- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Modeling Perturbations in Protein Filaments at the Micro and Meso Scale Using NAMD and PTools/Heligeom
Published: Vol 11, Iss 14, Jul 20, 2021 DOI: 10.21769/BioProtoc.4097 Views: 3499
Reviewed by: Mostafa RahnamaRAMESH KUDIRASonal Patel PatelAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Protein filaments are dynamic entities that respond to external stimuli by slightly or substantially modifying the internal binding geometries between successive protomers. This results in overall changes in the filament architecture, which are difficult to model due to the helical character of the system. Here, we describe how distortions in RecA nucleofilaments and their consequences on the filament-DNA and bound DNA-DNA interactions at different stages of the homologous recombination process can be modeled using the PTools/Heligeom software and subsequent molecular dynamics simulation with NAMD. Modeling methods dealing with helical macromolecular objects typically rely on symmetric assemblies and take advantage of known symmetry descriptors. Other methods dealing with single objects, such as MMTK or VMD, do not integrate the specificities of regular assemblies. By basing the model building on binding geometries at the protomer-protomer level, PTools/Heligeom frees the building process from a priori knowledge of the system topology and enables irregular architectures and symmetry disruption to be accounted for.
Graphical abstract:
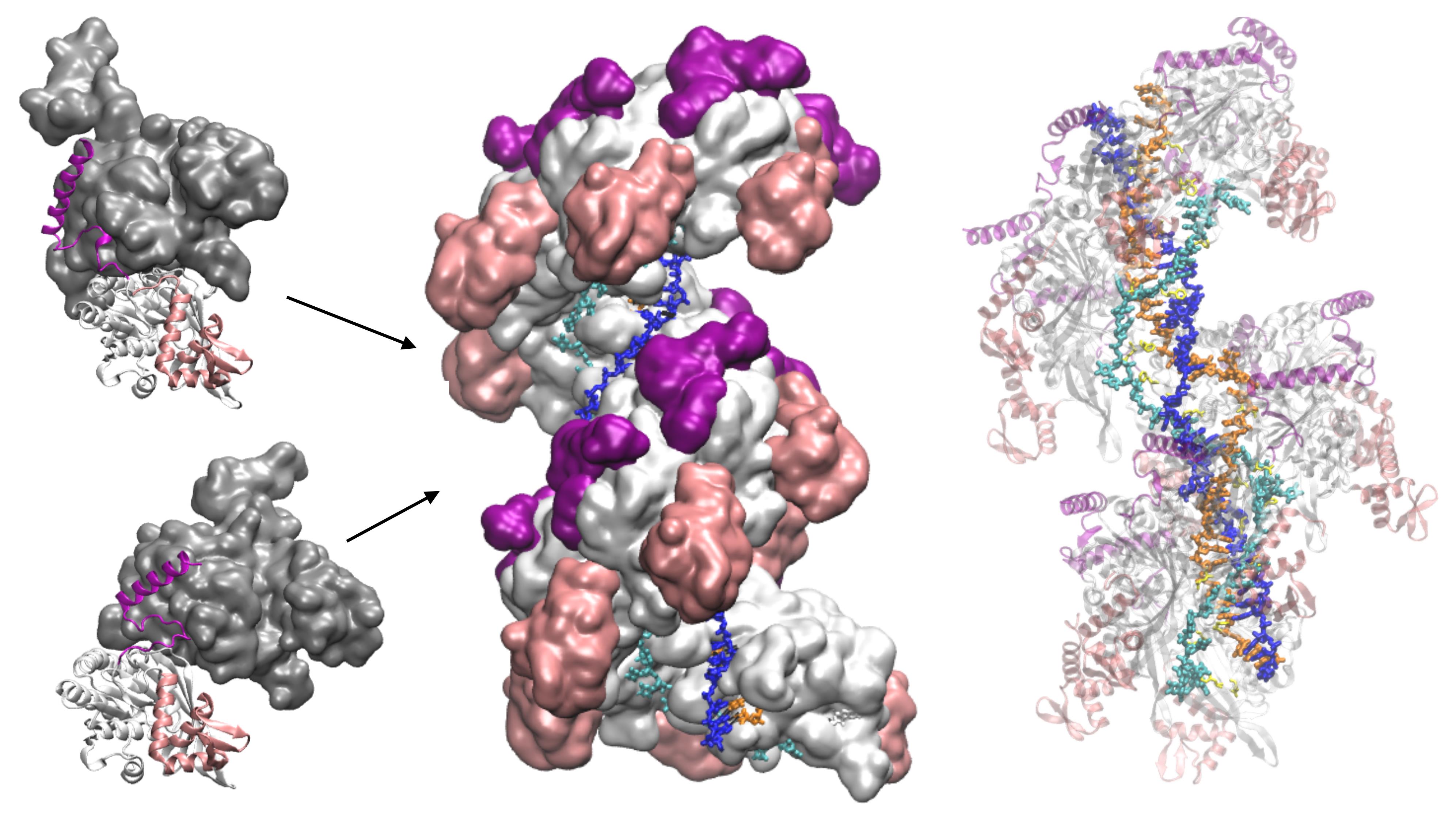
Model of ATP hydrolysis-induced distortions in the recombinant nucleoprotein, obtained by combining RecA-DNA and two RecA-RecA binding geometries.
Background
Oligomeric filamentous assemblies, made of the quasi-regular repetition of macromolecular protomers, are essential components of the cell. Filaments from the cytoskeleton support cell walls or form intracellular transport networks. Other types of filaments are involved in the maintenance, segregation, and repair of DNA (Ghosal and Löwe, 2015). These processes involve dynamic interactions between two generalized helices, the DNA helix and the filament helix, generally accompanied by changes in the DNA linking number and sometimes with coordinated changes in the filament topology. Elucidation of the structure of protein filaments using structural biology methods such as X-ray crystallography or Nuclear Magnetic Resonance has long been highly challenging due to their large size and difficulty in forming crystals. With the recent advances in Cryo-Electron Microscopy, many filament structures are being unveiled; however, filament plasticity and dynamic events are still difficult to capture, and model building remains necessary to decipher how filaments respond to external stimuli. Methods that have been proposed for the modeling of filaments are mostly based on specific docking approaches (Inbar et al., 2005; Casciari et al., 2006; Karaca et al., 2010; Esquivel-Rodriguez et al., 2012) and generally take advantage of known symmetry characteristics (Eisenstein et al., 1997; Berchanski et al., 2003 and 2005; Comeau and Camacho, 2005; Pierce et al., 2005; Schneidman-Duhovny et al., 2005). These methods are not appropriate for investigating structural perturbations in the filament, either global (changes in helical characteristics) or local (symmetry disruption), that appear as responses to external stimuli.
The Heligeom module (Boyer et al., 2015) of the PTools library software (Saladin et al., 2009) has been developed to meet the challenge of modeling helical protein filaments, their interactions with other molecules such as DNA, and their structural responses to external stimuli, either diffuse (changes in longitudinal or torsional stress, following changes in salt concentration or pH), or focused (local molecule binding or interface modifications). Heligeom derives the geometry of oligomeric assemblies from the binding geometry between pairs of neighboring protomers, which offers the possibility to combine several binding modes within the same filament. The PTools library itself gathers a collection of tools to manipulate macromolecules or selected regions of these macromolecules in atomic or coarse-grained representation. While it shares several functionalities with other libraries such as MMTK (Hinsen, 2000) or the VMD Script Library (Humphrey et al., 1996), certain functionalities such as the easy use of screw transformations make it particularly well suited to dealing with helical or quasi-helical assemblies.
Here, we present the application of PTools/Heligeom to model an irregular form of the RecA nucleofilament complex that is active in homologous recombination and includes one to three DNA strands (Boyer et al., 2015). Distortion in the filament results from the introduction of an alternative binding geometry of RecA-RecA interaction at the center of a two-turn helical nucleofilament (twelve monomers). Whereas the principal RecA-RecA binding mode corresponds to the favorable binding geometry between two RecA monomers in the presence of ATP, the alternative binding mode corresponds to the geometry in the presence of ADP. Both binding geometries have been solved by crystallography. The model therefore represents the result of one ATP molecule being hydrolyzed in the filament center. All three DNA strands participate in a strand exchange process. Strand 1 is a damaged DNA strand on which the filament has polymerized; it is bound to DNA-binding site I in the filament. Strand 3 is homologous to strand 1 and occupies the secondary binding site (site II). Strand 2 is complementary to both strand 1 and strand 3. During the strand exchange process catalyzed by the filament, strand 1 captures strand 2, which was initially paired with strand 1. We model three states of the system: the initial state, where only strand 1 is bound to site I (one bound strand); the annealed state, where strand 2 has paired with strand 1 at site I (two bound strands); and the state resulting from strand exchange, where strands 1 and 2 are at site I and strand 3 is at site II (three bound strands). The protocol presented here can be used for any oligomeric system in which three-dimensional structures are available for at least two protein-protein binding geometries obtained under two different conditions, for example, with different cofactors, and present regions of structural similarity. The protocol enables exploration of transient intermediate geometries that are difficult to access experimentally. It is particularly well suited to the study of "collaborative protein filaments," as described in Ghosal and Lowe (2015).
Equipment
Linux workstation (Dell) running a Linux distribution Ubuntu 18.04.5 LTS (GNU/Linux 4.15.0-118-generic x86_64)
Access to a scalar supercomputer (petaflop performance) for molecular dynamics simulations
Software
PTools (Saladin et al., 2009) is a python/C++ library dedicated to the manipulation, assembly, and analysis of macromolecular complexes. It is freely available in the GitHub repository at the address https://github.com/ptools/ptools/tree/develop; it is licensed under the GNU General Public License v3.0. The Heligeom module relates protomer-protomer binding geometries to the geometry of large oligomeric assemblies (Boyer et al., 2015). Heligeom basic functions consist of analyzing protomer interfaces in terms of the helical parameters (Figure 1A) of associated screw objects – pitch (P), number of monomers per turn (N), handedness, see Figure 1A, right – and constructing helices with the desired number of protomers. The helix can optionally be aligned to the Z axis at the origin. Figure 1B shows an example of the workflow of PTools/Heligeom functions that are commonly used in this work. These include selection functions (chains, residue numbers or types, atoms numbers or types), the superposition function that outputs a transformation matrix, and the mat_trans_to_screw function that produces the screw object corresponding to the transformation matrix.
The NAMD software (Scalable Molecular Dynamics, Mackerell et al., 2004; Phillips et al., 2005; http://www.ks.uiuc.edu/Research/namd/) is used to perform energy minimization and molecular dynamics (MD) simulations.
VMD (Humphrey et al., 1996) is a molecular visualization program: VMD can display and analyze large biomolecular systems and their MD trajectories using 3-D graphics and built-in scripting https://www.ks.uiuc.edu/Research/vmd/script_library/.
NAMD and VMD are both developed at the NIH Center for Macromolecular Modeling & Bioinformatics, University of Illinois at Urbana-Champaign, by the Theoretical and Computational Biophysics Group. Both are distributed free of charge with source code.
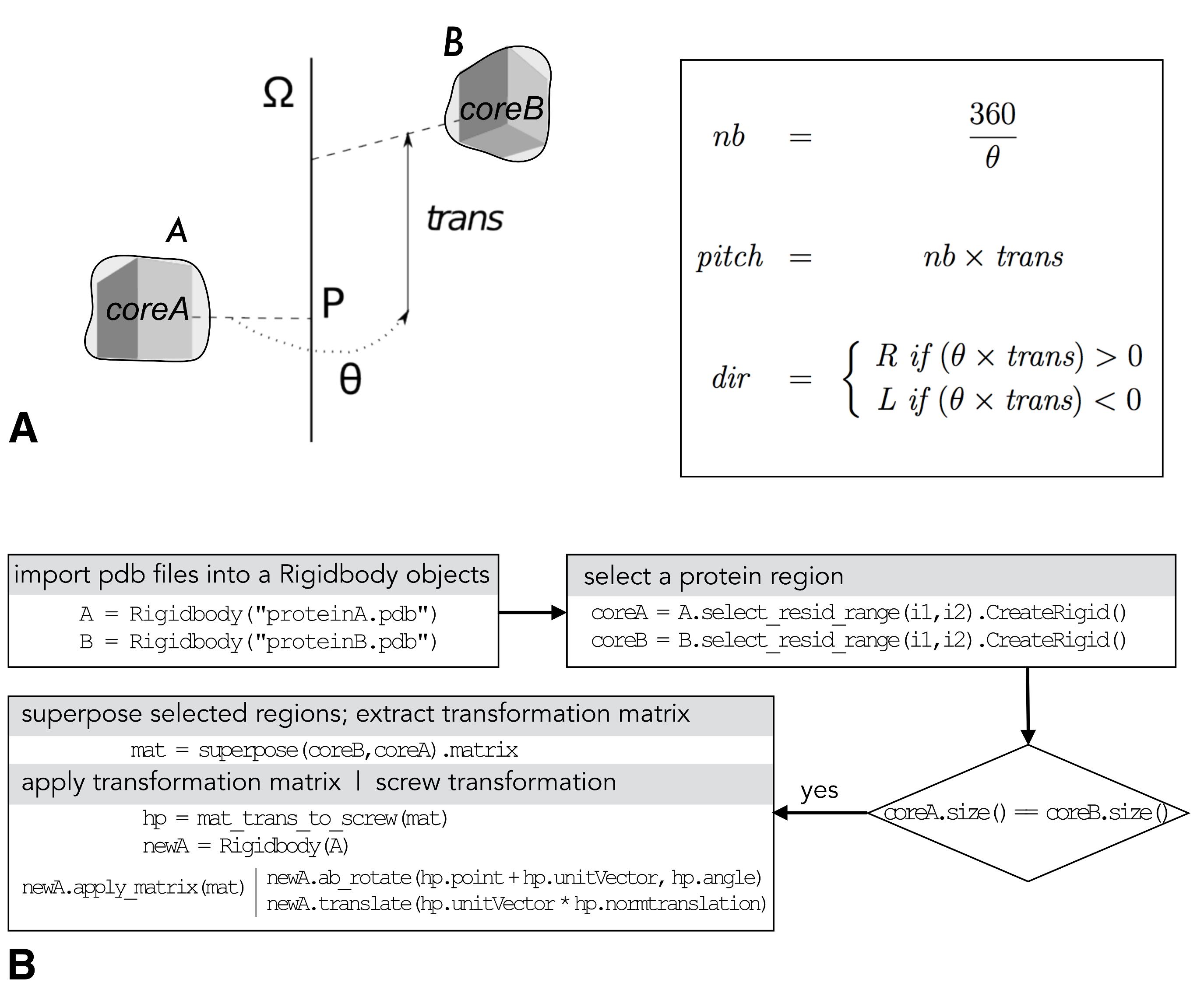
Figure 1. PTools/Heligeom. (A) Left: scheme of a screw transformation between two proteins A and B, with structurally similar core regions coreA and core2; right: calculation of the number of monomers per turn (nb), the pitch and the handedness (R: right, L: left) from the screw parameters θ and trans. (B) Typical PTools operations that enable the combination of available binding geometries. This includes creating Rigidbody objects from the .pdb files ("import .pdb files…"), defining structurally superposable regions with identical sizes using the select_resid_range attribute ("select a protein region"), defining mat, the transformation matrix that superposes the defined regions using the superpose function ("superpose selected regions…"), and defining the screw transformation hp equivalent to mat; example of how to use the screw object hp to create a new Rigidbody object according to the desired screw transformation is given under "apply transformation matrix…"
Procedure
Filament construction
Preparing building blocks
Binding geometries are defined by pairs of interacting RecA monomers extracted from the RecA crystal structures with PDB codes 2REB (RecA bound to ADP molecules) and 3CMW (RecA bound to ATP and two DNA strands at site I). From each of these structure files, manually extract two neighboring monomers to define the corresponding binding geometry (monomers with different binding geometries are schematized in Figure 2A). In the case of 3CMW, in which binding geometries slightly differ along the 5-monomer filament, select central monomers (position 3 and 4) to avoid possible end effects. Make sure that the two monomers have the same number of residues and are structurally close (RMSD on CA atoms < 3 Å); otherwise, select structurally similar regions using the commands given in reference (Saladin et al., 2009) and Figure 1B. The pair of monomers extracted from the 2REB structure will be denoted RA and RB, and those extracted from 3CMW, CA and CB (Figure 2A). The corresponding new .pdb files should be named RA.pdb, RB.pdb, CA.pdb, CB.pdb (see Note 1).
Building blocks are formed by monomer CA (resp. CB) with bound ATP/Mg2+ together with one, two, or three bound DNA segments. To define the DNA segments that are part of the building blocks, proceed as follows: for each of the two strands at site I, extract the three nucleotides that are bound to CA (resp. CB) in the 3CMW structure (the stoichiometry of RecA/DNA association is 3 nucleotides/monomer); note that determination of the residue numbers of the target nucleotides may require visual inspection of the 3CMW structure using VMD. The strand at site II is taken from our model published in Yang et al. (2015) (.pdb file is available upon request): select one RecA monomer and the bound DNA. Then, using PTools commands (Figure 1B), superpose the RecA monomer onto CA (resp. CB), calculate the transformation matrix associated with the superposition, and apply this transformation matrix to the DNA segment. Finally, for each number of strands, generate a .pdb file, named CA_x.pdb where x = 1, 2, or 3, that contains the CA monomer and its associated ATP molecule, magnesium ion, and DNA segment(s).
Assembling building blocks
Construct a 6-monomer filament segment with geometry characteristic to 3CMW using PTools/Heligeom (see Figure 2B) via the command line:
$ ptools heligeom -n 6 -o BLOCK1.pdb -Z CA_x.pdb CB_x.pdb
In a python script into which the PTools package has been imported, construct the Rigidbody objects corresponding to RA, RB, and BLOCK1 (resp. RA, RB, and BL1). Select chain A of BL1 and store it in a Rigidbody object B1. Select chain F of BL1 and store it in a Rigidbody object B6. Superpose RA onto RB and output the corresponding transformation matrix mat1 (Figure 2A). Apply mat1 onto B6, thereby generating a monomer B7 that adjoins B6 following the binding geometry characteristic to 2REB (Figure 2C). Superpose B1 onto B7 and calculate the transformation matrix mat2 (Figure 2D); apply mat2 onto BL1, thus generating a new 6-monomer filament segment BL2 adjacent to BL1, with the binding geometry between the last unit of BL1 and the first unit of BL2 being 2REB type (Figure 2E and 2F).
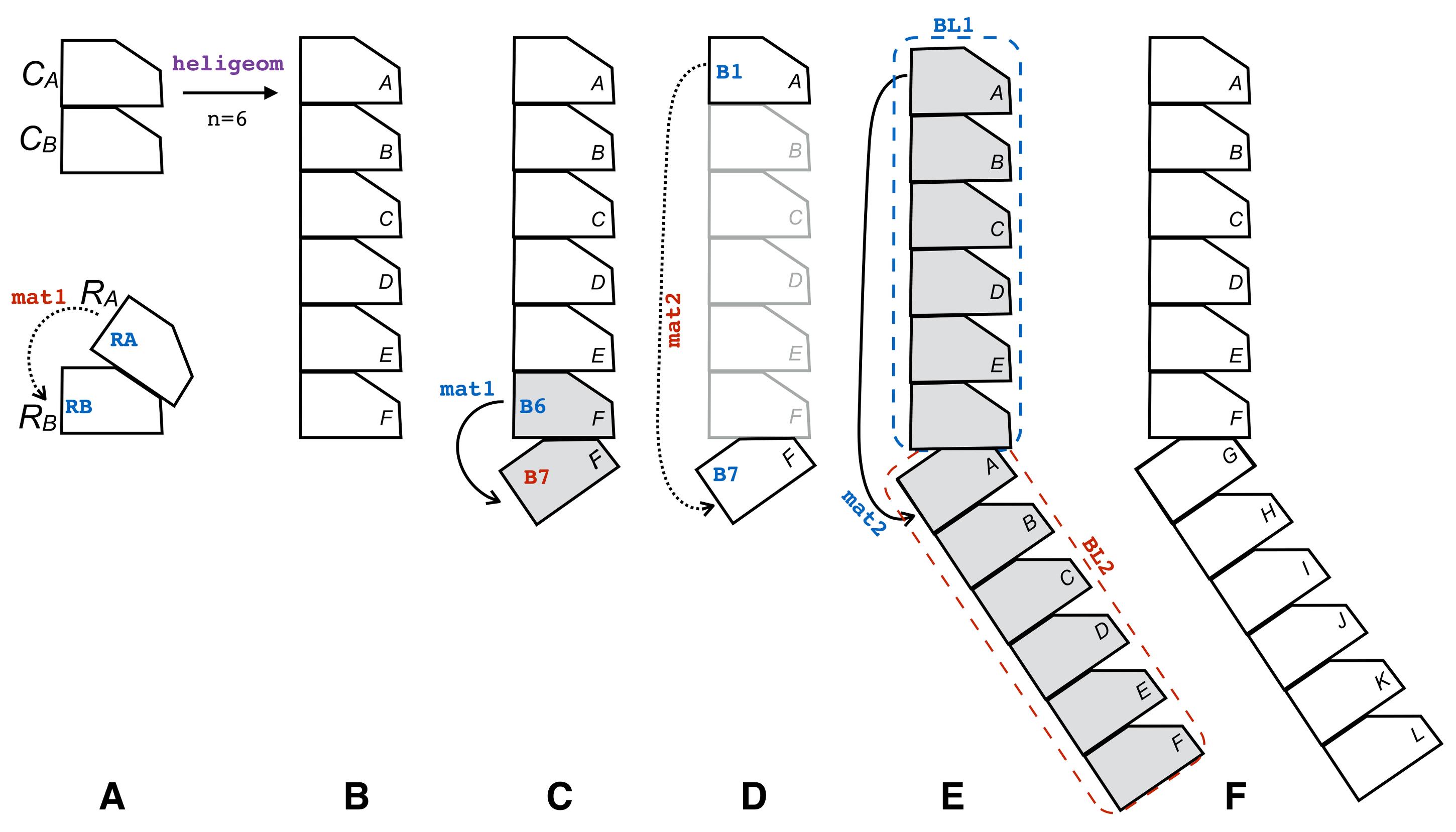
Figure 2. Construction of a composite filament. (A) Schematic representation of the building blocks that define two RecA-RecA binding modes: (top) the CA-CB type and (bottom) the RA-RB type; calculation of the transformation matrix mat1 that relates Rigidbody objects RA and RB (arrow). (B) Applying the heligeom command line to building blocks CA and CB produces a 6-unit helix with interfaces of the CA-CB type (Step A2a). (C) Generation of Rigidbody B7 from B6 by applying mat1; B6 and B7 interact via a RA-RB interface. (D) Calculation of mat2 that transforms B1 into B7. (E) Applying mat2 to the BL1 block. (F) Final result (see text in A.2.b). PTools object names are written in blue when used and in red when defined. The process is illustrated in the accompanying video at Figure2_video.mp4.Refining the junction between building blocks
Adjust the interface flexible groups by replacing the N-terminal domain (one helix and associated linker, residues 1 to 37) of Rigidbody B7 with the equivalent region from RA. To this end, create an empty Rigidbody object BL2_adjusted. Select the Cα atoms of residue numbers 38 to 156, 165 to 193, and 213 to 333 of RA (non-flexible regions) and store them in the Rigidbody SRA. Perform the same selection for B7, stored in SB7; superpose SRA onto SB7 and output the transformation matrix mat3. Select the N-terminal domain of RA, residues 1 to 37, and store the selection in NT; store residues 38 to 333 of B7 in CB7; store chains B, C, D, E, and F of BL2 in BL2_BtoF. Apply mat3 to NT and store the result in NT7. Successively concatenate NT7, CB7, and BL2_BtoF to BL2_adjusted; concatenate BL2_adjusted to BL1. Output the structure in PDB format, named FINAL_x.pdb, where x = 1, 2, 3 is the number of DNA strands. Three final structures have now been generated.
Process the final structures in such a way as to group the DNA fragments, label them with a specific chain name, group the ATP molecules and change their chain name, and name the protein chains from A to L (Figure 2C, right); replace the ATP/Mg2+ bound to chain F with ADP. This can be done within the python/PTools script before outputting the .pdb files in the previous paragraph or manually by processing the .pdb files using a text editor.
Closing DNA chains with restrained minimization for each file FINAL_x.pdb, x = 1, 2, 3
Using Recipe 1, roughly reposition the trinucleotide DNA segment T3F from strand 3 associated with monomer F. This segment presents numerous steric clashes with monomer G following the change in filament axis orientation, consecutive to the modification of the F-G interface geometry. The change in orientation also produces an important rupture of site II continuity between T3F and the DNA segment T3G of strand 3 associated with monomer G.
Generate a topology .psf file using the «Automatic PSF builder» VMD extension.
Create a constraint file from the FINAL_x.pdb file for NAMD minimization by adding «1.00» to column 62 of the restrained atoms (P for the DNA chains and CA for proteins); the script given as Recipe 2 below can be used to this end. The resulting file FINAL_x_cstr.pdb needs to be modified in order to free the phosphate atoms situated at the junction between chains F and G for energy minimization, where the helix symmetry has been broken, by replacing «1.00» with «0.00» for these specific phosphate atoms (three phosphate atoms are freed per DNA chain).
Perform 5000 steps of conjugate gradient energy minimization using NAMD2.10. This enables closure of the DNA backbones by restoring the distances between covalently linked atoms according to the topology file and releasing small steric clashes if any.
Stability and evolution of the filament model: molecular dynamics simulations
Simulation preparation for each file FINAL_x.pdb, x = 1, 2, 3
Immerse the structure in a water box using the VMD extension «Add Solvation Box»; use a TIP3P water model and a box padding value of 10 Â.
Add Na+ and Cl- ions to the water box using the VMD extension «Add Ions»; select the option «Neutralize and set NaCl concentration to 0.15 mol/L».
Simulation runs for each of the three systems using NAMD2.10
Given the large size of the systems (about 450,000 atoms), this step needs to be performed on distributed computer nodes, for example, in a computer center. The simulation follows standard NAMD protocols.
Simulation conditions: use the CHARMM 27 force field including CMAP corrections; use periodic boundary conditions, with the particle mess Ewald method to account for long range interactions and a smooth switch of van der Waals interactions between 10 and 12 Å; set time steps to 2 fs using the SHAKE algorithm; control the temperature and pressure using a Langevin dynamics scheme and a Nose-Hoover Langevin piston.
Simulation run: perform 5000 steps of conjugate gradient minimization followed by a progressive heating stage up to 300K, a long equilibration phase of 30 ns, and a 100 ns production phase. During heating and equilibration, set restraints on the P and CA atoms of the files, with the force constant decreasing from 0.5 to 0.05 kcal·mol-1·Å-2. Do not use restraints during the production phase. Perform three independent simulations for the three-stranded system using identical conditions.
Data analysis
Results of the following analysis steps can be found in the Supplemental Information (SI) of Boyer et al. (2015).
Conventional MD analysis. Check the stability of the energy and temperature during the molecular dynamics simulation trajectories using VMD tools. Reduce the size of the trajectory files by eliminating the water molecules. Plot the time evolution of root-mean-square fluctuation (RMSF) or deviation (RMSD) values of the system or selected regions of the system, taken on the Cα atoms: whole filament, filament center, DNA center (Boyer et al., 2015, Figure SI-7). To separate the fluctuations of each monomer from those of the entire filament, calculate the RMSD and RMSF values along the trajectory for each individual RecA monomer, after superposition restricted to this monomer; plot the evolution of the average value of the resulting RMSDs, taken on all monomers (Boyer et al., 2015, Figure SI-7); individually plot the RMSF corresponding to each monomer (Boyer et al., 2015, Figure SI-5). Visually inspect the final structures of trajectories using VMD to select atoms or groups of atoms of interest that modify their contacts with other groups as a result of the interface modification; plot the time evolution of these distances; calculate the corresponding distances in regular filaments and incorporate this value into the plot as a horizontal line as a reference (Boyer et al., 2015, Figure SI-3).
Specific MD analysis for a filamentous assembly. Calculate the width of the groove entrance at the level of each monomer, as described in Boyer et al. (2015) (Materials and Methods); plot the time evolution of the width value for each monomer and then plot the value measured in regular filaments as in Boyer et al. (2015), Figure SI-2. Using PTools utilities, calculate the evolution, along the trajectory, of the fNAT values for each non-terminal interface, where fNAT is the fraction of pair contacts from the starting structure that are conserved in a given snapshot (a contact being defined as a pair of residues from each interacting monomer that are distant by less than 5 Å, and pair contacts being restricted to the non-flexible regions of the interface, as defined above in Step A3a) (Boyer et al., 2015, Figure SI-6).
DNA analysis. Evolution of the interaction between DNA strands is monitored in two ways: by measuring the shortest phosphate-phosphate inter-strand distances between each pair of strands for each snapshot (see Boyer et al., 2015, Figure SI-9 for details) or by calculating the time evolution of pairing distances between three base pairs, selected by visual inspection using VMD (Boyer et al., 2015, Figure 4).
Notes
In the 3CMW .pdb file, the residues are numbered from 1 to 333 for the first monomer, from 1001 to 1333 for the second, from 2001 to 2333 for the third, etc. It is advisable to edit the files CA.pdb and CB.pdb in order to have their residue numbering within the range of 1-333. Care must be taken that all modifications of .pdb files respect the .pdb file format.
The models generated in this work were deposited into ModelArchive https://www.modelarchive.org/ with codes ma-900pk, ma-l1kfl and ma-eaaa9, and are publicly available at the following URLs: https://www.modelarchive.org/doi/10.5452/[code] (where [code] is either ma-900pk, ma-l1kfl or ma-eaaa9).
Recipes
The T3F trinucleotide segment defined in Step A4a can be repositioned by following the commands below, where the chain index for strand 3 is "P," the T3F nucleotides are numbered from 16 to 18 in the 5’-3’ direction, nucleotide number 15 is associated with monomer E, and nucleotide 19 is the first nucleotide of T3G in the 5’ direction. The backbone discontinuity occurs between nucleotides 18 and 19. The script below defines three points, O, A, and B, where O is positioned on atom P of nucleotide 15, A on atom O3' of nucleotide 19, and B on atom P of nucleotide 18. T3F will be rotated by 40° around the axis (O,u), where u is the normalized cross product between vectors OA and OB:
$ python
>>> from ptools import *
>>> fila = Rigidbody("FINAL_3.pdb")
>>> strP = fila.select_chain_id("P").create_rigid()
>>> t3f = strP.select_res_range(15,18).create_rigid()
>>> atom=(strP.select_res_range(15,15) & \
strP.select_atom_type("P")).create_rigid()
>>> O = atom.get_coords(0)
>>> atom=(strP.select_res_range(19,19) & \
strP.select_atom_type("O3'")).create_rigid()
>>> A = atom.get_coords(0)
>>> atom=(strP.select_res_range(18,18) & \
strP.select_atom_type("P")).create_rigid()
>>> B = atom.get_coords(0)
>>> OA = A - O
>>> OB = B - O
>>> u = Coord3D()
>>> u.x = OA.y * OB.z - OA.z * OB.y
>>> u.y = OA.z * OB.x - OA.x * OB.z
>>> u.z = OA.x * OB.z - OA.z * OB.x
>>> u.normalize()
>>> newt3f = Rigidbody(t3f)
>>> newt3f.ab_rotate(O,O+u,40*3.14/180.)
>>> write_pdb(newt3f,"new_T3F.pdb")
The file FINAL_3.pdb can then be edited to incorporate new_T3F.pdb in place of T3F.
The following Linux command can be used to generate a constraint file for NAMD in order to restrain the displacement of P and CA atoms during energy minimization, heating, and the beginning of equilibration (see Step A4c):
$ awk '$3 == "P" || $3 == "CA" { \
> printf "%s 1.00%s\n",substr($0,1,61), substr($0,67)}; \
> $3 != "P" && $3 != "CA" ' FINAL_x.pdb > FINAL_x_cstr.pdb
This command labels all "P" and "CA" atoms of the file FINAL_x.pdb. In order for the minimization process to be able to close the DNA backbones, phosphate atoms from the junction regions must be released by replacing the "1.00" in columns 63 to 66 with "0.00". For example, for strand 3, if one keeps the notations from Recipe 1, phosphates 15 to 20 of chain "P" will be released.
Acknowledgments
The authors wish to acknowledge the ‘Initiative d’Excellence’ program of the French State for funding [DYNAMO, ANR-11-LABX-0011-01]. The original research paper from which this protocol is derived was published by Boyer et al. (2019). We thank Raquel de Miranda for assistance in preparing the video describing RecA filament preparation.
Competing interests
The authors certify that they have no competing interests.
Ethics
The protocol presented here did not use any human or animal subjects.
References
- Berchanski, A. and Eisenstein, M. (2003). Construction of molecular assemblies via docking: modeling of tetramers with D2 symmetry. Proteins 53(4): 817-829.
- Berchanski, A., Segal, D. and Eisenstein, M. (2005). Modeling oligomers with Cn or Dn symmetry: application to CAPRI target 10. Proteins 60(2): 202-206.
- Boyer, B., Ezelin, J., Poulain, P., Saladin, A., Zacharias, M., Robert, C. H. and Prevost, C. (2015). An integrative approach to the study of filamentous oligomeric assemblies, with application to RecA. PLoS One 10(3): e0116414.
- Boyer, B., Danilowicz, C., Prentiss, M. and Prevost, C. (2019). Weaving DNA strands: structural insight on ATP hydrolysis in RecA-induced homologous recombination. Nucleic Acids Res 47(15): 7798-7808.
- Casciari, D., Seeber, M. and Fanelli, F. (2006). Quaternary structure predictions of transmembrane proteins starting from the monomer: a docking-based approach. BMC Bioinformatics 7: 340.
- Comeau, S. R. and Camacho, C. J. (2005). Predicting oligomeric assemblies: N-mers a primer. J Struct Biol 150(3): 233-244.
- Eisenstein, M., Shariv, I., Koren, G., Friesem, A. A. and Katchalski-Katzir, E. (1997). Modeling supra-molecular helices: extension of the molecular surface recognition algorithm and application to the protein coat of the tobacco mosaic virus. J Mol Biol 266(1): 135-143.
- Esquivel-Rodriguez, J., Yang, Y. D. and Kihara, D. (2012). Multi-LZerD: multiple protein docking for asymmetric complexes. Proteins 80(7): 1818-1833.
- Ghosal, D. and Lowe, J. (2015). Collaborative protein filaments. EMBO J 34(18): 2312-2320.
- Hinsen, K. J. (2000). The molecular modeling toolkit: a new approach to molecular simulations. J Comput Chem 21(2): 79-85.
- Humphrey, W., Dalke, A. and Schulten, K. (1996). VMD: visual molecular dynamics. J Mol Graph 14(1): 33-38, 27-38.
- Inbar, Y., Benyamini, H., Nussinov, R. and Wolfson, H. J. (2005). Prediction of multimolecular assemblies by multiple docking. J Mol Biol 349(2): 435-447.
- Karaca, E., Melquiond, A. S., de Vries, S. J., Kastritis, P. L. and Bonvin, A. M. (2010). Building macromolecular assemblies by information-driven docking: introducing the HADDOCK multibody docking server. Mol Cell Proteomics 9(8): 1784-1794.
- Mackerell, A. D., Jr., Feig, M. and Brooks, C. L., 3rd (2004). Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem 25(11): 1400-1415.
- Phillips, J. C., Braun, R., Wang, W., Gumbart, J., Tajkhorshid, E., Villa, E., Chipot, C., Skeel, R. D., Kale, L. and Schulten, K. (2005). Scalable molecular dynamics with NAMD. J Comput Chem 26(16): 1781-1802.
- Pierce, B., Tong, W. and Weng, Z. (2005). M-ZDOCK: a grid-based approach for Cn symmetric multimer docking. Bioinformatics 21(8): 1472-1478.
- Saladin, A., Fiorucci, S., Poulain, P., Prevost, C. and Zacharias, M. (2009). PTools: an opensource molecular docking library. BMC Struct Biol 9: 27.
- Schneidman-Duhovny, D., Inbar, Y., Nussinov, R. and Wolfson, H. J. (2005). Geometry-based flexible and symmetric protein docking. Proteins 60(2): 224-231.
- Yang, D., Boyer, B., Prevost, C., Danilowicz, C. and Prentiss, M. (2015). Integrating multi-scale data on homologous recombination into a new recognition mechanism based on simulations of the RecA-ssDNA/dsDNA structure. Nucleic Acids Res 43(21): 10251-10263.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Boyer, B., Laurent, B., Robert, C. H. and Prévost, C. (2021). Modeling Perturbations in Protein Filaments at the Micro and Meso Scale Using NAMD and PTools/Heligeom. Bio-protocol 11(14): e4097. DOI: 10.21769/BioProtoc.4097.
Category
Biophysics > Macromolecular simulations
Biochemistry > Protein > Structure
Molecular Biology > DNA
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.