- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Efficient and Rapid Analysis of Polysomes and Ribosomal Subunits in Cells and Tissues Using Ribo Mega-SEC
Published: Vol 11, Iss 15, Aug 5, 2021 DOI: 10.21769/BioProtoc.4106 Views: 6387
Reviewed by: Koshi ImamiAlison GallowayKif Liakath-Ali
Original research article
The authors used this protocol in:

Aug 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Polysome profile analysis is a popular method for separating polysomes and ribosomal subunits and is typically achieved using a sucrose density gradient (SDG). This has remained the gold standard method since ribosomes were first discovered; however, this method is time-consuming and requires multiple steps from making the gradient and long ultracentrifugation to collecting and analyzing the fractions. Each of these steps in the SDG workflow can introduce potential technical variation that affects the reproducibility of gradient profiles between samples. To address these limitations, we have developed a flexible, alternative approach for analyzing polysomes and ribosomal subunits based on size-exclusion chromatography (SEC), termed ‘Ribo Mega-SEC.’ In comparison with the SDG method, Ribo Mega-SEC involves a single step using ultra-high-performance liquid chromatography (uHPLC). The entire workflow, from injecting the lysate to collecting the fractions, can be performed in as little as 15 min, with high reproducibility. By varying the pore size of the SEC column, polysomes and ribosomal subunits can be separated using extracts from either human or mouse cultured cell lines or from tissue samples, Drosophila embryos, or budding yeast. The resulting separated fractions are suitable for analysis using a wide range of subsequent analytical techniques including mass spectrometry (MS)-based proteomics, RNA-Seq, electron microscopy (EM), and multiple biochemical assays.
Keywords: PolysomeBackground
Sucrose density gradient (SDG) fractionation is still the most frequently used method across species for separating polysomes and ribosomal subunits since the discovery of ribosomes in the 1950s (Britten and Roberts, 1960; Rheinberger, 2004). However, the SDG approach has several well-known limitations; for example, it is a time-consuming procedure that includes a long ultracentrifugation step, which can also result in the loss of proteins weakly associated with the ribosomes (Simsek et al., 2017). Moreover, the multiple steps involved in the SDG workflow introduce potential sources of technical variation, with concomitant effects on the overall reproducibility, resolution, and accuracy. Furthermore, both the length of the SDG workflow and the recovery of fractionated samples in the presence of high sucrose, which must be removed before many types of analytical procedures, are problematic for combining it with downstream high-throughput analyses.
To overcome these issues and enable wider access of polysome fractionation to researchers outside the protein translation field, we have recently developed the Ribo Mega-SEC method for the separation of polysomes and ribosomal subunits using high-resolution size-exclusion chromatography (SEC) with ultra-high-performance liquid chromatography (uHPLC) (Yoshikawa et al., 2018). We have shown that polysomes and ribosomal subunits can be separated in extracts from human and mouse cultured cell lines and tissues, Drosophila embryos, and budding yeast.
Ribo Mega-SEC is rapid, highly reproducible, and compatible with most subsequent analytical techniques including MS-based proteomics analysis, RNA-Seq, (cryo-)EM, and most types of biochemical assays. Notably, physiological buffers rather than sucrose solutions can be used throughout, facilitating the analysis of fractionated samples. For example, Ribo Mega-SEC allows the convenient enrichment of a pool of ‘heavy polysomes,’ which consists of more than three ribosomes and translates mRNAs efficiently (Liang et al., 2018). Therefore, this can be used for polysome profiling to assess changes in translation efficiency of mRNA populations under different conditions in combination with RNA-Seq. Ribo Mega-SEC can also be used to check polysome/monosome (P/M) ratios, providing an alternative measurement of translational activity.
In addition to the separation of ribosomes and translation complexes, the specific setup and conditions for performing Ribo Mega-SEC can be customized readily by users to optimize analysis depending on experimental aims. On the other hand, despite the advantages of speed, accuracy, and high reproducibility, polysomes are predominantly eluted in a single major peak using Ribo Mega-SEC. Therefore, as previously discussed (Yoshikawa et al., 2018), if a comparison of different n-mer polysome species is critical for a specific experiment, such as ‘TrIP-Seq’ or ‘Poly-Ribo-Seq’ (Aspden et al., 2014; Floor and Doudna 2016), performing SDG analysis may still be preferable in those specific applications.
In this manuscript, we provide a detailed step-by-step protocol including practical ‘tips’ for performing Ribo Mega-SEC analysis successfully.
Thing to consider before starting
Sample preparation
Lysis buffer: We recommend the standard method of polysome extraction, as used in traditional SDG analysis. The lysis buffer contains the standard reagents used to stabilize polysomes (RNase inhibitors and Mg2+) (Klein et al., 2004; Panda et al., 2017; Poria and Ray, 2017; Shi et al., 2017; Simsek et al., 2017; Imami et al., 2018).
Detergent: The separation principle of SEC is effectively sorting molecules based on their size and shape, which is compatible with the physiological buffer used in cell lysis also being used for subsequent SEC separation. A typical buffer for polysome extraction contains a detergent, e.g., 0.5-1% Triton X-100 or NP-40; however, these detergents have a low critical micelle concentration (CMC) (Triton X-100: 0.0155%; NP-40: 0.0179%). The micelle formed above the CMC is relatively large (~90 kDa) (https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma/Product_Information_Sheet/1/detergent-selection-guide.pdf). Moreover, these detergents have a strong absorbance at 280 nm, which results in background noise during SEC. We therefore selected CHAPS as the detergent of choice for Ribo Mega-SEC because: 1) the CMC is sufficiently high (0.4920-0.6150%) to be used effectively in SEC separation buffers below the CMC; 2) the micelle size is small (6,150 Da); and 3) the absorbance at 280 nm is weak. CHAPS is also known to solubilize protein complexes without causing major denaturation (Hjelmeland, 1980). Moreover, we confirmed that the recovery efficiency of polysomes and ribosomal subunits was similar for lysates prepared from HeLa cells using either 1% Triton X-100 or 1% CHAPS (Yoshikawa et al., 2018). Therefore, we use 1% CHAPS for cell lysis and 0.3% CHAPS for the SEC running buffer. However, in principle, any detergent with a sufficiently high CMC is suitable for use in SEC separation.
Translation elongation inhibitor (Cycloheximide (CHX), Emetine): In our initial experiments, we treated cells with CHX to ‘freeze’ the polysome structure on mRNAs following standard methods of polysome extraction. As CHX is a reversible translation elongation inhibitor, it should be included in all solutions; otherwise, the effect is reduced (Schneider-Poetsch et al., 2010). Emetine is another translation elongation inhibitor but it is irreversible (David et al., 2012). However, considering that there are potential side-effects of treating cells with translation inhibitors (Gerashchenko and Gladyshev 2014; Santos et al., 2019), we now prefer not to use any translation elongation inhibitors for preparing polysomes from mammalian cells (see Figure 3). Please note, however, that CHX should be added for preparing polysomes from budding yeast.
The amount of material for fractionation
For analysis of polysomes and ribosomes, we usually inject lysates containing 10-20 µg RNA (usually equivalent to ~100-200 µg protein). Therefore, for a typical analysis, lysate prepared from cells cultured in a 60-mm dish (~3.0 × 106 cells) is sufficient. For SEC analysis, we recommend injecting up to 100 µg RNA (equivalent to ~1 mg protein) in 200 µl, since injecting very large quantities (e.g., more than 2 mg protein in 200 µl) can affect column performance and lead to clogging (please note that the SEC column is an analytical not preparative column). If larger amounts of fractionated material are required for downstream analysis, we recommend achieving this by performing multiple runs, i.e., injecting smaller amounts of the same lysate multiple times onto the same SEC column, rather than injecting the same total amount in one large, concentrated sample. The resulting equivalent fractions from each individual run can then be pooled and used for further analysis. The very high reproducibility of the uHPLC fractionation system ensures that each aliquot is directly comparable, and equivalent fractions from separate runs are, in our experience, near identical.
Choice of column
The 80S ribosome in human cells has a molecular weight of ~4.3 MDa and a diameter of 250-300 Å. The very large size of polysome complexes, which contain multiple ribosomes on a single mRNA, is outside the effective separation range of the typical ~300-500 Å pore size SEC columns commonly used for separating individual proteins and smaller complexes, e.g., Superose 6 column. Ribosome and polysome complexes simply emerge in the void volume of these columns; therefore, we use a 2,000 Å pore size SEC column for the separation of polysomes and ribosomal subunits isolated from human cells. This large pore size column successfully resolves complexes in the polysome size range, with essentially no protein peaks detected in the void volume (Yoshikawa et al., 2018). This 2,000 Å SEC column can also be used for the separation of lysates from tissue samples (Yoshikawa et al., 2018).
We note that there is a trade-off in the choice of SEC column depending on the resolution required in a specific experiment. Thus, the use of a 1,000 Å pore size SEC column provides a higher resolution separation between 60S and 40S mammalian ribosomal subunits, as well as between the 40S subunit and smaller protein complexes, but results in the 80S peak almost overlapping the polysome peak (Yoshikawa et al., 2018). For analysis of translation complexes in other species, we find that the 1,000 Å SEC column is suitable for extracts from either Drosophila embryos or budding yeast (Figure 1).
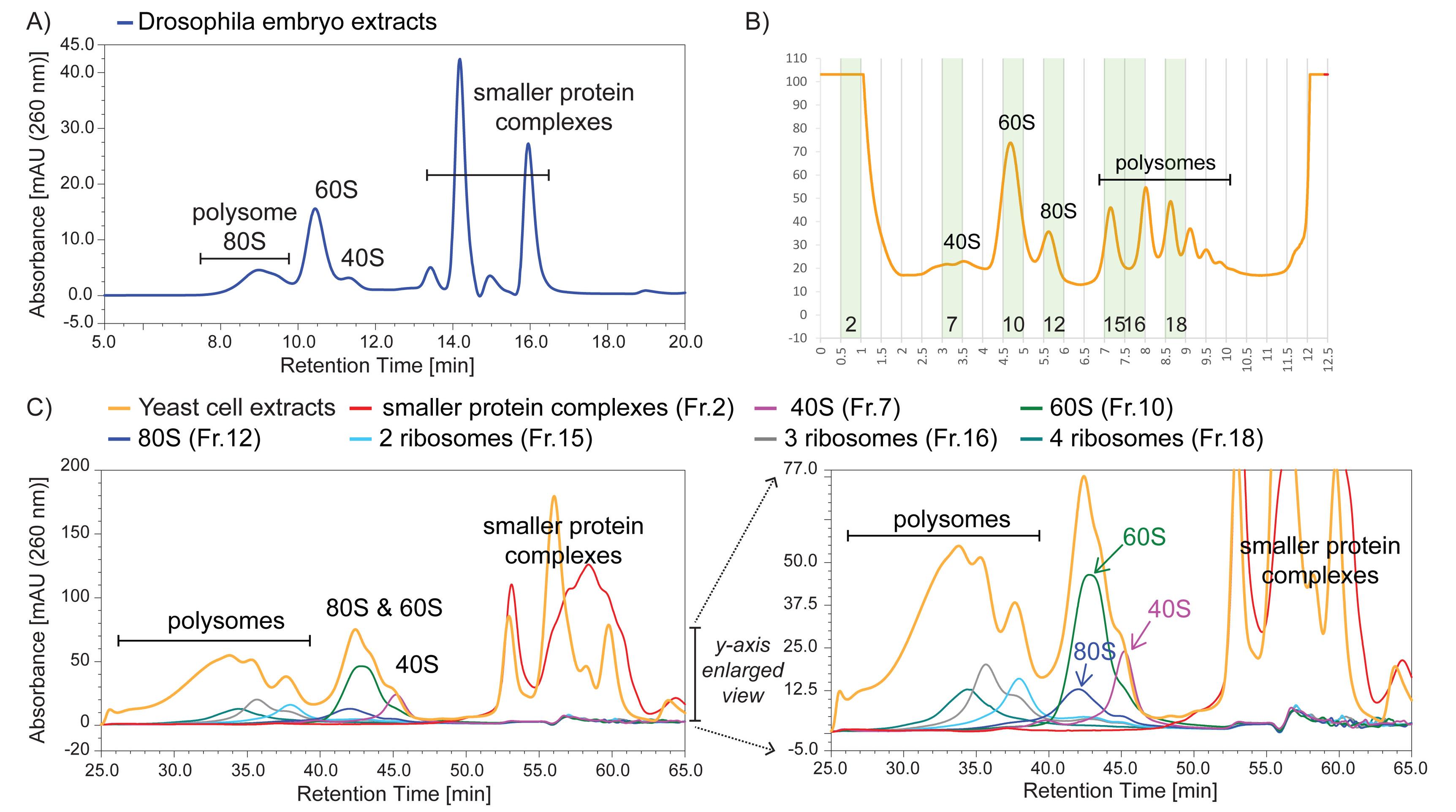
Figure 1. Ribo Mega-SEC profiles of Drosophila embryo and budding yeast cell extracts using a 1,000 Å pore size SEC column. A) Drosophila embryo extracts were analyzed using a flow rate of 0.8 ml/min. B) Yeast cell extracts were analyzed by sucrose density gradient fractionation, and the highlighted fractions were collected for subsequent Ribo Mega-SEC analysis. C) Yeast cell extracts that were used for the sucrose density gradient analysis, or the fractions collected in Figure 2C, were injected onto a 1,000 Å pore size SEC column using a flow rate of 0.2 ml/min. The right panel shows a y-axis enlarged view of the left panel.Column performance evaluation
To evaluate the performance of 2,000 Å and 1,000 Å pore size SEC columns, we use three different materials as size standards: 1) BSA; 2) DNA HyperLadder 1 kb; and 3) HeLa cell lysates. While BSA elutes at the penetration limit where ‘molecules below a certain size completely penetrate the pores and tend to elute almost in the same position’ (https://www.shimadzu.com/an/service-support/technical-support/analysis-basics/basic/55/55intro.html), several DNA fragments elute between the exclusion limit and the penetration limit (Figure 2). Moreover, injecting HeLa cell lysates containing 10 µg RNA onto the SEC column provides the ‘standard’ separation of polysomes, 80S, 60S, 40S, and other smaller protein complexes (Figure 2). We monitor the following parameters to determine the column performance: 1) shape, 2) intensity, 3) resolution, and 4) peak retention time.
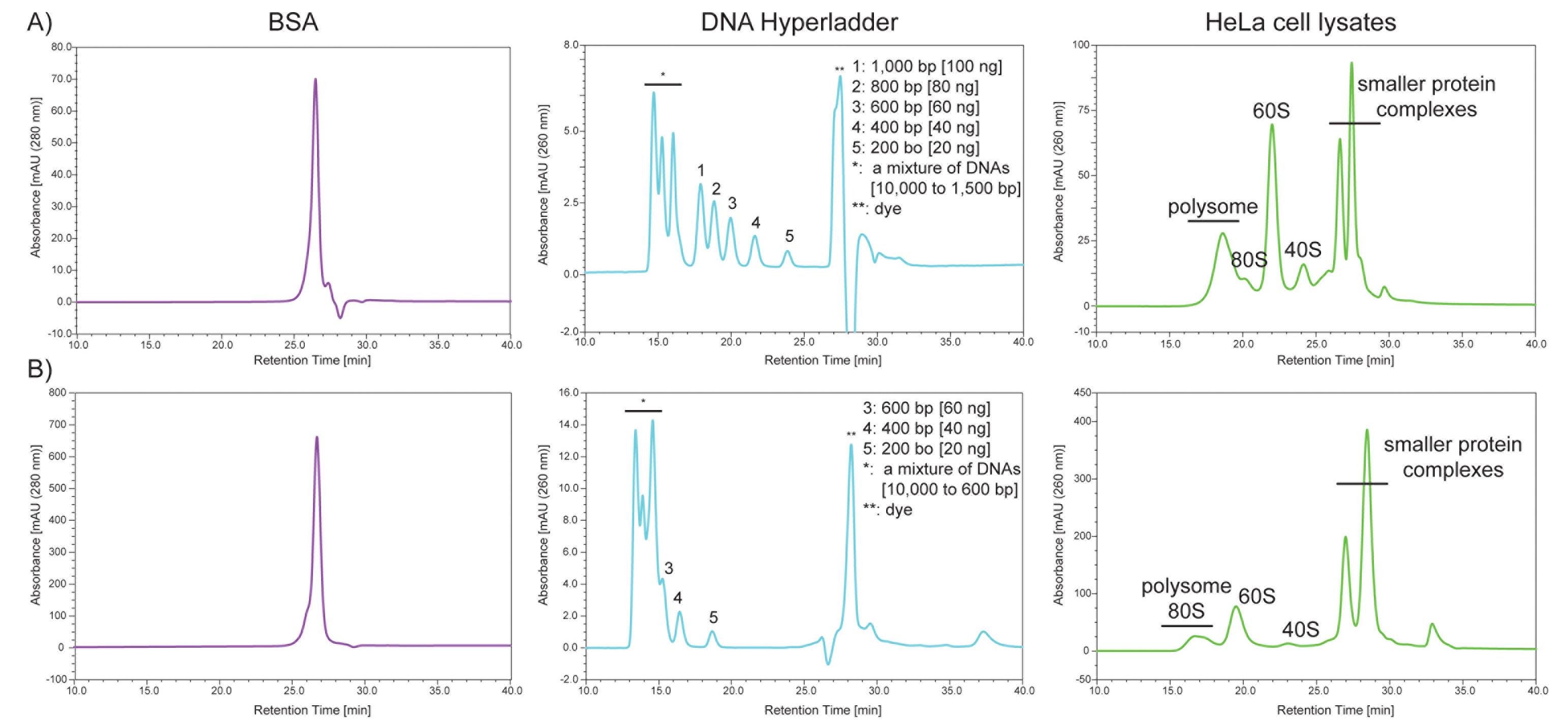
Figure 2. Typical separation profiles of the standards by SEC using a 2,000 Å pore size SEC column (A) or a 1,000 Å pore size SEC column (B) with a flow rate of 0.4 ml/minFlow rate, column pressure, and resolution
In a typical experiment, we analyze samples using a 2,000 Å pore size SEC column with a flow rate of 0.2 ml/min, a separation time of 60 min, and an operating pressure of ~40 bar at 5°C in the column compartment. This provides high-resolution separation of large complexes. When using a column flow rate of 0.8 ml/min, the separation time is reduced to 15 min with an operating pressure of ~160 bar at 5°C. Considering the pressure, a fast protein liquid chromatography (FPLC) instrument can also be used for Ribo Mega-SEC. We recommend using a column flow rate of 0.8 ml/min for initial screening, then 0.4 ml/min for most subsequent detailed analyses.
In addition to the column flow rate, injection volume is another important factor affecting the column resolution. It is recommended that the injection volume is less than 2% of the total column volume. In our uHPLC system, the maximum injection volume is set to 200 µl, which is equal to 1.4% of the column volume of the Agilent Bio SEC-5 column (7.8 mm × 300 mm). For comparison of samples between conditions, we prepare lysates with the same total concentration so that the same volume can be injected with the same amount of protein and RNA.
Sample Normalization
Normalization of sample amounts, by measuring the total protein and/or RNA content in the cell/tissue extracts, enables the reliable detection of differences in the relative levels of polysomes and ribosomal subunits across conditions. For example, decreased polysome levels, with a corresponding increase in the peaks for 80S ribosome and 60S and 40S subunits, were detected in lysates from amino acid-starved mammalian cells in comparison with cells cultured under normal growth conditions (Yoshikawa et al., 2018). An alternative approach is to use cell number for normalization, which provides information on changes in both proportion and abundance (Brenes et al., 2021; Galloway et al., 2021).
Materials and Reagents
Agilent Bio SEC-5 HPLC column (7.8 × 300 mm, 5 μm particles) 1,000 Å (Agilent, catalog number: 5190-2536) or 2,000 Å (Agilent, catalog number: 5190-2541)
Vials (e.g., Agilent, catalog number: 5184-3550)
Caps (e.g., Agilent, catalog number: 5182-0723)
Deep-well Plate 96/1,000 µl, Protein LoBind (Eppendorf, catalog number: 0030504208)
Ultrafree-MC Centrifugal Filter, 0.45-µm pore size, hydrophilic PVDF, 0.5-ml volume, non-sterile (Merck Millipore, catalog number: UFC30HVNB)
Stericups (Merck Millipore)
1.5-ml tubes (e.g., Sarstedt, catalog number: 72.690.001)
100-mm cell culture dishes (e.g., Corning, catalog number: 430791)
Cell scraper
Embryo collection cages (Flystuff.com, catalog number: 59-101)
w1118 or any other flies of the required genotype
Ice bucket
DPBS, no calcium, no magnesium (Thermo Fisher Scientific, catalog number: 14190144)
Tris-buffered saline (TBS)
HEPES
NaOH
Tris
Trisodium citrate dehydrate (Merck, Sigma-Aldrich, catalog number: C8532)
NaCl
MgCl2·6H2O
EDTA disodium salt
CHAPS, Sol-Grade (Anatrace, catalog number: C316S)
Glycerol
Dithiothreitol (DTT)
Heparin sodium salt from porcine intestinal mucosa (Merck, Sigma-Aldrich, catalog number: H3393)
Glass beads, acid-washed, 425-600 μm (Merck, Sigma-Aldrich, catalog number: G8772)
Cycloheximide (Merck, Sigma-Aldrich, catalog number: C7698)
Sodium dodecyl sulfate (SDS)
Pepsin (Merck, Sigma-Aldrich, catalog number: P7125)
RNase A (Merck, Sigma-Aldrich, catalog number: R5000)
10 mg/ml BSA solution
Trichloroacetic acid solution (Merck, Sigma-Aldrich, catalog number: T0699)
TRI Reagent (Merck, Sigma-Aldrich, catalog number: T3934)
SUPERase•InTM RNase Inhibitor (20 U/μl) (Thermo Fisher Scientific, catalog number: AM2694)
cOmpleteTM, EDTA-free Protease Inhibitor Cocktail (Roche, catalog number: 11873580001)
PierceTM 660 nm Protein Assay Kit (Thermo Fisher Scientific, catalog number: 22662)
HyperLadder 1 kb (BIOLIBE, catalog number: BIO-33053)
2× SSC, 1% SDS solution
Filtered Milli-Q (MQ) water
Filtered 20% ethanol (EtOH)
Apple juice agar (CSH_Protocols 2011) dispensed in 100-mm plates
Polysome extraction buffer (see Recipes)
SEC running buffer (see Recipes)
50× protease inhibitor solution (see Recipes)
Pepsin solution (see Recipes)
RNase A solution (see Recipes)
20× SSC (see Recipes)
Standard HeLa cell lysates (see Recipes)
Note: If the vendor/manufacturer is not described in the standard laboratory chemicals above, these can be obtained from Merck (Sigma-Aldrich), VWR, or Formedium.
Equipment
Ultimate 3000 Bio-RS HPLC with Diode Array Detector (Thermo Fisher Scientific)
Vortex mixer (e.g., Scientific Industries, Vortex-Genie 2)
Refrigerated microcentrifuge (e.g., Thermo Fisher Scientific, FrescoTM 17 Microcentrifuge)
Eppendorf BioPhotometer
Biospec Mini-Beadbeater-24
Software
Chromeleon software (Thermo Fisher Scientific)
Procedure
uHPLC setup
Prepare and filter all solutions and SEC running buffer (see Recipes): SEC running buffer in line ‘A,’ MQ water in line ‘B’, and 20% EtOH in line ‘C’.
Purge the pump with the default settings (3 ml/min, 5 min).
In the column compartment, attach a 2,000 Å pore size SEC column for mammalian samples or a 1,000 Å pore size SEC column for drosophila or yeast samples.
Flush the column with at least 2 column volumes (CV) of MQ water, using a flow rate of 0.2-0.8 ml/min at room temperature.
Set the temperature of the column compartment at 5°C and that of the autosampler/fraction collection compartment at 4°C (Note 1).
Equilibrate the column by flushing with at least 2 CV SEC buffer at a flow rate of 0.8 ml/min.
Start a blank run to normalize the baseline at 260 and 280 nm.
Check an operating pressure of ~160 bar at 5°C using a flow rate of 0.8 ml/min.
Inject 100 µl 1 mg/ml BSA solution to block the sites of non-specific interactions.
Inject 10 µl 1 mg/ml BSA solution to run as part of the standard and check the separation (Figure 2).
Inject 10 µl HyperLadder solution to run as part of the standard and check the separation (Figure 2).
Inject standard HeLa cell lysates containing 10 µg RNA (Figure 2) (Note 2).
Start another blank run.
Sample Preparation (all centrifugations at 4°C)
Lysates of human cell lines
Wash cells in one 100-mm dish (~80% confluence; approximately ~5 × 106 cells) twice with ice-cold PBS.
Harvest cells by scraping in 1 ml ice-cold PBS and collect them in a 1.5-ml tube on ice.
Centrifuge at 500 × g for 5 min and remove the supernatant carefully using an aspirator.
Rinse cells with 1 ml ice-cold PBS if needed.
Cell pellets can either be snap-frozen in liquid nitrogen and stored at -80°C or immediately used for lysate preparation (Note 4).
Lyse cells by pipetting, vortexing for 10 s in 250 μl polysome extraction buffer, and incubating on ice for 15 min.
Centrifuge at 17,000 × g for 20 min to pellet the debris.
Collect and filter the supernatants through 0.45-μm Ultrafree-MC HV centrifugal filter units, spinning at 12,000 × g for 2-5 min (Note 3).
Transfer the filtrates into new 1.5-ml tubes and leave on ice.
Lysates can be snap-frozen in liquid nitrogen and stored at -80°C (Note 4).
Measure RNA and protein concentrations in the filtrates using a BioPhotometer and a PierceTM 660 nm Protein Assay Kit, respectively.
Equalize the concentrations of the samples across the conditions by diluting in polysome extraction buffer (Note 5).
Transfer lysates into a vial (12 × 32 mm), close the cap, and keep on ice.
(Optional) Place 5 µl protease inhibitors (50× stock solution) and 2 U RNase inhibitors in each well of a 96 deep-well plate.
Lysates of Drosophila embryos
Set up egg collection cups with w1118 or any other required genotype of flies. Change the apple juice agar plates 2-4 times every day for 2 days.
On the day of embryo collection, change the apple juice agar plate twice after 30-min collections. Discard the plates.
Subsequently, collect embryos in a fresh plate for 30 min at 25°C.
Age the collected embryos for an additional 40 min. At this point, the embryos will be 40-70-min-old and therefore will still be in the syncytial stage.
Dechorionate the collected and aged embryos with 5% bleach until most of the embryos rise to the surface of the solution.
Wash the embryos thoroughly with tap water and dry on a tissue.
Collect the dechorionated embryos using a paintbrush and transfer them to a 1.5-ml tube.
At this point, embryos can either be snap-frozen and stored at -80°C or processed further.
Add 400 μl polysome extraction buffer, lyse the embryos with a pestle that fits into the 1.5-ml tube, and incubate on ice for 15 min.
Follow the steps ‘g to n’ described in the ‘Lysates of human cell lines.
Lysates of budding yeasts
Grow yeast cultures on YPD or selective media. Take a sweep of the yeast strain of interest grown on a YPD plate with clean toothpick and inoculate a 5-ml culture.
Grow overnight to the early stationary phase. In the morning, inoculate a 500-ml culture with 5 ml overnight culture and grow in a 2-L Erlenmeyer glass flask at 30°C until it reaches OD600 ~1.0.
To stabilize the polysomes before harvesting, add cycloheximide (CHX) at a final concentration of 0.1 mg/ml and incubate for 10 min on ice.
Spin the cells for 10 min at 4,000 × g and discard the supernatant.
Wash the cell pellet with ice-cold TBS containing 0.1 mg/ml CHX.
Resuspend the pellet in 10 ml polysome extraction buffer containing 0.1 mg/ml CHX.
Separate the resuspended cells into 1-ml aliquots in 2-ml tubes.
Cells can be snap-frozen in liquid nitrogen and stored at -80°C (Note 4).
Add an equal volume of glass beads to the tube and mix.
Disrupt the cells in a glass bead beater at 4°C; 2 min bead beating with 2 min interval on ice.
Repeat this process 6 times.
Take 1 μl disrupted cells and dilute to 10 μl; check under the microscope for ghosting of cells.
Centrifuge the disrupted cells at 6,000 × g, 4°C for 5 min.
Transfer the supernatant to a pre-cooled 1.5-ml microcentrifuge tube.
Clarify the lysates by two successive centrifugations at 11,300 × g, 4°C for 5 min, each time transferring the supernatant carefully to a new pre-cooled 1.5-ml tube.
Lysates can be snap-frozen in liquid nitrogen and stored at -80°C (Note 4).
Follow steps ‘k to n’ described in the ‘Lysates of human cell lines.’
Ribo Mega-SEC run
Place the vial and the plate in the autosampler/fraction collection compartment.
Inject 1/5 volume to an equal volume of the lysates and decide the time at which to collect the fractions based on the chromatogram.
Set the time for fraction collection in the sequence (Note 6).
Analyze the samples using the sequence set up.
Sample preparation from Ribo Mega-SEC fractions
For western blotting, precipitate the proteins by trichloroacetic acid (TCA) precipitation followed by an ethanol wash or methanol/chloroform precipitation and resuspend the protein pellets in SDS sample buffer. Where necessary, add Tris to the samples to adjust the pH.
To prepare samples for proteomics analysis, digest RNA in the samples with benzonase, reduce and alkylate the proteins, and clean up the proteins using FASP (Wisniewski et al., 2009; Potriquet et al., 2017), which efficiently removes salts and detergents such as heparin and CHAPS. SP3-based protein clean up (Hughes et al., 2014) is an alternative approach that can be used if samples do not need to contain heparin for separating molecules or protein complexes other than ribosomes.
To prepare RNA from the fractions, extract RNA using TRIzol LS (Thermo) or an equivalent product. Please note that RNA needs to be re-purified by LiCl precipitation for RNA-Seq or RT-qPCR to remove heparin (Jung et al., 1997).
Column maintenance after analyzing samples
Flush with MQ water using at least 2 CV, with a flow rate of 0.2-0.8 ml/min at room temperature (Note 7, Note 8).
Flush with 20% EtOH using at least 2 CV, with a flow rate of 0.2-0.4 ml/min at room temperature.
Take the column from the uHPLC and store it according to the manufacturer’s instructions.
Column cleanup
Flush with MQ water using at least 5 CV, with a flow rate of 0.2-0.8 ml/min at room temperature.
Flush with pepsin solution using at least 5 CV, with a flow rate of 0.2-0.8 ml/min at 37°C; incubate the column for 1 h at 37°C without flow (Note 9).
Flush with RNase A solution using at least 5 CV, with a flow rate of 0.2-0.8 ml/min at 37°C; incubate the column for 1 h at 37°C without flow (Note 9).
Flush with 2× SSC, 1% SDS solution using at least 10 CV, with a flow rate of 0.2-0.8 ml/min at 50°C.
Rinse the column with at least 10 CV MQ water, using a flow rate of 0.2-0.8 ml/min at room temperature.
Data analysis
We use the Chromeleon software to run a uHPLC and to analyze data. To compare the data, we draw a mean profile together with standard deviation across biological replicates [see our original paper (Yoshikawa et al., 2018)]. We also use the area under the curve calculated by the Chromeleon software (Figure 3).
As an example, we compared polysomes by Ribo Mega-SEC using lysates from HeLa cells treated with or without CHX or Emetine. Better recovery was seen from Emetine-treated cells as compared with CHX-treated cells (Figure 3). We also detected sufficient yield of polysomes for most experiments from untreated cells, despite the yield being lower than from either CHX- or Emetine-treated cells (Figure 3).
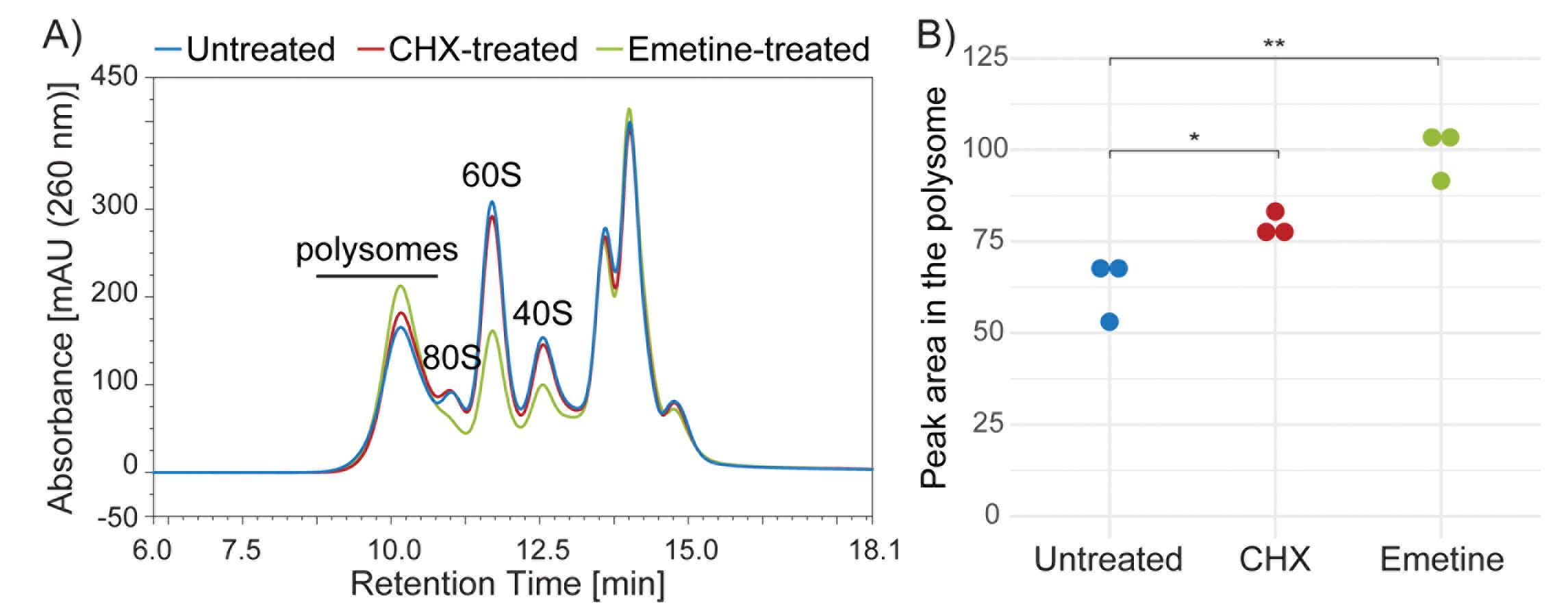
Figure 3. SEC separation profiles of lysates prepared from untreated, Cycloheximide (CHX)-treated or Emetine-treated HeLa cells. A) Lysates prepared from untreated HeLa cells (blue line) or cells treated with CHX (red line) or Emetine (green) were analyzed by Ribo Mega-SEC using a 2,000 Å SEC column with a flow rate of 0.8 ml/min. B) Quantitation of the area under the curve in the polysome region shown in Figure 1A. Dots indicate biological replicates. *P < 0.05, **P < 0.01.
Notes
We usually use the ‘Set Delay’ option to conduct steps 6-12 during the night, such that we can start analyzing samples the next morning.
To check reproducibility, injecting HeLa cell lysates at least twice is advisable when enough sample is available.
Filters tend to become clogged when lysates contain lipids. Where necessary, lysates remaining on the top of the filter unit need to be transferred to a new filter.
Snap-frozen lysates or cells can be kept for at least 6 months at -80°C.
Injection volume affects peak resolution; therefore, an equal volume should be injected across conditions.
We typically collect either 12, 24, or 48 fractions, ranging from polysomes to ‘small RNPs.’
Running two blanks, followed by injecting BSA and HyperLadder solution, is advisable to check the column condition. We typically see a good separation for up to ~50 injections of the lysates containing 20 µg RNA.
When new samples are to be analyzed the next day, the column is flushed with at least 2 CV MQ water and re-equilibrated with SEC buffer.
We found that rRNAs tend to interact with the fused silica in an SEC column. As rRNAs are protected by ribosomal proteins, we digest proteins using pepsin prior to RNase digestion to remove RNAs.
Recipes
Polysome extraction buffer
20 mM HEPES-NaOH (pH 7.4)
130 mM NaCl
10 mM MgCl2
5% glycerol
1% CHAPS
0.2 mg/ml heparin
Filter either through a high PVDF filter or using a Stericup to sterilize and remove any contaminating RNase traces. Store for up to 6 months at 4°C.
Add before use:
2.5 mM DTT (from a 1 M stock solution stored at -20°C)
20 U SUPERase in RNase inhibitor/1 ml polysome extraction buffer
cOmplete EDTA-free protease inhibitor (from 50× stock solution stored at -20°C; see Recipes for ‘50× protease inhibitor solution’ below)
SEC running buffer
20 mM HEPES-NaOH (pH 7.4)
60 mM NaCl
10 mM MgCl2
5% glycerol
0.3% CHAPS
0.2 mg/ml heparin
2.5 mM DTT
Filter either through a high PVDF filter or using a Stericup to sterilize and remove any contaminating RNase traces. Ideally, always prepare fresh, but it can be stored at room temperature for 2 days.
50× protease inhibitor solution
1 tablet cOmpleteTM, EDTA-free protease inhibitor cocktail
0.5 ml MQ water
Resuspend well in a 1.5-ml tube and store at -20°C.
Pepsin solution
20 μg/ml pepsin
0.1 M acetic acid
0.5 M NaCl
Filter either through a high PVDF filter or using a Stericup to sterilize and remove any contaminating RNase traces. Always prepare fresh.
RNase A solution
20 μg/ml RNase A
10 mM Tris-HCl (pH 7.4)
0.5 M NaCl
Filter either through a high PVDF filter or using a Stericup to sterilize and remove any contaminating RNase traces. Always prepare fresh.
20× SSC
3 M NaCl
0.3 M trisodium citrate dehydrate
2× SSC, 1% SDS solution
Filter either through a high PVDF filter or using a Stericup to sterilize and remove any contaminating RNase traces. Store at room temperature.
Acknowledgments
This protocol was adapted with minor modification from previous study published by Yoshikawa et al. (2018). We thank our colleagues in the Lamond laboratory for valuable suggestions. This research was supported by the following grants: Naito Foundation Grant for Studying Overseas (2013-413 to HY); Uehara Memorial Foundation Postdoctoral Fellowship (201430061 to HY); European Union's Horizon 2020 Research and Innovation Programme under the Marie Sklodowska-Curie Individual Fellowship (657087 to HY); and Tenovus Scotland Small Pilot grants (round 2017 to HY).
Competing interests
The authors declare no competing interests.
References
- Aspden, J. L., Eyre-Walker, Y. C., Phillips, R. J., Amin, U., Mumtaz, M. A., Brocard, M. and Couso, J. P. (2014). Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. Elife 3: e03528.
- Brenes, A. J., Yoshikawa, H., Bensaddek, D., Mirauta, B., Seaton, D., Hukelmann, J. L., Jiang, H., Stegle, O. and Lamond, A. I. (2021). Erosion of human X chromosome inactivation causes major remodeling of the iPSC proteome. Cell Rep 35(4): 109032.
- Britten, R. J. and Roberts, R. B. (1960). High-Resolution Density Gradient Sedimentation Analysis. Science 131(3392): 32-33.
- CSH_Protocols. (2011). Drosophila apple juice-agar plates. Cold Spring Harb Protoc 2011(9): pdb.rec065672-pdb.rec065672.
- David, A., Dolan, B. P., Hickman, H. D., Knowlton, J. J., Clavarino, G., Pierre, P., Bennink, J. R. and Yewdell, J. W. (2012). Nuclear translation visualized by ribosome-bound nascent chain puromycylation. J Cell Biol 197(1): 45-57.
- Floor, S. N. and Doudna, J. A. (2016). Tunable protein synthesis by transcript isoforms in human cells. Elife 5: e10921.
- Galloway, A., Kaskar, A., Ditsova, D., Atrih, A., Yoshikawa, H., Gomez-Moreira, C., Suska, O., Warminski, M., Grzela, R., Lamond, A. I., Darzynkiewicz, E., Jemielity, J. and Cowling, V. H. (2021) Upregulation of RNA cap methyltransferase RNMT drives ribosome biogenesis during T cell activation. Nucleic Acids Res 49 (12): 6722-6738
- Gerashchenko, M. V. and Gladyshev, V. N. (2014). Translation inhibitors cause abnormalities in ribosome profiling experiments. Nucleic Acids Res 42(17): e134.
- Hjelmeland, L. M. (1980). A nondenaturing zwitterionic detergent for membrane biochemistry: design and synthesis. Proc Natl Acad Sci U S A 77(11): 6368-6370.
- Hughes, C. S., Foehr, S., Garfield, D. A., Furlong, E. E., Steinmetz, L. M. and Krijgsveld, J. (2014). Ultrasensitive proteome analysis using paramagnetic bead technology. Mol Syst Biol 10(10): 757.
- Imami, K., Milek, M., Bogdanow, B., Yasuda, T., Kastelic, N., Zauber, H., Ishihama, Y., Landthaler, M. and Selbach, M. (2018). Phosphorylation of the Ribosomal Protein RPL12/uL11 Affects Translation during Mitosis. Mol Cell 72(1): 84-98 e89.
- Jung, R., Lubcke, C., Wagener, C. and Neumaier, M. (1997). Reversal of RT-PCR inhibition observed in heparinized clinical specimens. Biotechniques 23(1): 24, 26, 28.
- Klein, D. J., Moore, P. B. and Steitz, T. A. (2004). The contribution of metal ions to the structural stability of the large ribosomal subunit. RNA 10(9): 1366-1379.
- Liang, S., Bellato, H. M., Lorent, J., Lupinacci, F. C. S., Oertlin, C., van Hoef, V., Andrade, V. P., Roffe, M., Masvidal, L., Hajj, G. N. M. and Larsson, O. (2018). Polysome-profiling in small tissue samples. Nucleic Acids Res 46(1): e3.
- Panda, A. C., Martindale, J. L. and Gorospe, M. (2017). Polysome Fractionation to Analyze mRNA Distribution Profiles. Bio-protocol 7(3): e2126.
- Poria, D. K. and Ray P. S. (2017). Polysome Analysis. Bio-protocol 7(6): e2192.
- Potriquet, J., Laohaviroj, M., Bethony, J. M. and Mulvenna, J. (2017). A modified FASP protocol for high-throughput preparation of protein samples for mass spectrometry. PLoS One 12(7): e0175967.
- Rheinberger, H. J. (2004). A history of protein biosynthesis and ribosome research. In: Protein synthesis and ribosome structure (pp. 1-51). Wiley-VCH Verlag, Weinheim, Germany.
- Santos, D. A., Shi, L., Tu, B. P. and Weissman, J. S. (2019). Cycloheximide can distort measurements of mRNA levels and translation efficiency. Nucleic Acids Res 47(10): 4974-4985.
- Schneider-Poetsch, T., Ju, J., Eyler, D. E., Dang, Y., Bhat, S., Merrick, W. C., Green, R., Shen, B. and Liu, J. O. (2010). Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol 6(3): 209-217.
- Shi, Z., Fujii, K., Kovary, K. M., Genuth, N. R., Rost, H. L., Teruel, M. N. and Barna, M. (2017). Heterogeneous Ribosomes Preferentially Translate Distinct Subpools of mRNAs Genome-wide. Mol Cell 67(1): 71-83 e77.
- Simsek, D., Tiu, G. C., Flynn, R. A., Byeon, G. W., Leppek, K., Xu, A. F., Chang, H. Y. and Barna, M. (2017). The Mammalian Ribo-interactome Reveals Ribosome Functional Diversity and Heterogeneity. Cell 169(6): 1051-1065 e1018.
- Wisniewski, J. R., Zougman, A., Nagaraj, N. and Mann, M. (2009). Universal sample preparation method for proteome analysis. Nat Methods 6(5): 359-362.
- Yoshikawa, H., Larance, M., Harney, D. J., Sundaramoorthy, R., Ly, T., Owen-Hughes, T. and Lamond, A. I. (2018). Efficient analysis of mammalian polysomes in cells and tissues using Ribo Mega-SEC. Elife 7: e36530.
Article Information
Copyright
![]() Yoshikawa et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Yoshikawa et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Yoshikawa, H., Sundaramoorthy, R., Mariyappa, D., Jiang, H. and Lamond, A. I. (2021). Efficient and Rapid Analysis of Polysomes and Ribosomal Subunits in Cells and Tissues Using Ribo Mega-SEC. Bio-protocol 11(15): e4106. DOI: 10.21769/BioProtoc.4106.
- Yoshikawa, H., Larance, M., Harney, D. J., Sundaramoorthy, R., Ly, T., Owen-Hughes, T. and Lamond, A. I. (2018). Efficient analysis of mammalian polysomes in cells and tissues using Ribo Mega-SEC. Elife 7: e36530.
Category
Biochemistry > Protein > Synthesis
Biochemistry > RNA > mRNA translation
Molecular Biology > RNA > RNA-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.