- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Protocol for Spontaneous and Chaperonin-assisted in vitro Refolding of a Slow-folding Mutant of GFP, sGFP
(*contributed equally to this work) Published: Vol 11, Iss 14, Jul 20, 2021 DOI: 10.21769/BioProtoc.4099 Views: 3644
Reviewed by: Emilia KrypotouAli Asghar KermaniAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Understanding the folding pathway of any protein is of utmost importance for deciphering the folding problems under adverse conditions. We can obtain important information about the folding pathway by monitoring the folding of any protein from its unfolded state. It is usually very difficult to monitor the folding process in real time as the process is generally very fast, and we need a suitable read out. In this protocol, we have solved this issue by using a protein that is non-fluorescent in its unfolded state but fluoresces in its native state after folding. The kinetics of refolding can be monitored by following the increase in fluorescence in real time. Previously, this was generally achieved by either monitoring a protein’s enzymatic activity or measuring the tryptophan fluorescence, where the signal output depends on well-described enzymatic activity or the frequency of tryptophan residues present in the proteins, respectively. Here, we describe a simple and real-time assay to monitor the refolding of sGFP, a recently described slow-folding mutant of yeGFP (yeast enhanced GFP). We unfold this protein using chemical denaturant and refold in a suitable buffer, monitoring the increase in fluorescence over time. GFP is fluorescent only when correctly folded; thus, using this technique, we can measure the true rate of protein refolding by following the increase in fluorescence over time. Therefore, sGFP can be used as an ideal model to study the in vitro protein folding process. Accordingly, the effects of different conditions and molecules on the protein folding pathway can be efficiently studied using sGFP as a model protein.
Graphical abstract: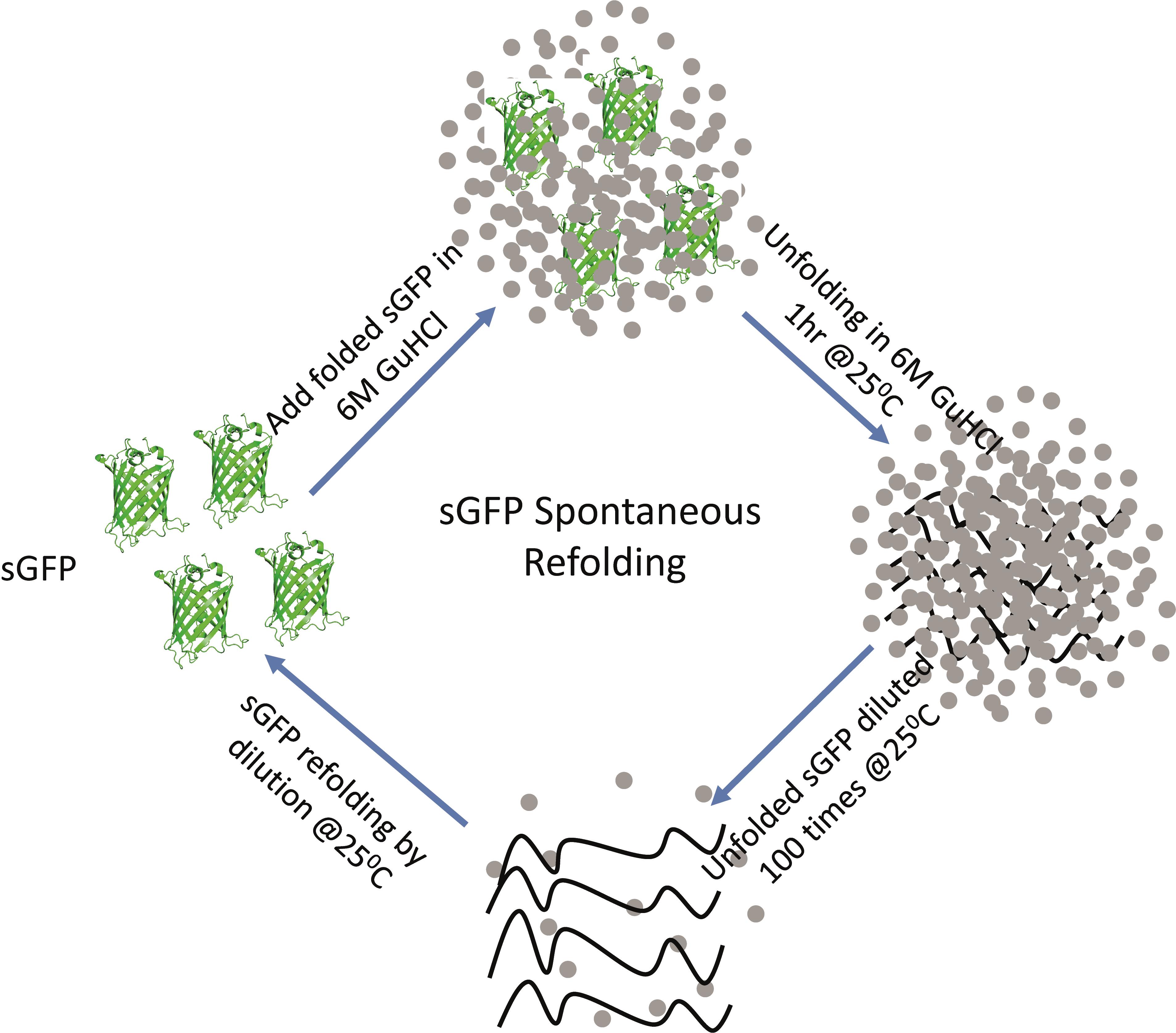
Schematic of the steps involved in the sGFP refolding pathway. Native sGFP is unfolded by chemical denaturation using 6 M GuHCl at 25°C for 1 hour and then refolded in refolding buffer by 100-fold dilution.
Background
Deciphering the folding pathway of proteins is one of the most explored topics in biology to date. Once synthesized, a nascent polypeptide chain is folded into its native structure, which is essential for its biological function. These polypeptide chains, especially the unfolded chains of small single-domain proteins, often fold spontaneously without any active assistance from other proteins. In contrast, larger multi-domain proteins frequently require folding assistance from proteins, commonly known as molecular chaperones, to fold into their native structure. The process of protein folding from nascent chains to correctly folded native states is extremely fast on a biologically relevant time scale. Thus, in order to study the steps of the folding pathway, we need to monitor the folding process using a slow-folding protein in a cell-free system, which is achieved by mimicking the cellular environment in vitro using suitable buffers. In an in vitro experiment, we unfold the test protein using a suitable denaturant and then refold it in a folding-favorable environment. Currently, the available protocols for measuring refolding rates are either based on measuring the enzymatic activity of proteins after they are refolded or monitoring the intrinsic tryptophan fluorescence of the protein (Weissman et al., 1994; Zahn et al., 1994; Makio et al., 1999; Doyle et al., 2003; Taniguchi et al., 2004; Kerner et al., 2005; Sharma et al., 2008; Malhotra et al., 2014; Jain et al., 2018). While measuring the refolding efficiency by estimating the enzymatic activity is one of the most suitable ways of checking the folded and active states of the protein, it largely depends on its well-known enzymatic activity. In the case of proteins without well-described enzymatic activity or activity that depends on the functional form of the protein, which may not form immediately after it is refolded, the estimation of refolding efficiency becomes difficult (Rospert et al., 1993). Another way of evaluating in vitro refolding is by measuring the tryptophan fluorescence, which again is largely dependent on the number of tryptophan residues and their location within the native structure (Chakraborty et al., 2010). We have devised an in vitro refolding assay of a recently identified in vivo substrate of the GroEL/ES chaperonin system (Sadat et al., 2020). In this protocol, we describe the methodology to monitor the refolding of a slow-folding yeGFP mutant protein, hereafter referred to as sGFP, in real time. We unfold the sGFP protein using guanidine hydrochloride as a chemical denaturant and subsequently refold the unfolded protein in a suitable refolding buffer in the presence and absence of chaperones (Sadat et al., 2020). The potential issues that are frequently encountered during in vitro refolding experiments are: (a) achieving complete unfolding of the protein in a reversible manner; (b) measuring the exact time taken by the unfolded protein to fold into its native form; and (c) accurately measuring the refolding rate, since most proteins fold very fast and this becomes a potential problem for measuring the in vitro refolding rate of a protein. Using sGFP successfully evades these potential problems in monitoring the refolding process in real time; therefore, our protocol can be a very useful guide for measuring the in vitro refolding rate of fluorescent proteins possessing similar characteristics to GFP.
Materials and Reagents
Tips: Sterile pipette tips (low-retention tips)
Falcon, 15-ml
Sterile microcentrifuge tubes (1.5-ml)
Wash bottle
Liquid discard beaker
Kimwipe tissue paper (Kimberly-Clark, catalog number: 34120)
Microcentrifuge tube rack
Falcon rack
Fluorescence cuvette (3.0 mm path length) (Hellma Analytics, catalog number: Z802336)
0.22-μm filter (Millex, catalog number: SLGP099RS)
10-ml syringe
MilliQ water (deionized water)
Methanol (100%) (HIMEDIA, catalog number: AS061-2.5L)
Guanidine hydrochloride (MP, catalog number: 105696)
Magnesium chloride (HIMEDIA MB, catalog number: 040-500G)
Potassium chloride (Sigma, catalog number: 793590)
Tris (ultra-pure) (MP, catalog number: 103133)
HCl (HMEDIA, catalog number: AS004-2.5L)
ATP (Sigma, catalog number: A2383)
DTT (MP, catalog number: 100597)
Purified protein (sGFP)
Purified chaperones and co-chaperones (GroEL, GroES)
sGFP (Sadat et al., 2020)
Buffer A (see Recipes)
Equipment
Pipettes: 2.5-μl, 10-μl, 200-μl, 1-ml
Centrifuge
Fluorescence spectrophotometer (Fluorolog 3 Spectrofluorometer, Jobin Yvon, Horiba)
Dry bath
pH meter
Procedure
sGFP spontaneous refolding
Protein Unfolding
Add 2.5 μl sGFP (20 μM) to a microcentrifuge tube containing 7.5 μl 8 M GuHCl (final GuHCl concentration in the unfolding reaction is 6 M) in buffer A (see Recipes). Place the tube in a dry bath for 1 h at 25°C. This will lead to complete unfolding of the protein.
Protein Refolding
Switch on the Fluorolog instrument (Fluorescence spectrofluorometer). Keep the instrument switched ON for 45 min prior to fluorescence measurement, so that the lamp is uniformly heated and fluctuations are minimal.
Set the instrument to emission spectra mode. Keep the excitation wavelength at 350 nm and the emission spectra in the 365-430 nm range. After 45 min, add MilliQ H2O to a quartz microcuvette and take a Raman Spectra. If you get an emission peak at 397 nm, the spectrofluorometer is working fine.
Set the instrument to kinetic acquisition mode. Set the excitation wavelength to 480 nm and the emission wavelength to 515 nm. Set to anti-photobleaching mode. Set the collection interval to 20 s and the integration time to 0.7 s. The total time for the kinetics is set to 1,800 s. Set the temperature to 25°C. The slit-width is kept at 2 nm for excitation and 5 nm for emission.
Clean a quartz cuvette (Hellma Analytics, 10 mm path length) with 100% methanol and then MilliQ water, and wipe using a kimwipe. Take 2 μl (200 nM final) completely unfolded protein, dilute 100-fold in Buffer A (198 μl), and mix thoroughly by pipetting (total reaction volume 200 μl) in the cuvette.
Start the kinetics acquisition as soon as the mixing is finished (keep an equal delay in this step).
sGFP refolding typically follows a two-state pathway. The unfolded proteins are refolded in refolding buffer to their native state in a single step. In the refolding experiment, we monitor the increase in GFP fluorescence by exciting the refolding reaction at 480 nm and measuring the fluorescence emission at 515 nm. GFP fluoresces only after folding into its native state; therefore, GFP fluorescence is the readout for the amount of folded protein in our refolding experiment. The refolding kinetics trace obtained after the refolding reaction can be best fitted using nonlinear regression by a single exponential kinetics equation. The kinetics data are analyzed using the Origin software.
A mathematical model to fit the refolding kinetics:
The model for fitting the data obtained from refolding experiments is a mathematical equivalent of two-state folding kinetics. The one-phase exponential decay equation is as follows:
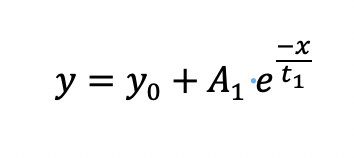
where y0 is the offset, A1 is the amplitude, and t1 is the time constant. The apparent rate of the refolding reaction is obtained by taking the reciprocal of the time constant. A typical refolding kinetics plot is shown in Figure 1a.
Chaperonin-assisted sGFP refolding
Protein Unfolding
Unfold sGFP as described earlier in Step A1.
Protein Refolding
a, b, c follow steps A2a, b, c as described for spontaneous refolding.
Take 2 μl (200 nM) completely unfolded protein and dilute 100-fold in Buffer A containing GroEL (400 nM tetradecamers of GroEL). Add GroES (800 nM heptamers of GroES), such that the ratio of substrate to GroEL toGroES is 1:2:4. The refolding is initiated by the addition of 2 mM ATP (final concentration); the total reaction volume is 200 μl. Mix the reaction thoroughly by pipetting slowly.
Start the kinetics acquisition as soon as the mixing is finished (keep an equal delay in this step).
A representative refolding kinetics plot is shown in Figure 1a. Figure 1b, 1c, 1d explains the various steps involved in the data analysis for calculating the refolding rate. 1/t1 gives the refolding rate/second.
Same as Step A2f.
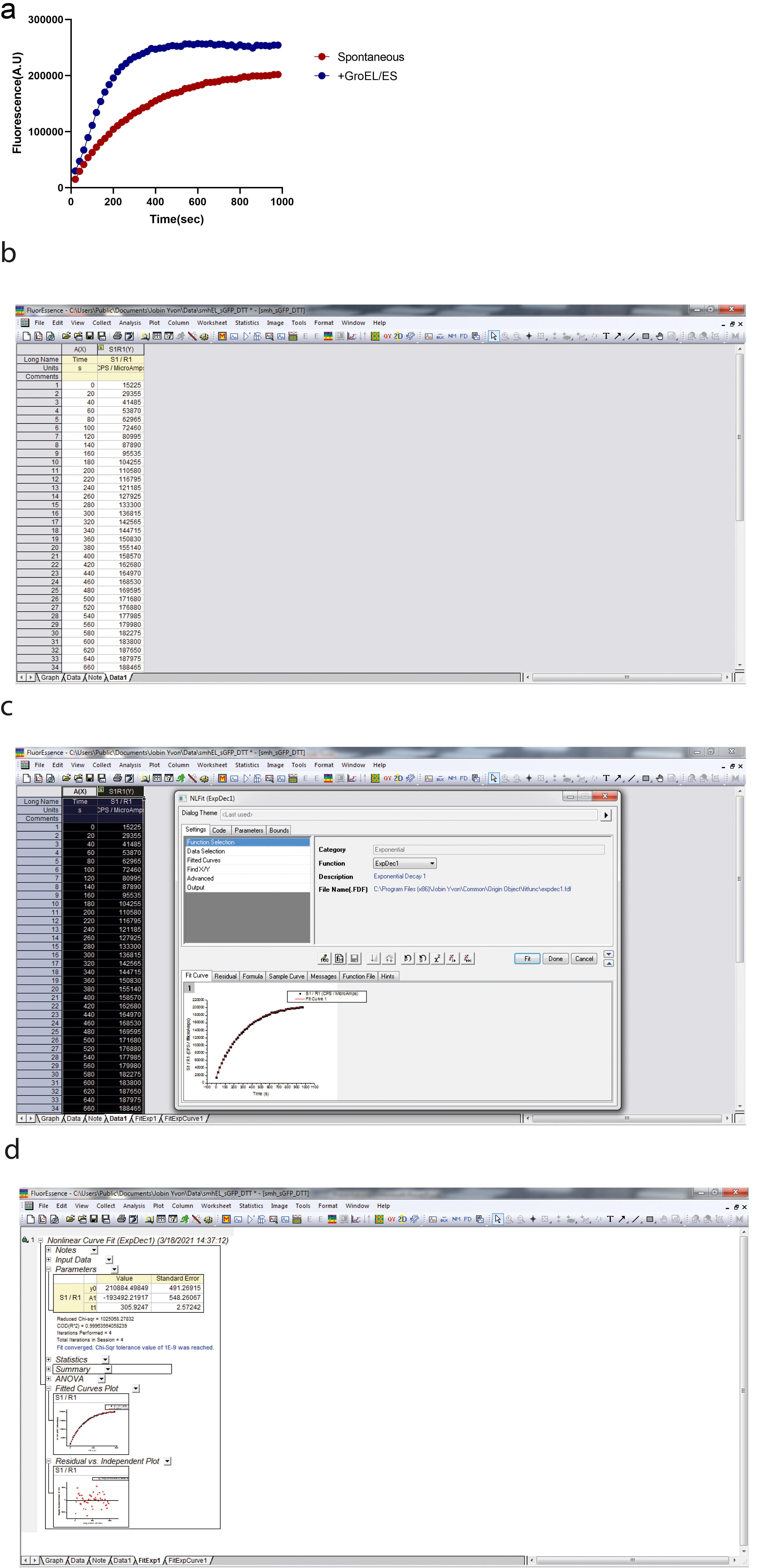
Figure 1. sGFP refolding plot. a.Plot showing the kinetics of sGFP refolding as measured by an increase in GFP fluorescence. sGFP was unfolded in 6 M GuHCl in Buffer A at 25°C for 1 h. Subsequently, unfolded sGFP was refolded by 100-fold dilution in Buffer A. b. The output screen after refolding. c. This screen appears by clicking on analysis after selecting the columns for time and fluorescence. d. This screen appears after the fitting by selecting ‘expdec1’ in the function section.
Notes
While this method can be used for monitoring the refolding of any fluorescent protein, we must keep in mind that only those fluorescent proteins that start to fluoresce as soon as they are folded are exclusively suitable for measuring refolding kinetics using the described protocol. As described, sGFP fluoresces as soon as it is folded into its native state; hence, we actually measure the rate of increase in sGFP fluorescence, which corresponds to the refolding rate of the protein. Therefore, this is an indirect method of calculating the refolding rate of sGFP. We assume that there are no such limitations in using this method for measuring the refolding rate of other fluorescent model proteins by similar spontaneous or chaperonin-assisted in vitro refolding assays. This protocol can be easily modified, especially the instrument settings (slit-width, excitation-emission wavelengths, total time of kinetic measurements), time of unfolding, concentrations of GuHCl, and other parameters depending on the properties of the model fluorescent protein used. In the case of chaperone-assisted refolding experiments, the molar ratio of substrate to chaperones in the refolding buffer must be optimized for the individual chaperone/co-chaperones used for the refolding assay.
Recipes
Buffer A
20 mM Tris pH 7.4
20 mM KCl
5 mM MgCl2
2 mM DTT
Filtered using a 0.22-μm filter and a 10-ml syringe
Acknowledgments
K.M. acknowledges the funding from the Department of Biotechnology (DBT), Government of India (grant number BT/PR28386/BRB/10/1671/2018) and the Science and Engineering Research Board (SERB), Government of India, for Core Research Grant (SERB/CRG/2019/006281) and SNU core funding. The work in the IGIB Lab was supported by a Swarnajayanthi Fellowship Grant from DST, and partly by BSC0124 from CSIR. Instrument support was also obtained from Wellcome Trust-DBT India Alliance and CSIR. KM, acknowledges SNU, and AS and ST acknowledge CSIR and CSIR-IGIB for infrastructural support. AS acknowledges DBT.
Competing interests
The authors declare no competing interests.
References
- Chakraborty, K., Chatila, M., Sinha, J., Shi, Q., Poschner, B. C., Sikor, M., Jiang, G., Lamb, D. C., Hartl, F. U. and Hayer-Hartl, M. (2010). Chaperonin-catalyzed rescue of kinetically trapped states in protein folding. Cell 142(1): 112-122.
- Doyle, S. M., Anderson, E., Zhu, D., Braswell, E. H. and Teschke, C. M. (2003). Rapid unfolding of a domain populates an aggregation-prone intermediate that can be recognized by GroEL. J Mol Biol 332(4): 937-951.
- Jain, N., Knowles, T. J., Lund, P. A. and Chaudhuri, T. K. (2018). Minichaperone (GroEL191-345) mediated folding of MalZ proceeds by binding and release of native and functional intermediates. Biochim Biophys Acta Proteins Proteom 1866(9): 941-951.
- Kerner, M. J., Naylor, D. J., Ishihama, Y., Maier, T., Chang, H. C., Stines, A. P., Georgopoulos, C., Frishman, D., Hayer-Hartl, M., Mann, M. and Hartl, F. U. (2005). Proteome-wide analysis of chaperonin-dependent protein folding in Escherichia coli. Cell 122(2): 209-220.
- Makio, T., Arai, M. and Kuwajima, K. (1999). Chaperonin-affected refolding of alpha-lactalbumin: effects of nucleotides and the co-chaperonin GroES. J Mol Biol 293(1): 125-137.
- Malhotra, P. and Udgaonkar, J. B. (2014). High-energy intermediates in protein unfolding characterized by thiol labeling under nativelike conditions. Biochemistry 53(22): 3608-3620.
- Rospert, S., Glick, B. S., Jeno, P., Schatz, G., Todd, M. J., Lorimer, G. H. and Viitanen, P. V. (1993). Identification and functional analysis of chaperonin 10, the groES homolog from yeast mitochondria. Proc Natl Acad Sci U S A 90(23): 10967-10971.
- Sadat, A., Tiwari, S., Verma, K., Ray, A., Ali, M., Upadhyay, V., Singh, A., Chaphalkar, A., Ghosh, A., Chakraborty, R., Chakraborty, K. and Mapa, K. (2020). GROEL/ES Buffers Entropic Traps in Folding Pathway during Evolution of a Model Substrate. J Mol Biol 432(20): 5649-5664.
- Sharma, S., Chakraborty, K., Muller, B. K., Astola, N., Tang, Y. C., Lamb, D. C., Hayer-Hartl, M. and Hartl, F. U. (2008). Monitoring protein conformation along the pathway of chaperonin-assisted folding. Cell 133(1): 142-153.
- Taniguchi, M., Yoshimi, T., Hongo, K., Mizobata, T. and Kawata, Y. (2004). Stopped-flow fluorescence analysis of the conformational changes in the GroEL apical domain: relationships between movements in the apical domain and the quaternary structure of GroEL. J Biol Chem 279(16): 16368-16376.
- Weissman, J. S., Kashi, Y., Fenton, W. A. and Horwich, A. L. (1994). GroEL-mediated protein folding proceeds by multiple rounds of binding and release of nonnative forms. Cell 78(4): 693-702.
- Zahn, R., Axmann, S. E., Rucknagel, K. P., Jaeger, E., Laminet, A. A. and Pluckthun, A. (1994). Thermodynamic partitioning model for hydrophobic binding of polypeptides by GroEL. I. GroEL recognizes the signal sequences of beta-lactamase precursor. J Mol Biol 242(2): 150-164.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Sadat, A., Tiwari, S. and Mapa, K. (2021). Protocol for Spontaneous and Chaperonin-assisted in vitro Refolding of a Slow-folding Mutant of GFP, sGFP. Bio-protocol 11(14): e4099. DOI: 10.21769/BioProtoc.4099.
Category
Biochemistry > Protein > Modification
Biochemistry > Protein > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.