at room temperature.
Remove PFA-PBS solution and gently replenish with 1.5 ml of PBS to wash the myofibres. Incubate for 5 min and repeat the wash with fresh PBS. Lysine or BSA may be added to more efficiently inhibit the PFA. Fixed myofibres can be stored at 4°C for at least 2 weeks.
Remove PBS, wash and replace with freshly prepared Hoechst 33342 dye solution diluted in PBS to stain myofibre nuclei. Incubate for 15 min and replace with fresh PBS (as in Step C5). Myofibres are now ready to be mounted on glass slides for detailed analysis.
Use a water-repellent pap pen to outline a rectangular area (size depending on the size of the coverslip, e.g., 50 mm × 22 mm) on several glass slides. Under the microscope, use a clean smaller diameter pipette, pre-rinsed with BSA-PBS to collect myofibres from the 2 ml tube and transfer them onto the prepared glass slide. We suggest limiting to 10/15 myofibres per slide to facilitate handling.
Remove as much PBS as possible from the glass slide with a pipette to ease myofibre adhesion and reduce the risk of damage and/or loss. A 200 µl micropipette tip wrapped in aluminium foil and pre-immersed in BSA-PBS can be used to carefully reposition myofibres after PBS removal. We suggest being quick as residual liquid, along with immersed myofibres, will dry out rapidly.
Place two/three drops of glycerol-based mounting medium on the glass slide, position a 50 mm × 22 mm coverslip with an edge on the slide touching glycerol and gently lower the coverslip to avoid trapping air bubbles that can mis-position or sweep away myofibres. Wait 5 min to allow the mounting medium to spread beneath the coverslip.
Seal the coverslip and secure to the glass slide by brushing on a small amount of nail varnish, first at the corners and then seal the edges.
Mounted myofibres can be photographed using an epifluorescence or confocal microscope.
Using imaging software, measure myofibre width (diameter) at a minimum of two different positions along each myofibre. Concomitantly, measure myofibre length and count the number of nuclei using Hoechst (Figure 2A).
Myofibre volume can be calculated with the formula reported in the Data analysis section and Figure 2A. An example of myofibre volume in Myog+/- adults from Ganassi et al. (2020) is shown (Figure 2B). Alternatively, confocal scanning may give full myofibre morphology profile (see Ganassi et al., 2020).
Myofibre Immunolabelling
Fixed myofibres (Step C5) can be processed for immunolabelling. Here, we deploy the transgenic fish TgBAC(pax7a:GFP)t32239Tg (Nüsslein-Volhard C.; MPI Tübingen) (Mahalwar et al., 2014) and provide a template immunolabelling protocol using anti-GFP antibody to detect MuSCs. Although GFP fluorescence encoded by the pax7a:GFP transgene does resist PFA fixation, we suggest enhancement using the anti-GFP antibody, especially if co-labelling with multiple antibodies and fluorophores (Ganassi et al., 2020).
Remove the PBS from the fixed myofibres with the BSA-PBS pre-rinsed smaller-diameter glass pipette and replace with 0.5% Triton-X100 detergent in PBS (PBSTx; see Recipe 6). Incubate for 15 min to permeabilise the cell membranes of both myofibres and associated MuSCs.
Remove PBSTx and gently add a blocking solution of 10% normal goat serum (NGS) in PBS to block non-specific antibody binding. Incubate for at least 30 min, occasionally tilting the tube (see Note 6). Alternatively, 5% NGS in PBS solution can be used to incubate for 1 h.
Prepare antibody solution by diluting the anti-GFP primary antibody in 0.1% Triton-X100 detergent PBS solution (PBSTx0.1) containing 2% NGS. Remove blocking solution from tube and gently add the primary antibody solution. Incubate overnight (16 h) at 4°C.
Remove the primary antibody solution and replace with fresh PBSTx0.1 to wash myofibres for 5 min (see Note 7). Primary antibody solution can be stored at 4°C and re-used reliably within one week (perhaps longer if 0.002% sodium azide in PBS is added).
Wash myofibres three times for 5 min each using PBSTx0.1 with occasional gentle tilting of the tube.
Dilute fluorochrome-conjugated (e.g., Alexa Fluor 488) secondary antibodies and Hoechst 33342 dye solution (10 µg/ml final) in PBSTx0.1 and incubate for at least 60 min at room temperature, protected from light, with occasional tube swirling. Secondary antibody solution can be stored at 4°C in the dark and re-used reliably within one week or longer if 0.002% sodium azide in PBS is added.
Transfer myofibres onto a prepared glass slide and mount under a coverslip as described in Steps C7-C11. An example of a pax7a:GFP MuSC on a myofibre immunolabelled for GFP is shown in Figure 2C and can be found in Ganassi et al. (2020). Store slides at 4°C in the dark; GFP fluorescence lasts for up to 14 days.
Myofibre-derived MuSC Culture and Immunolabelling
Coat the desired number of wells of a 24-well plate by rinsing with Matrigel solution (see Recipe 7). Be sure to completely cover the surface of each well. Immediately remove excess solution using a sterile pipette and return the Matrigel solution to 4°C to avoid precocious gelling. Place the prepared plate in a 28.5°C 5% CO2 incubator for 30-45 min to allow Matrigel gelling.
Prepare the proliferation medium (PM; see Recipe 8) and pre-warm at 28.5°C in the incubator prior to aliquoting 200 µl per Matrigel-coated well.
Transfer myofibres with a pipette into a new 100 mm dish containing 5 ml of 40% FBS-cDMEM solution. Each transfer of myofibres leads to concomitant carry-over of nearly 150-200 µl of cDMEM from the original dish that dilutes the serum concentration. At the end of the transfer, the final 100 mm dish now contains about 10 ml of DMEM with approximately 20% FSB, thus mimicking PM.
Gently swirl the 100 mm dish to gather myofibres at its centre. Use the small diameter glass pipette BSA-PBS pre-rinsed to transfer approximately 90-100 freshly isolated myofibres into each Matrigel-coated well. Ensure that the myofibres are evenly spaced across the well by placing the plate on a flat surface and moving laterally in cross-like orthogonal directions several times.
Place the 24-well plate(s) in the incubator and culture the myofibres undisturbed for at least 48 h. During the initial 24 h, myofibres can easily be dislodged, impacting MuSC activation, proliferation, migration and adhesion to the culture plate. Even opening/closing the incubator door can cause vibrations that dislodge myofibres.
While a fraction of MuSCs activates within 24 h after plating, migrate off the myofibre and adhere to the Matrigel coating (see Note 8), we suggest waiting 48 h before analysis on MuSC. MPCs/myoblasts can be followed in culture and assayed for proliferation or differentiation capacity over time (Figure 2D).
At 48 h, cells can be immunolabelled with anti-Desmin antibody to confirm the purity of the myogenic population. Myofibre plating usually yields almost pure myogenic progenitors in culture (Figure 2E). The original data is presented in Figure 4-figure supplement 1 (Ganassi et al., 2020), and a similar calculation can be found in Supplementary Figure 6g (Ganassi et al., 2018). Cell fixation is described below (Step E12).
Cells can be collected at desired time point(s) for RNA extraction and gene expression analysis by RT-qPCR (see Note 9 and Figure 4 in Ganassi et al., 2020).
Analysis of MuSC proliferation using 5-ethynyl-2′-deoxyuridine (EdU) is best performed no earlier than 2 days (48 h) from myofibre plating. Dilute EdU to a final concentration of 10 µM in fresh pre-warmed PM (EdU-PM).
Remove PM from culture well and rinse vigorously twice with freshly prepared PM to remove plated myofibres. At this time point, most myofibres should be either floating or loosely adhering to Matrigel and thus easily removed.
Remove PM and quickly rinse twice with PBS. Replace PBS with EdU-PM solution and place for 2-8 h in 28.5°C 5% CO2 incubator. Duration of incubation can be changed according to the experimental design; the example shown here refers to 8 h treatment.
At the end of incubation, remove EdU-PM solution, wash vigorously twice with PBS and fix with PFA-PBS (4% PFA in PBS) for 15 min. If next step is immunolabelling, continue as described above for myofibres (Steps D2-D6).
After the final wash in PBSTx0.1, perform click chemistry to reveal EdU incorporation following the manufacturer’s instructions.
Remove PBS, wash and replace with freshly prepared Hoechst 33342 dye solution diluted in PBS to stain nuclei. Incubate for 15 min.
Wash twice with fresh PBS, then replenish each well with 300 µl of PBS. The cells are now ready to be visualised using an inverted epifluorescence microscope (Figure 2F). Cells can be stored at 4°C (2-4 weeks) by replacing PBS with PBS containing 0.002% sodium azide to prevent microbial growth.
Dynamics of MuSC/MPC proliferation can be followed over time, as reported in Figure 2F (adapted from Figures 4E and 4F [Ganassi et al., 2020]).
Zebrafish myoblasts can also be induced to differentiate to evaluate the myogenic program. After 96 h from initial plating, remove PM and wash twice with sterile PBS to eliminate serum residues. Remove PBS and add 500 µl of differentiation medium (DM; see Recipe 9) to each well. Replace DM every 48 h. We previously assessed myogenic differentiation by culturing zebrafish primary myoblasts in low serum medium for 5 days prior to immunolabelling for structural components such as Myosins (Figure 2D). Figure 2G shows myotubes differentiated for 5 days and co-immunolabelled for myosin heavy chain (MyHC) using MF20 and A4.1025 concomitantly (Blagden et al., 1997) and counterstained with Hoechst 33342 to assess differentiation and cell fusion (original data is in Figure 8 in Ganassi et al., 2018) and Figure 4 - Supplement 1E (Ganassi et al., 2020).
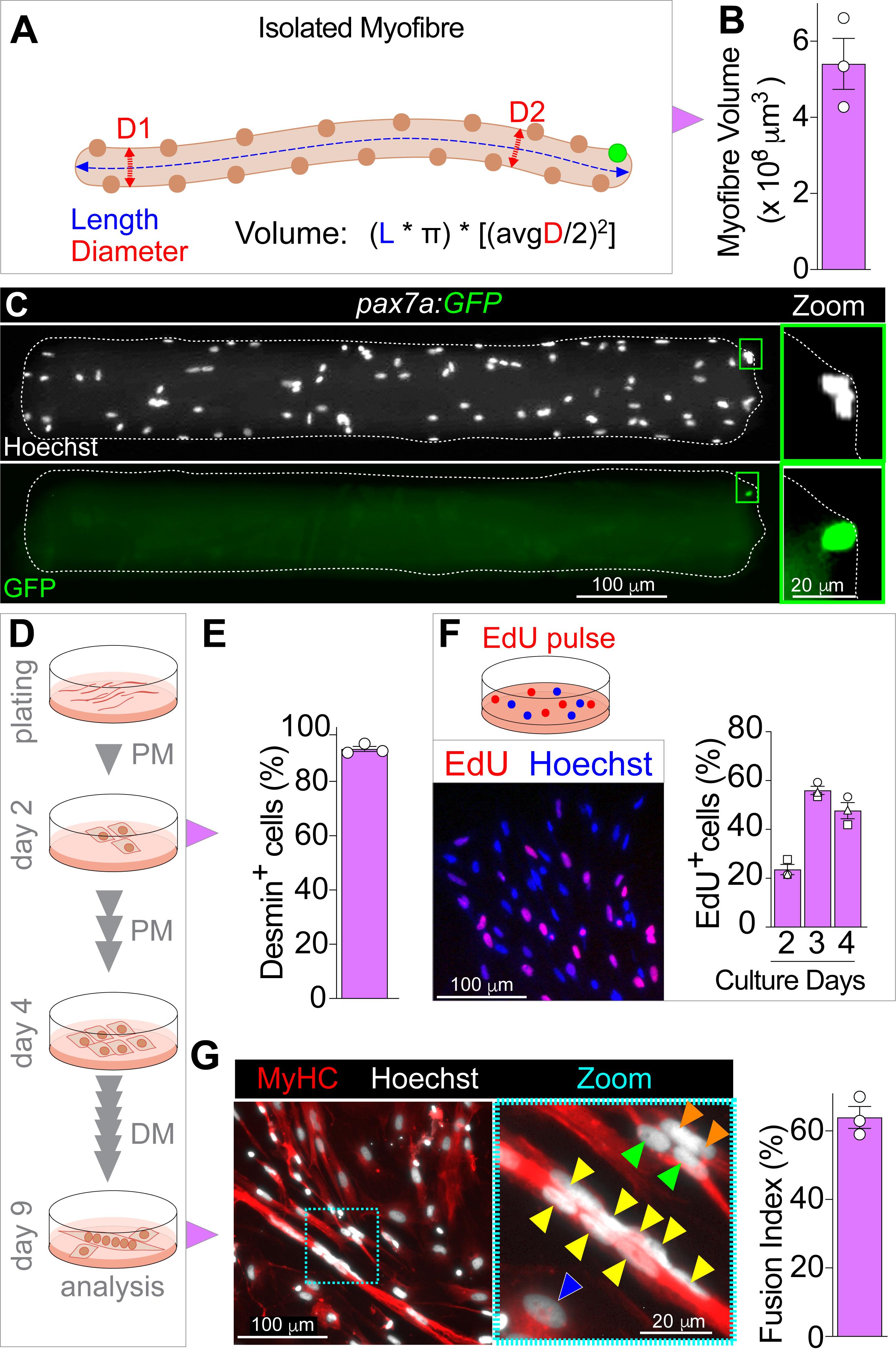
Figure 2. Analysis of Isolated Myofibres and Cultured MuSCs. A. Diagram of myofibre average diameter (myofibre width, red arrowed dashed line) and unit length (blue arrowed dashed line) measurements to calculate myofibre volume. Avg, indicates arithmetic mean of diameter measure across myofibre length (D1 and D2). B. Example results of 8-month-old adult myofibre volume (adapted from Ganassi et al., 2020). Symbols represent average values from 20-30 myofibres from each of three different fish. C.TgBAC(pax7a:GFP)t32239Tg (pax7a:GFP) myofibre immunolabelled for GFP (green) reveals the position of a MuSC (green rectangle, magnified at right) near the myofibre-end and myofibre nuclei counterstained with Hoechst 33342 (white). D. Schematic of myofibre plating, MuSCs/myoblasts proliferation (PM; proliferation medium), expansion and myotube differentiation protocol (DM; differentiation medium) with indicated timing, medium change and analysis. E. Example calculation of fraction of cells that were Desmin+ (myoblasts) two days after myofibre plating. F. MuSC-derived myoblasts can be EdU pulsed (red) two days after myofibre plating in proliferation medium. Nuclei were counterstained with Hoechst 33342 (blue). G. Representative image of differentiated multinucleated myotubes or mononucleated cells containing MyHC (red) after 5 days of culture in differentiation medium, coloured arrowheads indicate nuclei within the same cell (from Ganassi et al., 2020). Cyan dashed rectangle indicates the magnified area in the right panel. Nuclei were counterstained with Hoechst 33342 (white). Example results of fusion index (adapted from Ganassi et al., 2018). All graphs report mean ± SEM, and symbols represent biological replicates.
Data analysis
Use the measured average myofibre width (avg diameter) and length to calculate the volume of the myofibre following the formula:
[(Length × 𝜋) × [(average Diameter/2)2] (Figure 2A),
where ‘average’ is the arithmetic mean of diameter measurements at a minimum of two different positions along the myofibre length. Values can be compared with chosen statistical analysis (e.g., unpaired two-tailed t-test). Graphs were produced in GraphPad Prism 8 (see Software).
Myofibre volume and number of nuclei can be combined to calculate the myonuclear domain using the formula: [(Length × 𝜋) × [(average Diameter/2)2]/Number of myofibre nuclei]. Alternatively, average myofibre width and length can be used to calculate the surface area of the myofibre following the formula: Length × 𝜋 × avg Diameter. The Surface Area Domain Size (SADS), the notional SA occupied by each myofibre nucleus, is calculated using the formula: SA/number of myofibre nuclei. Examples of the calculation are available in Ganassi et al. (2020) or in Brack et al. (2005) for mouse myofibres.
Notes
For zebrafish weighing 0.3-0.6 g, euthanasia is usually reached within 5-10 min after incubation in ice-cold 0.3 mg/ml tricaine solution. However, this is only indicative timing and must be determined empirically and according to local guidelines.
It is essential to wash and thoroughly sterilise equipment and dissecting tools to avoid microbial contamination.
Removing fish fins is not essential but facilitates handling and reduces the risk of microbial contamination.
It is important to batch-test replacement reagents, such as Collagenase, against existing, optimised components. There are variable amounts of proteases in batches of Collagenase, but Collagenase with neutral protease around 53 U and clostripain at approximately 0.6 U is ideal, as described before (Rosenblatt et al., 1995).
Adult (8-15 months old) zebrafish trunk muscle is usually digested after 2 h. Although longer incubations (≥ 3 h) have a marginal effect on myofibre viability, shorter incubation may reduce digestion efficiency, hindering the isolation of viable myofibers. The precise time depends upon both the age and size of the fish and the activity of the batch of Collagenase used and should be determined empirically.
The normal serum used for blocking should derive from the species in which the secondary antibody was raised.
Myofibres can be stained with fluorochrome-conjugated toxins to detect the subcellular structure, such as filamentous actin (F-actin) using Phalloidin (ThermoFisher, A12379) or neuromuscular junction (acetylcholine receptor) using α-Bungarotoxin (ThermoFisher, B35451).
If myofibres/MuSCs are to be cultured for longer periods, replace half the volume of the medium with fresh medium every 48 h.
For gene expression analysis, collect cells by removing medium and washing twice with PBS. Incubate with the appropriate volume (e.g., 200 µl for a 24-well plate well) of Accutase® reagent to detach the cells from Matrigel for 10 min (or until complete detachment of all cells; check under a microscope, but this should not take longer than 15 min) at 28.5°C, 5% CO2 (see Ganassi et al., 2020). Collect cells in a 1.5 ml clear tube, pellet by centrifugation at 200 × g at 4°C and wash once in PBS. Pelleted cells are now ready for RNA extraction and analysis or can be stored at -80°C.
Recipes
1% Virkon
Prepare 1% Virkon by dissolving 5 g of powder in 500 ml of deionised sterile water.
1% Virkon solution can be aliquoted and stored at -20°C for at least 2 months. Virkon powder can be stored at room temperature.
P/S-PBS
Prepare 10% vol/vol Penicillin/Streptomycin solution in PBS (P/S-PBS) and aliquot 25 ml into a 100 mm plastic Petri plate.
BSA-PBS
Prepare 5% BSA solution in sterile PBS (BSA-PBS) and heat-inactivate at 60°C for 60 min before filtering through a 0.45 μm syringe filter.
cDMEM (complete DMEM)
Prepare cDMEM by adding Penicillin/Streptomycin solution at 1% vol/vol and gentamicin to 50 µg/ml to DMEM. Prepared cDMEM can be stored at 4°C for 2-3 weeks.
Collagenase-cDMEM
Immediately before dissection, prepare a 0.2% collagenase solution in cDMEM. In the tissue culture hood, filter-sterilise the Collagenase-cDMEM solution using a sterile syringe with a 0.2 μm filter. Consider approximately 2 ml Collagenase-cDMEM for each fish (two fillets) and aliquot into separate 7 ml bijou tubes.
PBSTx
Prepare PBSTx (0.5%) by diluting 100% Triton X100 in sterile PBS. Solutions containing Triton X-100 can be prepared in advance, but for long term storage, use PBS containing 0.002% sodium azide instead of PBS and wrap the tube/bottle in aluminium foil to protect from light.
Matrigel solution
Defrost Matrigel stock overnight at 4°C. Dilute it to 1 mg/ml in DMEM and aliquot into 2 ml micro-centrifuge tubes. It is essential to complete this step on ice or the Matrigel will gel and form lumps. Aliquots of diluted Matrigel can be stored at 4°C for up to 2 weeks or frozen at -20°C for longer term storage.
Proliferation Medium (PM)
Prepare PM by supplementing DMEM with 1% Penicillin/Streptomycin, 10 µg/ml gentamicin and 20% FBS. Pre-warm PM to 28.5°C in the incubator. We recommend preparing PM fresh prior to starting muscle dissection/dissociation and to use within 2 to 3 days.
Differentiation Medium (DM)
Prepare DM by supplementing DMEM with 1% Penicillin/Streptomycin, 10 µg/ml gentamicin and 2% horse serum. Pre-warm DM to 28.5°C in the incubator. We recommend preparing fresh DM as needed and using it within 2 to 3 days.
Acknowledgments
We thank all members of the Hughes lab and Bruno Correia da Silva and his staff for fish care. This work is supported by grants from the Medical Research Council to S.M.H. (MRC Programme Grants G1001029 and MR/N021231/1) and P.S.Z. (MR/P023215/1 and MR/S002472/1). The present protocol was developed and used in Ganassi et al. (2018) and Ganassi et al. (2020).
Competing interests
The authors declare that they have no competing interests.
Ethics
All procedures were performed on adult zebrafish in accordance with the PPL license held under the UK Animals (Scientific Procedures) Act 1986, and later modifications conformed to all relevant guidelines and regulations. All lines used were reared at King's College London on a 14/10 h light/ dark cycle at 28.5°C, with staging and husbandry as described before (Westerfield, 2000).
References
- Alexander, M. S., Kawahara, G., Kho, A. T., Howell, M. H., Pusack, T. J., Myers, J. A., Montanaro, F., Zon, L. I., Guyon, J. R. and Kunkel, L. M. (2011). Isolation and transcriptome analysis of adult zebrafish cells enriched for skeletal muscle progenitors. Muscle Nerve 43(5): 741-750.
- Anderson, J. E., Wozniak, A. C. and Mizunoya, W. (2012). Single muscle-fiber isolation and culture for cellular, molecular, pharmacological, and evolutionary studies. Methods Mol Biol 798: 85-102.
- Bekoff, A. and Betz, W. (1977a). Properties of isolated adult rat muscle fibres maintained in tissue culture. J Physiol 271(2): 537-547.
- Bekoff, A. and Betz, W. J. (1977b). Physiological properties of dissociated muscle fibres obtained from innervated and denervated adult rat muscle. J Physiol 271(1): 25-40.
- Bischoff, R. (1975). Regeneration of single skeletal muscle fibers in vitro. Anat Rec 182(2): 215-235.
- Blagden, C. S., Currie, P. D., Ingham, P. W. and Hughes, S. M. (1997). Notochord induction of zebrafish slow muscle mediated by Sonic hedgehog. Genes Dev 11(17): 2163-2175.
- Brack, A. S., Bildsoe, H. and Hughes, S. M. (2005). Evidence that satellite cell decrement contributes to preferential decline in nuclear number from large fibres during murine age-related muscle atrophy. J Cell Sci 118(Pt 20): 4813-4821.
- Buckingham, M. and Relaix, F. (2015). PAX3 and PAX7 as upstream regulators of myogenesis. Semin Cell Dev Biol 44: 115-125.
- Cardasis, C. A. and Cooper, G. W. (1975a). An analysis of nuclear numbers in individual muscle fibers during differentiation and growth: a satellite cell-muscle fiber growth unit. J Exp Zool 191(3): 347-358.
- Cardasis, C. A. and Cooper, G. W. (1975b). A method for the chemical isolation of individual muscle fibers and its application to a study of the effect of denervation on the number of nuclei per muscle fiber. J Exp Zool 191(3): 333-346.
- Davies, M. L., Johnston, I.A. and van de Wal, J. (1995). Muscle Fibers in Rostral and Caudal Myotomes of the Atlantic Cod (Gadus morhua L.) Have Different Mechanical Properties. Physiol Zool 68(4): 673-697.
- Froehlich, J. M., Seiliez, I., Gabillard, J. C. and Biga, P. R. (2014). Preparation of primary myogenic precursor cell/myoblast cultures from basal vertebrate lineages. J Vis Exp(86): 51354.
- Fukada, S. I., Akimoto, T. and Sotiropoulos, A. (2020). Role of damage and management in muscle hypertrophy: Different behaviors of muscle stem cells in regeneration and hypertrophy. Biochim Biophys Acta Mol Cell Res 1867(9): 118742.
- Ganassi, M., Badodi, S., Ortuste Quiroga, H. P., Zammit, P. S., Hinits, Y. and Hughes, S. M. (2018). Myogenin promotes myocyte fusion to balance fibre number and size. Nat Commun 9(1): 4232.
- Ganassi, M., Badodi, S., Wanders, K., Zammit, P. S. and Hughes, S. M. (2020). Myogenin is an essential regulator of adult myofibre growth and muscle stem cell homeostasis. Elife 9: e60445.
- Hammond, C. L., Hinits, Y., Osborn, D. P., Minchin, J. E., Tettamanti, G. and Hughes, S. M. (2007). Signals and myogenic regulatory factors restrict pax3 and pax7 expression to dermomyotome-like tissue in zebrafish. Dev Biol 302(2): 504-521.
- Harper, C. and Lawrence, C. (2011). The Laboratory Zebrafish. Boca Raton: CRC Press.
- Hinits, Y., Osborn, D. P. and Hughes, S. M. (2009). Differential requirements for myogenic regulatory factors distinguish medial and lateral somitic, cranial and fin muscle fibre populations. Development 136(3): 403-414.
- Hinits, Y., Williams, V. C., Sweetman, D., Donn, T. M., Ma, T. P., Moens, C. B. and Hughes, S. M. (2011). Defective cranial skeletal development, larval lethality and haploinsufficiency in Myod mutant zebrafish. Dev Biol 358(1): 102-112.
- Hromowyk, K.J., Talbot, J.C., Martin, B.L., Janssen, P.M. and Amacher, S.L. (2020). Cell fusion is differentially regulated in zebrafish post-embryonic slow and fast muscle. Dev Bol 462(1): 85-100.
- Johnston, I. and Altringham, J. (1988). Muscle contraction in polar fishes: experiments with demembranated muscle fibres. Comp Biochem Physiol 90B(3): 547-555.
- Johnston, I. A., Abercromby, M., Vieira, V. L., Sigursteindottir, R. J., Kristjansson, B. K., Sibthorpe, D. and Skulason, S. (2004). Rapid evolution of muscle fibre number in post-glacial populations of Arctic charr Salvelinus alpinus. J Exp Biol 207(Pt 25): 4343-4360.
- Katz, B. (1961). The termination of the afferent nerve fibre in the muscle spindle of the frog. Philos Trans R Soc Lond B Biol Sci 243(703): 221-240.
- Mahalwar, P., Walderich, B., Singh, A. P. and Nüsslein-Volhard, C. (2014). Local reorganization of xanthophores fine-tunes and colors the striped pattern of zebrafish. Science 345(6202): 1362-1364.
- Mauro, A. (1961). Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol 9: 493-495.
- Moyle, L. A. and Zammit, P. S. (2014). Isolation, culture and immunostaining of skeletal muscle fibres to study myogenic progression in satellite cells. Methods Mol Biol 1210: 63-78.
- Osborn, D. P. S., Li, K., Cutty, S. J., Nelson, A. C., Wardle, F. C., Hinits, Y. and Hughes, S. M. (2020). Fgf-driven Tbx protein activities directly induce myf5 and myod to initiate zebrafish myogenesis. Development 147(8): dev184689.
- Pipalia, T. G., Koth, J., Roy, S. D., Hammond, C. L., Kawakami, K. and Hughes, S. M. (2016). Cellular dynamics of regeneration reveals role of two distinct Pax7 stem cell populations in larval zebrafish muscle repair. Dis Model Mech 9(6): 671-684.
- Purohit, G. and Dhawan, J. (2019). Adult Muscle Stem Cells: Exploring the Links Between Systemic and Cellular Metabolism. Front Cell Dev Biol 7: 312.
- Relaix, F. and Zammit, P. S. (2012). Satellite cells are essential for skeletal muscle regeneration: the cell on the edge returns centre stage. Development 139(16): 2845-2856.
- Rosenblatt, J. D., Lunt, A. I., Parry, D. J. and Partridge, T. A. (1995). Culturing satellite cells from living single muscle fiber explants. In Vitro Cell Dev Biol Anim 31(10): 773-779.
- Westerfield, M. (2000). The Zebrafish Book - A guide for the laboratory use of zebrafish (Danio rerio). University of Oregon Press.
- Zammit, P. S., Golding, J. P., Nagata, Y., Hudon, V., Partridge, T. A. and Beauchamp, J. R. (2004). Muscle satellite cells adopt divergent fates: a mechanism for self-renewal? J Cell Biol 166(3): 347-357.
- Zhang, H. and Anderson, J. E. (2014). Satellite cell activation and populations on single muscle-fiber cultures from adult zebrafish (Danio rerio). J Exp Biol 217(Pt 11): 1910-1917.
Article Information
Copyright
![]() Ganassi et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Ganassi et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Ganassi, M., Zammit, P. S. and Hughes, S. M. (2021). Isolation of Myofibres and Culture of Muscle Stem Cells from Adult Zebrafish. Bio-protocol 11(17): e4149. DOI: 10.21769/BioProtoc.4149.
- Ganassi, M., Badodi, S., Wanders, K., Zammit, P. S. and Hughes, S. M. (2020). Myogenin is an essential regulator of adult myofibre growth and muscle stem cell homeostasis. Elife 9: e60445.
Category
Developmental Biology > Cell growth and fate > Myofiber
Stem Cell > Adult stem cell > Muscle stem cell
Biological Sciences > Biological techniques
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.