- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Purification of Recombinant Wild Type and Mutant Ryanodine Receptors Expressed in HEK293 Cells
Published: Vol 11, Iss 15, Aug 5, 2021 DOI: 10.21769/BioProtoc.4112 Views: 3914
Reviewed by: Michael StowellYaping SunRAMESH KUDIRAAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2020
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
High quantities of purified ryanodine receptor (RyR), a large (2.26 MDa) intracellular homotetrameric membrane protein, can be obtained from heterologous expression in HEK293 cells and used for structure determination by cryo-EM. The advantage of using recombinant protein is that the variability due to post-translational modifications can be minimized, to which the high resolution of up to 2.4 Å achieved for RyR2 can be attributed (Iyer et al., 2020). In addition, recombinant protein expression enables the study of mutations that are deleterious when expressed homozygously in animals. Protein purification was achieved using two strategies, sucrose density gradient and affinity chromatography, which have previously been used for purification of RyR from tissue. The sucrose gradient method was developed from (Lee et al., 1994) and later adapted for cryo-EM (Samsó et al., 2005). The affinity chromatography method takes advantage of the high affinity of RyR for its ligand FKBP12/12.6, by using a construct between FKBP and streptavidin binding protein (SBP) (Cabra et al., 2016). While the sucrose gradient method can yield a higher protein concentration (≥ 2 mg/ml), the affinity purification method is faster. Both methods are suitable and applicable to the purification of recombinant proteins and were successfully used in the first 3D near-atomic reconstructions of RyRs purified from cells expressing disease mutants (Iyer et al., 2020). This purification protocol is also suitable for functional studies, such as single-channel analysis, that require pure RyR protein.
Keywords: RyRBackground
RyRs are intracellular Ca2+ channels located on sarcoplasmic and endoplasmic reticula and are present in nearly all cell types, including excitable cells such as neurons and muscles. In skeletal and cardiac muscles, excitation-contraction coupling takes place by activation of RyRs via dihydropyridine receptors on the T-tubular membrane (Samsó, 2017). Since RyRs are membrane-embedded proteins, their purification requires solubilization in detergent (e.g., CHAPS, Digitonin, Tween-20) within aqueous buffers; however, excessive or insufficient detergent can denature or leave the channels only partially solubilized, respectively. The FK506-binding proteins of 12 and 12.6 kDa (FKBP12 and FKBP12.6) are important RyR modulators, which bind to RyR with high affinity. Hence, detergent-solubilized RyR can be purified using GST-FKBP (or SBP-FKBP) affinity chromatography. However, FKBP exhibits reduced binding to RyR in vitro (Zissimopoulos et al., 2012), resulting in a lower than expected yield of pure RyR. Considering that RyR is the largest known ion channel, with a molecular weight of ~2.3 MDa (a tetramer of ~565 kDa monomers), sucrose gradient centrifugation and gel filtration are suitable for its purification, in which RyR elutes as a peak from the denser fraction (sucrose gradient) or as the first peak (gel filtration). In recent years, structures have been solved at near-atomic resolution for both RyR1 and RyR2, which were purified from skeletal or cardiac muscle (Gong et al., 2020). More than 300 identified RyR mutations cause a number of debilitating or fatal disorders, most of which result in a gain-of-function phenotype; therefore, their structure is of significant interest. However, transgenic animals carrying RyR mutations have a lower or zero survival rate, and obtaining homozygous animals for severe RyR mutations is exceptionally difficult or impossible; thus, purification of RyR from cells is an attractive solution. Among the different cell types, bacteria lack the endoplasmic reticulum compartment where RyR resides and are therefore unsuitable for RyR expression. Among eukaryotic systems, the most widely used are mammalian cells such as HEK293 cells, which can easily accommodate the 15,000-bp cDNA and possess the mammalian machinery for post-translational modifications. Ideally, the cells are stably transfected with a plasmid containing an inducible promoter, such as pcDNA5/FRT/TO, which was designed for use with the Flp-In-T-Rex system in our case. The main limitation of cell culture is the low mass yield as compared with that of muscle tissue. Other limitations include the requirement for high quantities of expensive supplies such as cell culture media and the time required for cell culture (2-3 weeks) as well as for the sucrose gradient centrifugation-based protocol (~3 days). We were the first to purify recombinant RyR at a high yield from HEK293 cells using sucrose gradient centrifugation and ion-exchange chromatography (Iyer et al., 2020). In the same work, we also used a one-step affinity chromatography that, although resulting in a lower yield, was faster and allowed complete purification in one day. Hence, purifying recombinant RyR from HEK293 cells using either of these two protocols is suitable for structural and functional studies that require high quantities of pure mutant RyR. Additionally, this protocol may serve as the starting point for purification of other membrane proteins of similar size, although the detergent solubilization conditions will need to be customized for the membrane protein of interest.
Materials and Reagents
150 × 20 mm cell culture dishes (USA Scientific, catalog number: CC7682-3614)
5 ml serological pipettes (VWR, catalog number: 89130-896)
30 ml plastic syringes (NORM-JECT, catalog number: 4100-X00V0)
15 ml centrifuge tubes (VWR, catalog number: 89039-664)
50 ml centrifuge tubes (VWR, catalog number: 525-0402)
3.5 ml centrifuge tubes (Beckman, catalog number: 349622)
25 × 89 mm centrifuge tubes (Beckman, catalog number: 344058)
0.45 µm filter (PALL, catalog number: 4652)
StrepTrap HP column (Cytiva, catalog number: 28-9075-46)
Cell scraper (USA Scientific, catalog number: CC7600-0220)
Dulbecco’s Modified Eagle’s Medium (DMEM) (VWR, catalog number: 45000-304)
Characterized fetal bovine serum, Canadian origin (Cytiva, catalog number: SH30396.02)
Bovine growth serum (U.S.) (Cytiva, catalog number: SH30541.03)
Penicillin-streptomycin (Thermo Fisher, catalog number: 15140122)
HiTrap Heparin HP (Cytiva, catalog number: 17-0406-01)
Doxycycline (Millipore, catalog number: 3382281)
D-Desthiobiotin (Sigma, catalog number: D1411)
Dithiothreitol (DTT, Millipore, catalog number: 233156)
PBS (Gibco, catalog number: 2052719)
SBP-FKBP12.6 construct (Cabra et al., 2016)
Imidazole (Sigma, catalog number: I2399)
Sucrose (Sigma, catalog number: S9378)
4-Morpholinepropanesulfonic acid (MOPS, Sigma, catalog number: M1254)
Sodium chloride (NaCl, Fisher, catalog number: S671-3)
3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS, Sigma, catalog number: 220201)
L-α-phosphatidylcholine (Sigma, catalog number: P3644)
10% Tween-20 (Millipore, catalog number: 655206)
Aprotinin (Thermo Fisher Scientific, catalog number: 78432)
Leupeptin (Thermo Fisher Scientific, catalog number: 78435)
Pefabloc (Thermo Fisher Scientific, catalog number: 78431)
Anti-RyR antibody (34C, Abcam ab2868)
Homogenization Buffer (see Recipes)
Solubilization Buffer I (see Recipes)
Solubilization Buffer II (see Recipes)
Dilution Buffer (see Recipes)
Wash Buffer I (see Recipes)
Wash Buffer II (see Recipes)
Wash Buffer III (see Recipes)
Elution Buffer I (see Recipes)
Elution Buffer II (see Recipes)
Equipment
Avanti J-E centrifuge (Beckman, rotor: F10BCI-6 × 500)
Optima MAX-TL ultracentrifuge (Beckman, rotor: TLA 100.3)
Optima LE-80K ultracentrifuge (Beckman, rotor: SW32 Ti)
Tabletop centrifuge (Eppendorf, model: 5415C)
Sonicator (Qsonica Model Q125, maximum power 125 watts)
Orbital shaker (Labnet Reciprocal 30)
pH meter (Mettler Toledo SevenEasy S20)
Peristaltic pump (Masterflex C/L, model: 77120-32)
−80°C laboratory freezer
Dounce homogenizer (10-ml Teflon plunger on glass (tight), Cole-Parmer)
Magnetic stirrer
Pipettes (Gilson Pipetman, models: P1000, P200, P20, P10)
NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific)
FluorChem E imaging system (ProteinSimple)
FEI Tecnai F20 transmission electron microscope (TEM)
Procedure
Purification of the vesicle fraction
Culture HEK293 cells, expressing the RyR construct with an inducible doxycycline promoter, on 150 × 20 mm dishes (~30-40 plates) in Dulbecco’s Modified Eagle’s Medium supplemented with fetal bovine serum (FBS, 2%), bovine growth serum (BGS, 7%), and penicillin-streptomycin. Fast and robust growth of HEK293 cells requires FBS containing growth hormones; a more cost-effective combination of FBS-BGS can be used. At 80-90% confluence, add doxycycline (2 µg/ml) and incubate for 3 days.
Harvest cells in PBS (~3 ml/plate) with the aid of a cell scraper, and create a single-cell suspension by pipetting up and down with a 5-ml serological pipette. Depending on the volume generated, collect cells in one or two 15-ml centrifuge tubes.
Centrifuge at 573 × g (Beckman Coulter Avanti J-E centrifuge, rotor F10BCI-6 × 500) for 10 min.
Discard the supernatant. Resuspend the pellet in Homogenization Buffer and homogenize using an ultrasonic probe (98% amplitude, around 3 pulses of 20 s in a Qsonica Q125 sonicator).
Centrifuge the homogenized pellet at 4,400 × g (Beckman Coulter Avanti J-E centrifuge, rotor F10BCI-6 × 500) for 10 min.
Transfer the supernatant to 3.5-ml centrifuge tubes.
Centrifuge at 100,000 × g (Beckman Coulter Optima MAX-TL ultracentrifuge, rotor TLA 100.3) for 60 min.
Discard the supernatant. Resuspend the resulting pellet in Homogenization Buffer using a Dounce homogenizer.
Determine the concentration of protein in the purified vesicles (i.e., typically ~40-60 mg/ml) by performing a Bradford assay on two 10-µl fractions and computing the average.
Freeze using liquid nitrogen and store at −80°C for subsequent protein purification.
Purification of recombinant RyR from vesicles using sucrose gradient centrifugation and ion-exchange chromatography
If starting from frozen purified vesicles (see Section A: Purification of vesicle fraction), thaw ~100 mg vesicles in a 37°C bath.
Add 9 ml Solubilization Buffer I without CHAPS to a 25-ml beaker.
Add the thawed vesicles to the beaker containing Solubilization Buffer I (without CHAPS).
Stir the solution at 300 rpm using a magnetic stirrer.
Carefully add 5 ml 5% CHAPS to the solubilization mixture while continuing to stir.
Cover the beaker with parafilm and continue stirring at 300 rpm for 25 min.
Centrifuge the solubilized protein at 100,000 × g (Beckman Coulter Optima MAX-TL Ultracentrifuge, rotor TLA 100.3) for 60 min.
Discard the pellet. The supernatant is ready for further RyR purification.
Prepare the following solutions: 10%, 12%, 14%, 16%, 18%, and 20% (w/v) sucrose in Solubilization Buffer I containing 0.5% (w/v) CHAPS-0.125% PC instead of 5% CHAPS-1.25% PC.
Pour 5 ml 20% sucrose into an Ultraclear Beckman centrifuge tube (25 × 89 mm).
Carefully add 6 ml 18%, 6 ml 16%, 6 ml 14%, 6 ml 12%, and 5 ml 10% sucrose solutions using a peristaltic pump, making sure that the liquid layers do not mix (Figure 1).
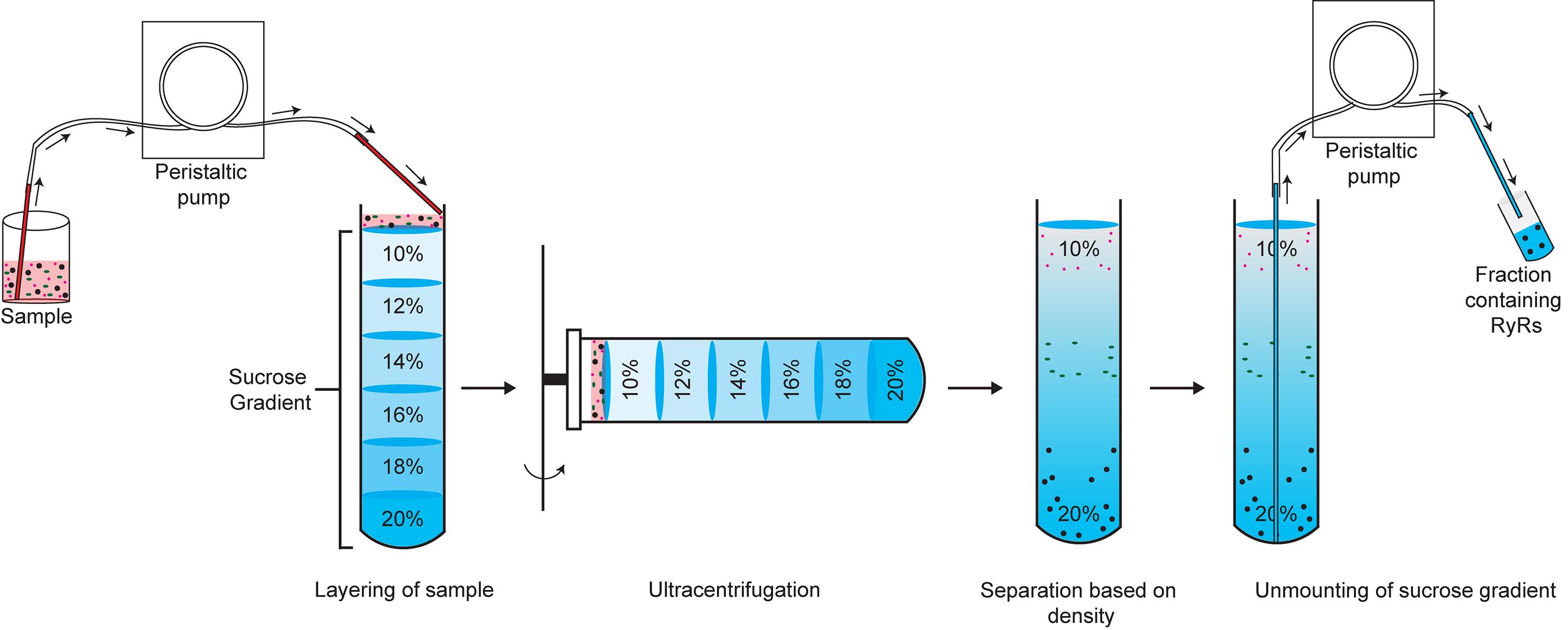
Figure 1. Schematic showing a discontinuous sucrose gradient with layers of sucrose of decreasing concentrations. The solubilized RyR sample is added on top of the 10% sucrose solution. Ultracentrifugation results in separation based on density, with RyR sedimenting near the bottom of the tube. The sucrose gradient is dismounted in 2-ml fractions starting from the bottom (denser) sucrose layer.Layer the solubilized RyR sample (up to 3.5 ml) on top of the 10% sucrose solution using the peristaltic pump (Figure 1).
Lift the tubes slowly and place them gently in a swing bucket rotor. Centrifuge at 120,000 × g (Beckman Coulter Optima LE-80K Ultracentrifuge, rotor SW32 Ti) for 23 h under no braking and low acceleration conditions.
Unmount the gradient (2.0-ml fractions) starting from the bottom using the peristaltic pump (Figure 1).
Analyze each fraction on a 10% SDS-PAGE gel stained with Coomassie Brilliant Blue or western blotting with a generic anti-RyR antibody (34C) to identify fractions containing pure RyR (Figure 2).
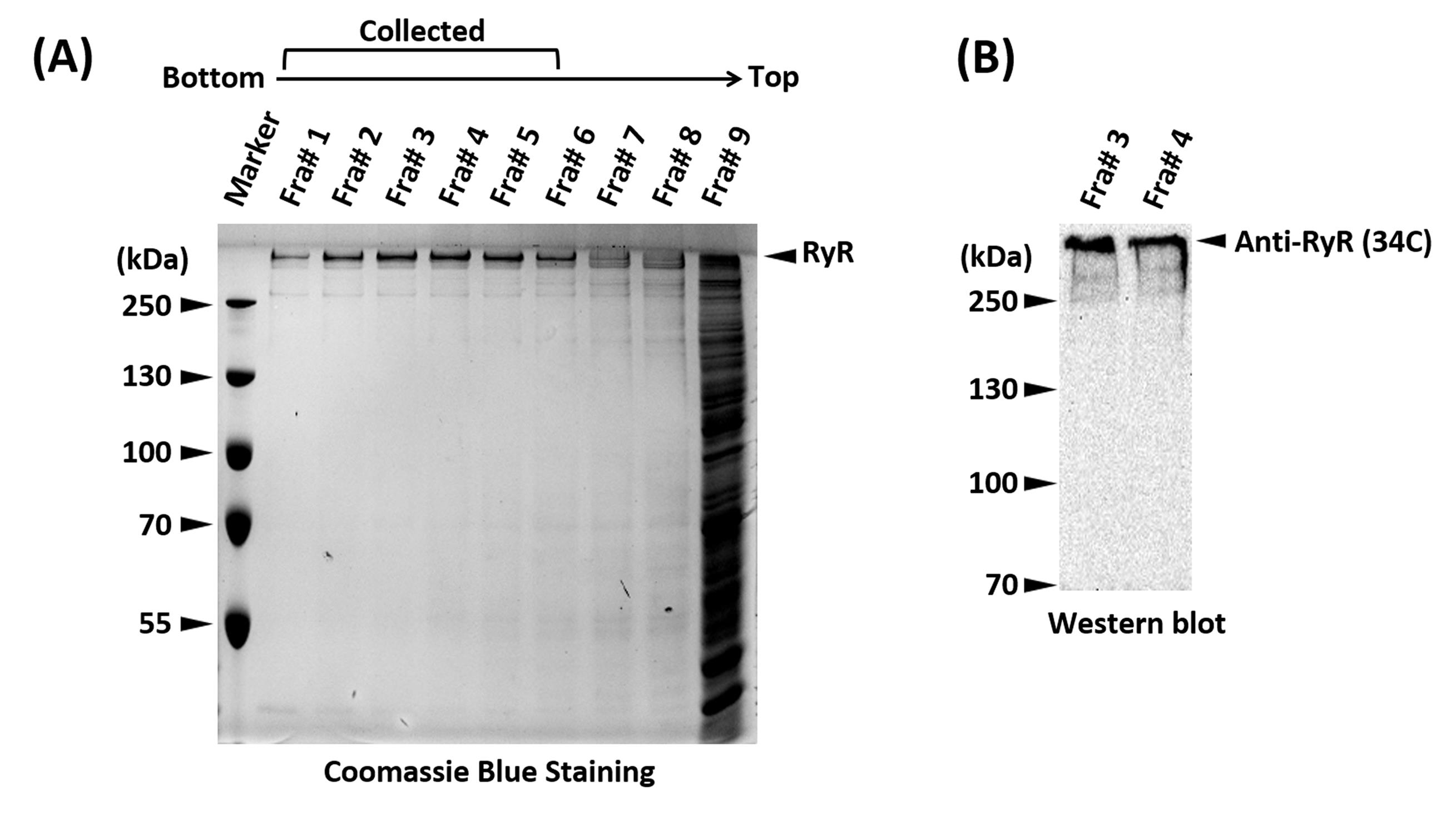
Figure 2. SDS-PAGE and western blot showing the sucrose gradient fractions and positive identification of the RyR. A. SDS-PAGE gel stained with Coomassie Brilliant Blue of the sucrose gradient fractions (Fra#9 represents a mixture of all the remaining fractions). Fractions 1-6, containing pure RyR, were pooled. The unusual high molecular weight of RyR (565 kDa per monomer) places its band well above the top molecular weight marker, a distinguishing trait that in most scenarios suffices for positive identification. B. Western blot of fractions #3 and #4 of a sucrose density gradient using an anti-RyR antibody (34C) to confirm the identity of RyR.Once identified, pool the fractions containing pure RyR.
Add DTT at a final concentration of 1 mM to the pooled purified RyR sample (to compensate for DTT decay).
Dilute the sample 5-fold with Dilution Buffer.
Connect a 1-ml HiTrap Heparin HP column (Amersham Biosciences) to a peristaltic pump (Figure 3).
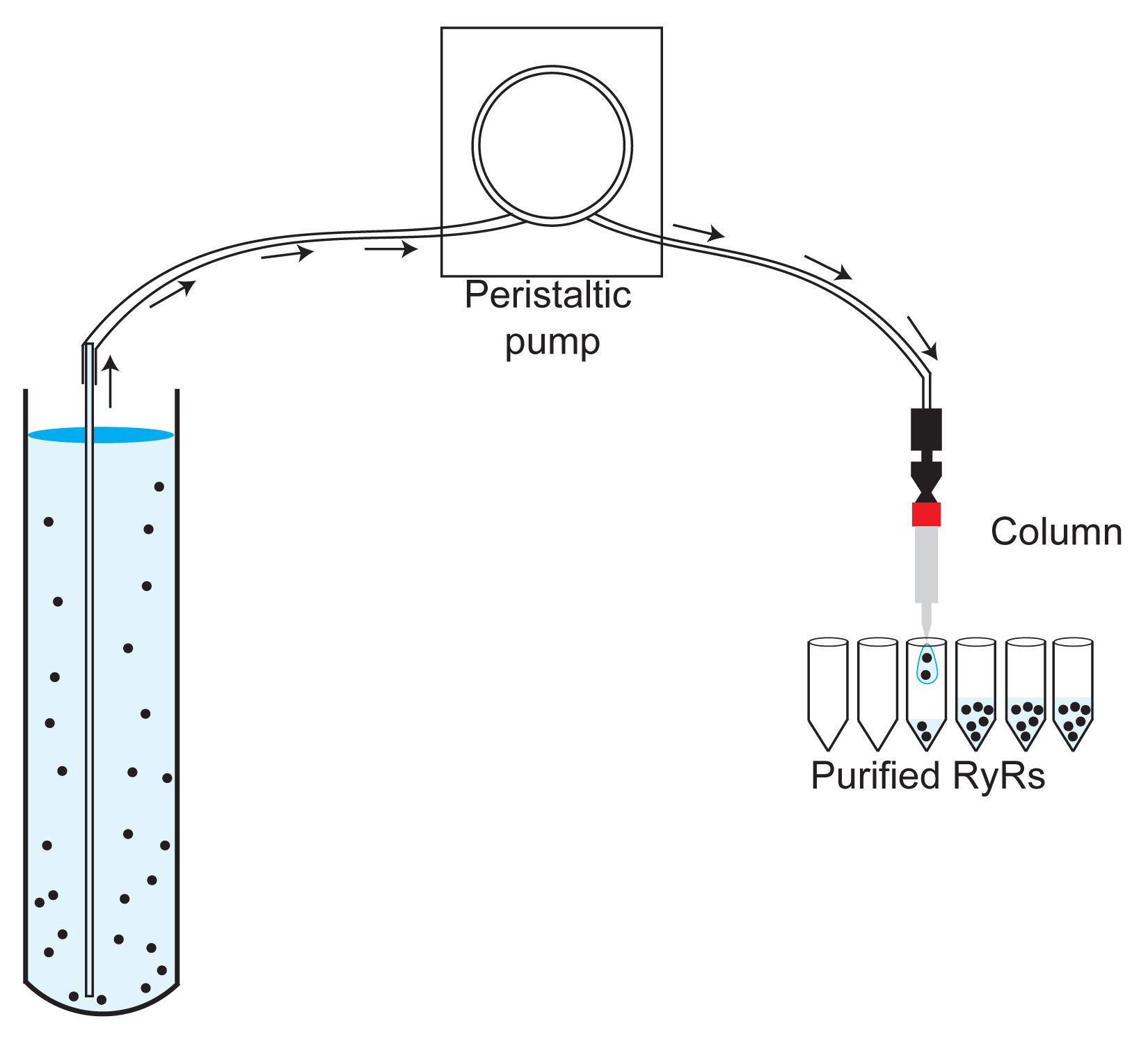
Figure 3. Image showing the assembly of the sample, the peristaltic pump, the 1-ml HiTrap Heparin HP (or 1-ml StrepTrap) column, and the tubes to collect the fractions.Wash the column with 20 ml deionized water (Milli-Q) (1 ml/min).
Equilibrate the column with 20 ml Wash Buffer I (1 ml/min).
Load the RyR sample on the column (0.5 ml/min).
Wash the column with 20 ml Wash Buffer I (1 ml/min).
Wash the column with 10 ml Wash Buffer II (1 ml/min).
Elute the sample with 10 ml Elution Buffer I (0.5 ml/min).
Manually collect eluted fractions in aliquots of 4 drops (~100 μl) in plastic 1.5-ml centrifuge tubes (Figure 3).
Determine the protein concentration of each fraction in a NanoDrop 2000 spectrophotometer using absorbance at 280 nm (extinction coefficient 2,271,320 M−1cm−1 for RyR).
Assess the purity and quality of the purified RyR by 10% SDS-PAGE (Figure 4) and negative staining EM (Figure 5).
Flash-freeze aliquots in liquid nitrogen and store at −80°C for subsequent cryo-EM grid preparation.
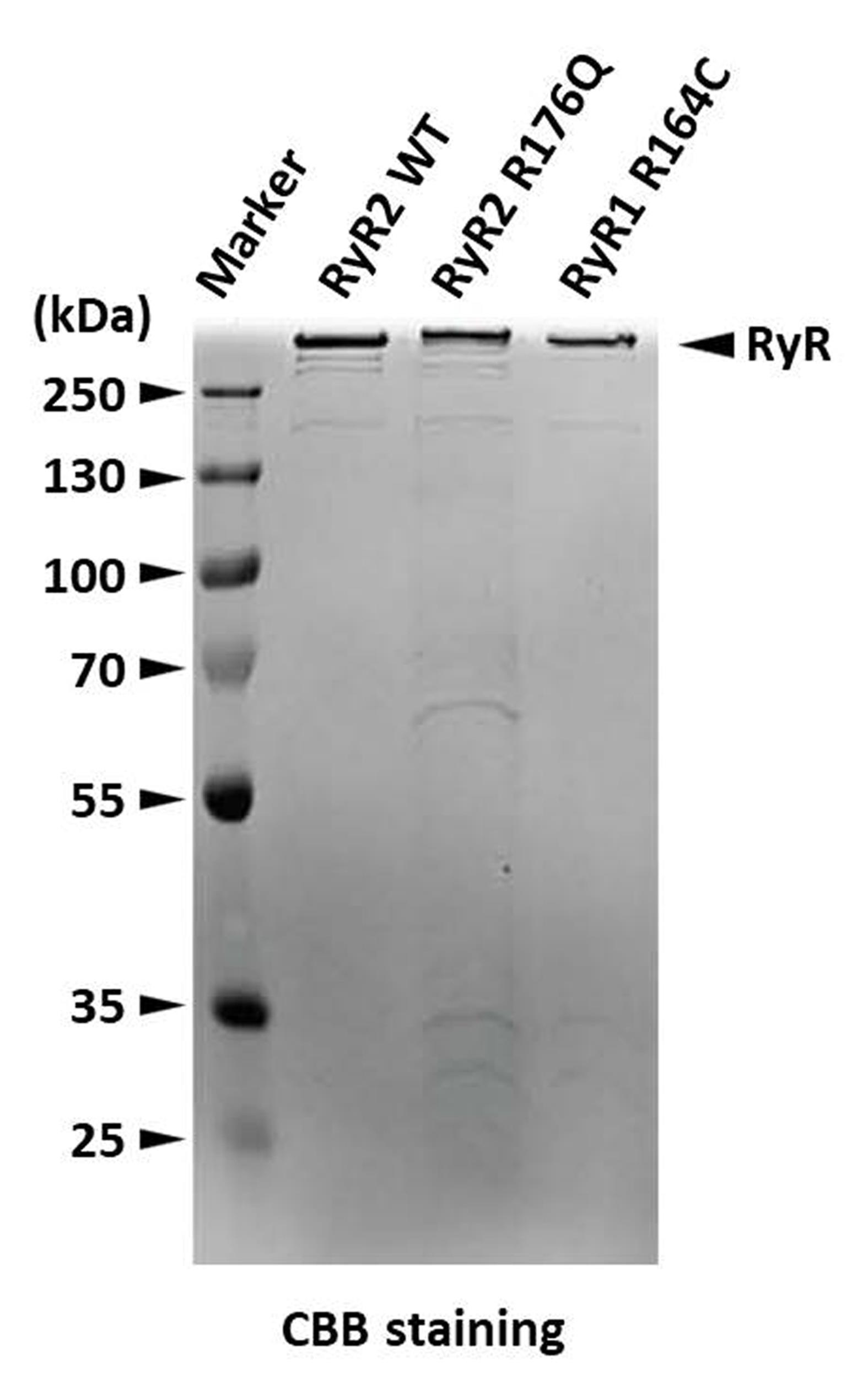
Figure 4. SDS-PAGE gel stained with Coomassie Brilliant Blue showing RyRs eluted from a StrepTrap affinity column (RyR2 WT) and from a Heparin-agarose affinity column (RyR2 R176Q and RyR1 R164C).
Figure 5. Micrograph of negatively stained RyRs purified from HEK293 cells (representative RyRs encircled in green). Negative staining was performed to check the quality of the purified proteins using established protocols (Ohi et al., 2004). Copper grids coated with a thin layer of carbon (CF300-Cu 300 mesh, Electron Microscopy Sciences) were first glow-discharged at 25 mA for 20 s. Purified RyRs (3.5 µl) were adsorbed onto the grid for 40 s, washed with deionized water, and stained with 0.75% uranyl formate; the excess stain was blotted off. Images were collected on an FEI Tecnai F20 TEM equipped with a GATAN Ultra Scan 4000 UHS (4k × 4k) camera at a magnification of 50,000×. Finer structural details of the RyR can be discerned in the enlarged images of individual RyRs corresponding to those highlighted in the micrograph.
Purification of recombinant RyR from vesicles by one-step affinity chromatography
If starting from frozen purified vesicles (see Section A: Purification of vesicle fraction), thaw ~20-30 mg vesicles using a 37°C bath.
Add 1 ml Solubilization Buffer II without CHAPS to a 10-ml beaker.
Add the thawed vesicles to the beaker containing Solubilization Buffer II (without CHAPS).
Stir the solution at 300 rpm using a magnetic stirrer.
Carefully add 0.5 ml 2% CHAPS to the solubilization mixture while continuing to stir.
Cover the beaker with parafilm and stir at 300 rpm for 20 min.
Centrifuge the solubilized protein at 100,000 × g (Beckman Coulter Optima MAX-TL Ultracentrifuge, rotor TLA 100.3) for 60 min.
Discard the pellet. Dilute the supernatant 5-fold with Dilution Buffer.
Centrifuge the solution at 16,000 × g (Eppendorf Centrifuge, Model 5415 C) for 10 min and discard the pellet.
Filter the supernatant using a 0.45-μm filter attached to a syringe.
Connect a 1-ml StrepTrap column (Cytiva) to a peristaltic pump (Figure 3).
Wash the column with 20 ml deionized water (Milli-Q) (1 ml/min).
Equilibrate the column with 20 ml Wash Buffer I (1 ml/min).
Load the recombinant SBP-FKBP12.6 (1:10 RyR: FKBP12.6 molar ratio) on the column (0.5 ml/min).
Wash the column with 20 ml Wash Buffer I (1 ml/min).
Load the filtered solubilized RyR sample on the column (0.5 ml/min).
Wash the column with 20 ml Wash Buffer I (1 ml/min).
Wash the column with 2 ml Wash Buffer III (1 ml/min).
Elute the sample with 5 ml Elution Buffer II containing 5 mM desthiobiotin (0.5 ml/min).
Manually collect eluted fractions in aliquots of 4 drops (~100 μl) in plastic 1.5-ml centrifuge tubes (Figure 3).
Determine the protein concentration of each fraction in a NanoDrop 2000 spectrophotometer using absorbance at 280 nm (extinction coefficient 2,271,320 M−1cm−1 for RyR).
Assess the purity and quality of the purified RyR by 10% SDS-PAGE (Figure 4) and negative staining EM (Figure 5).
Flash-freeze aliquots in liquid nitrogen and store at −80°C for subsequent cryo-EM grid preparation.
Notes
All procedures except for steps A1, A2, A9, A10, B1, B27-29, C1, and C21-C23 were carried out at 4°C.
Recipes
200× Protease Inhibitor (PI) Cocktail
1 mg/ml aprotinin
1 mg/ml leupeptin
0.5 mg/ml pefabloc
Homogenization Buffer
5 mM imidazole pH 7.4
10% (w/v) sucrose
PI
Solubilization Buffer I
20 mM Na MOPS (pH 7.4)
5% (w/v) CHAPS
1.25% (w/v) PC
1 M NaCl
2 mM DTT
PI
Solubilization Buffer II
20 mM Na MOPS (pH 7.4)
2% (w/v) CHAPS
0.5% (w/v) PC
1 M NaCl
2 mM DTT
PI
Dilution Buffer
20 mM Na MOPS (pH 7.4)
0.5% (w/v) CHAPS
0.125% (w/v) PC
2 mM DTT
PI
Wash Buffer I
20 mM Na MOPS (pH 7.4)
0.2 M NaCl
0.5% (w/v) CHAPS
0.125% (w/v) PC
2 mM DTT
PI
Wash Buffer II
20 mM Na MOPS (pH 7.4)
0.2 M NaCl
0.015% (w/v) Tween-20
2 mM DTT
PI
Wash Buffer III
20 mM Na MOPS (pH 7.4)
0.4 M NaCl
0.015% (w/v) Tween-20
2 mM DTT
PI
Elution Buffer I
20 mM Na MOPS (pH 7.4)
0.9 M NaCl
0.015% (w/v) Tween-20
2 mM DTT
Elution Buffer II
20 mM Na MOPS (pH 7.4)
0.4 M NaCl
0.015% (w/v) Tween-20
2 mM DTT
5 mM desthiobiotin
Acknowledgments
This work was supported by the NIH (R01 AR068431 and R01 HL133182); the Muscular Dystrophy Association (MDA352845 to M.S.); American Heart Association Post-Doctoral Fellowship (19POST34430178 to K.A.I.); the Japan Society for the Promotion of Science (19K07105 to N.K. and 19H03404 to T.M.); the Japan Agency for Medical Research and Development (19ek0109202 to N.K and JP20am0101080 to T.M.); and the Vehicle Racing Commemorative Foundation (to T.M.). This protocol was adapted, with minor modifications, from a previous study published by Iyer et al. (2020).
Competing interests
The authors declare no competing interests.
References
- Cabra, V., Murayama, T. and Samsó, M. (2016). Ultrastructural Analysis of Self-Associated RyR2s. Biophys J 110(12): 2651-2662.
- Gong, D., Yan, N. and Ledford, H. A. (2020). Structural Basis for the Modulation of Ryanodine Receptors. Trends Biochem Sci S0968-0004(20)30294-2.
- Iyer, K. A., Hu, Y., Nayak, A. R., Kurebayashi, N., Murayama, T. and Samsó, M. (2020). Structural mechanism of two gain-of-function cardiac and skeletal RyR mutations at an equivalent site by cryo-EM. Sci Adv 6(31): eabb2964.
- Lee, H. B., Xu, L. and Meissner, G. (1994). Reconstitution of the skeletal muscle ryanodine receptor-Ca2+ release channel protein complex into proteoliposomes. J Biol Chem 269(18): 13305-13312.
- Ohi, M., Li, Y., Cheng, Y. and Walz, T. (2004). Negative Staining and Image Classification - Powerful Tools in Modern Electron Microscopy. Biol Proced Online 6: 23-34.
- Samsó, M. (2017). A guide to the 3D structure of the ryanodine receptor type 1 by cryoEM. Protein Sci 26(1): 52-68.
- Samsó, M., Wagenknecht, T. and Allen, P. D. (2005). Internal structure and visualization of transmembrane domains of the RyR1 calcium release channel by cryo-EM. Nat Struct Mol Biol 12(6): 539-544.
- Zissimopoulos, S., Seifan, S., Maxwell, C., Williams, A.J. and Lai, F.A. (2012). Disparities in the association of the ryanodine receptor and the FK506-binding proteins in mammalian heart. J Cell Sci 125(7):1759-69.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Hu, Y., Iyer, K. A., Nayak, A. R., Kurebayashi, N., Murayama, T. and Samsó, M. (2021). Purification of Recombinant Wild Type and Mutant Ryanodine Receptors Expressed in HEK293 Cells. Bio-protocol 11(15): e4112. DOI: 10.21769/BioProtoc.4112.
- Iyer, K. A., Hu, Y., Nayak, A. R., Kurebayashi, N., Murayama, T. and Samsó, M. (2020). Structural mechanism of two gain-of-function cardiac and skeletal RyR mutations at an equivalent site by cryo-EM. Sci Adv 6(31): eabb2964.
Category
Biochemistry > Protein > Isolation and purification
Biological Sciences > Biological techniques
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.