- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Multi-color Bicistronic Biosensor to Compare the Translation Dynamics of Different Open Reading Frames at Single-molecule Resolution in Live Cells
Published: Vol 11, Iss 14, Jul 20, 2021 DOI: 10.21769/BioProtoc.4096 Views: 4073
Reviewed by: David PaulMarco Di Gioia
Original research article
The authors used this protocol in:

Dec 2020
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Here, we describe how to image and quantitate the translation dynamics of a bicistronic biosensor that we recently created to fairly compare cap-dependent and IRES-mediated translation at single-molecule resolution in live human cells. This technique employs a pair of complementary intrabodies loaded into living cells that co-translationally bind complementary epitopes in the two separate ORFs of the bicistronic biosensor. This causes the biosensor to fluoresce in different colors depending on which ORF/epitopes are translated. Using the biosensor together with high-resolution fluorescence microscopy and single-molecule tracking analysis allows for the quantitative comparison of translation dynamics between the two ORFs at a resolution of tens-of-nanometers in space and sub-seconds in time, which is not possible with more traditional GFP or luciferase reporters. Since both ORFs are on the same biosensor, they experience the same microenvironment, allowing a fair comparison of their relative translational activities. In this protocol, we describe how to get this assay up and running in cultured human cells so that translation dynamics can be studied under both normal and stressful cellular conditions. We also provide a number of useful tips and notes to help express components at appropriate levels inside cells for optimal live cell imaging.
Graphical abstract:
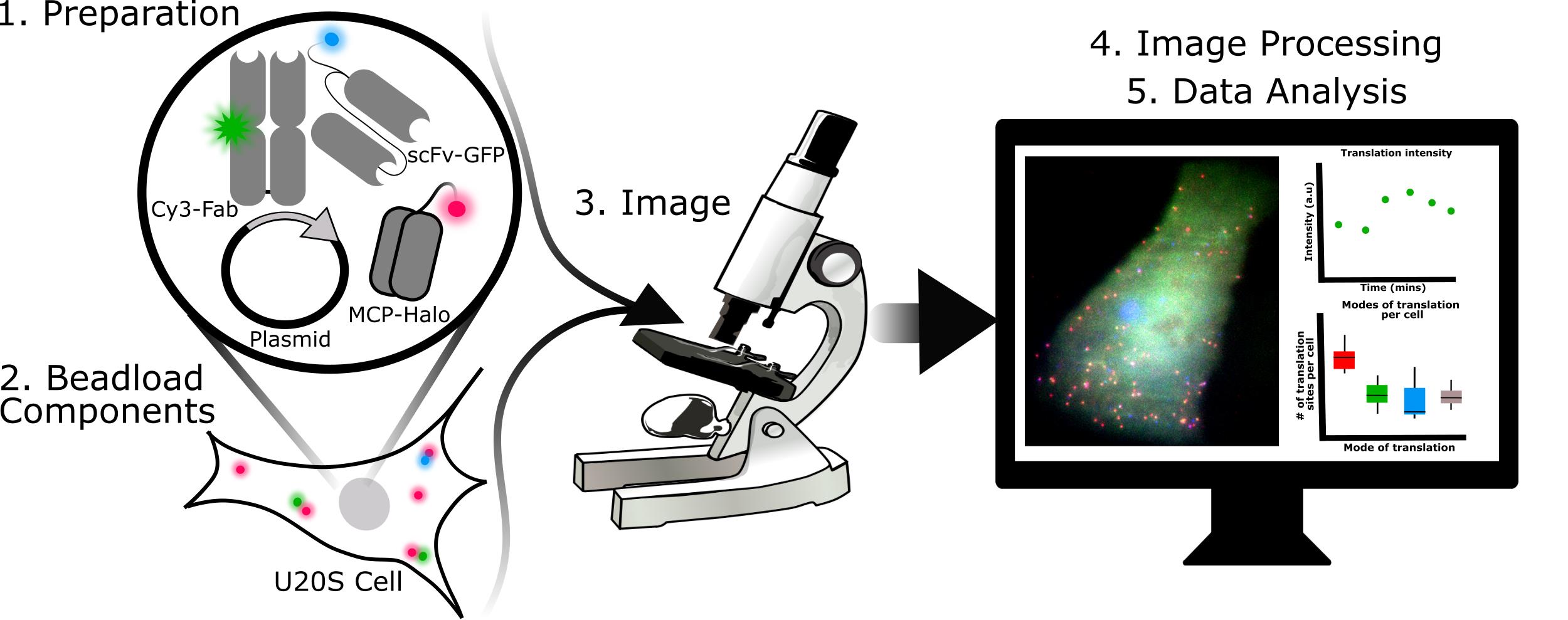
Steps required for 3-color single-molecule translation imaging and analysis.
Background
Recently, it has become possible to measure the translation dynamics of single mRNA molecules in live cells (Morisaki et al., 2016; Pichon et al., 2016; Wang et al., 2016; Wu et al., 2016; Yan et al., 2016). This technology allows for the quantitation of the heterogeneity of translation dynamics among mRNA (Morisaki and Stasevich, 2018). To image and quantitate single mRNA translation dynamics, fluorescent intrabodies such as Fab (Hayashi-Takanaka et al., 2011), scFv (Tanenbaum et al., 2014; Zhao et al., 2019), or nanobodies (Boersma et al., 2019) are required. These intrabodies bind and label repeated epitopes inserted at the N-terminus of a protein of interest. As the protein of interest is translated, the repeated epitopes emerge from the ribosome and are bound within seconds by the fluorescent intrabodies (Morisaki and Stasevich, 2018; Cialek et al., 2020). This strategy amplifies fluorescence within the translation sites at two levels: firstly, multiple fluorescent intrabodies can bind the repeated epitopes within a single nascent peptide chain at the same time; and secondly, multiple ribosomes can translate the mRNA in polysomes to produce multiple nascent peptide chains within a single translation site. These two levels of amplification produce bright fluorescent puncta that can be detected with single-molecule precision above the background using a sensitive fluorescence microscope. As ribosomes initiate, elongate, and terminate translation, the fluorescence intensity at individual translation sites fluctuates up or down, yielding insight into translation dynamics.
Over the last few years, complementary intrabodies and epitopes have been developed that make it possible to image translation in multiple colors at the same time. For example, our lab has developed anti-FLAG and anti-HA Fab that bind the classic FLAG and HA epitopes (Morisaki et al., 2016). More recently, we developed a genetically encodable scFv version of the anti-HA Fab that we call the anti-HA frankenbody (Zhao et al., 2019). Likewise, the Tanenbaum lab has developed the SunTag (SunTag epitopes + anti-SunTag scFv-GFP) and MoonTag (MoonTag epitopes + anti-MoonTag nanobody) systems (Tanenbaum et al., 2014; Boersma et al., 2019). Used together, these complementary imaging tools make it possible to compare the dynamics of different open reading frames (ORFs) at the single-molecule level within live cells.
Here, we describe how to use a 3-color bicistronic biosensor that we recently developed to compare translation kinetics at internal ribosomal entry sites (IRES) versus at the canonical 5’ cap (Figure 1a) (Koch et al., 2020). The biosensor encodes an MS2 tag to mark and track individual mRNA molecules (red) (Coulon et al., 2013; Pichon et al., 2020). When these mRNAs are expressed in cells, they light up in different colors depending on the nature of translation initiation. If translation is initiated at the cap, then FLAG epitopes are translated and bound by anti-FLAG Fab conjugated with Cy3 (green). If translation is initiated at the IRES, then SunTag epitopes are translated and bound by anti-SunTag scFv fused to eGFP (blue). This produces a variety of colorful puncta in cells that mark individual mRNA and their translation status (Figure 1a and 1c).
This basic imaging biosensor is unique because it enables quantitative comparisons between the translation dynamics of two ORFs at a spatial resolution on the tens-of-nanometers scale and temporal resolution on the sub-seconds scale. This is not possible with more traditional bulk assays that lack single-molecule resolution, including traditional luciferase and GFP reporters as well as bulk assays like western blotting. The biosensor is also versatile because a different IRES sequence can easily be inserted to study the translational dynamics of that IRES in diverse cell types and cellular conditions (Bornes Stéphanie et al., 2007; Firth and Brierley, 2012). Further, our 3-color assay can be extended to compare ORF translation dynamics on two separate mRNAs (Figure 1b). This can be achieved by simply splitting the bicistronic biosensor into two monocistronic biosensors, each encoding a probe/epitope pair and an MS2 tag. This setup is not only beneficial for comparing the translation dynamics of two different ORFs but also for fairly comparing different endogenous 5’ or 3’ UTRs or viral elements within single cells.
In this protocol, we focus mainly on the wet lab skills needed to express our biosensor in live human cells. In particular, since our biosensor fluoresces in three colors, multiple components need to be expressed within cells at appropriate levels. Here, we advocate the use of beadloading (McNeil and Warder, 1987; Hayashi-Takanaka et al., 2011) to achieve this with high efficiency and minimal effort. We describe how the various probes – including the MS2 coat protein used to label mRNA with an MS2 tag, Fab to label FLAG-tagged epitopes, and SunTag scFv to label SunTag epitopes – can be purified and loaded together in a single, simple step (Cialek et al., 2021; Morisaki et al., 2016; Koch et al., 2020). We find this approach to be very convenient, enabling the fine-tuning of probe levels, rapid testing, and combinatorial experimentation. Finally, we describe the basics of our imaging and analyses, including a specific test to verify translation, useful conditions for stressing cells while under the microscope, and basic codes to quantitate translation site intensities and distributions.
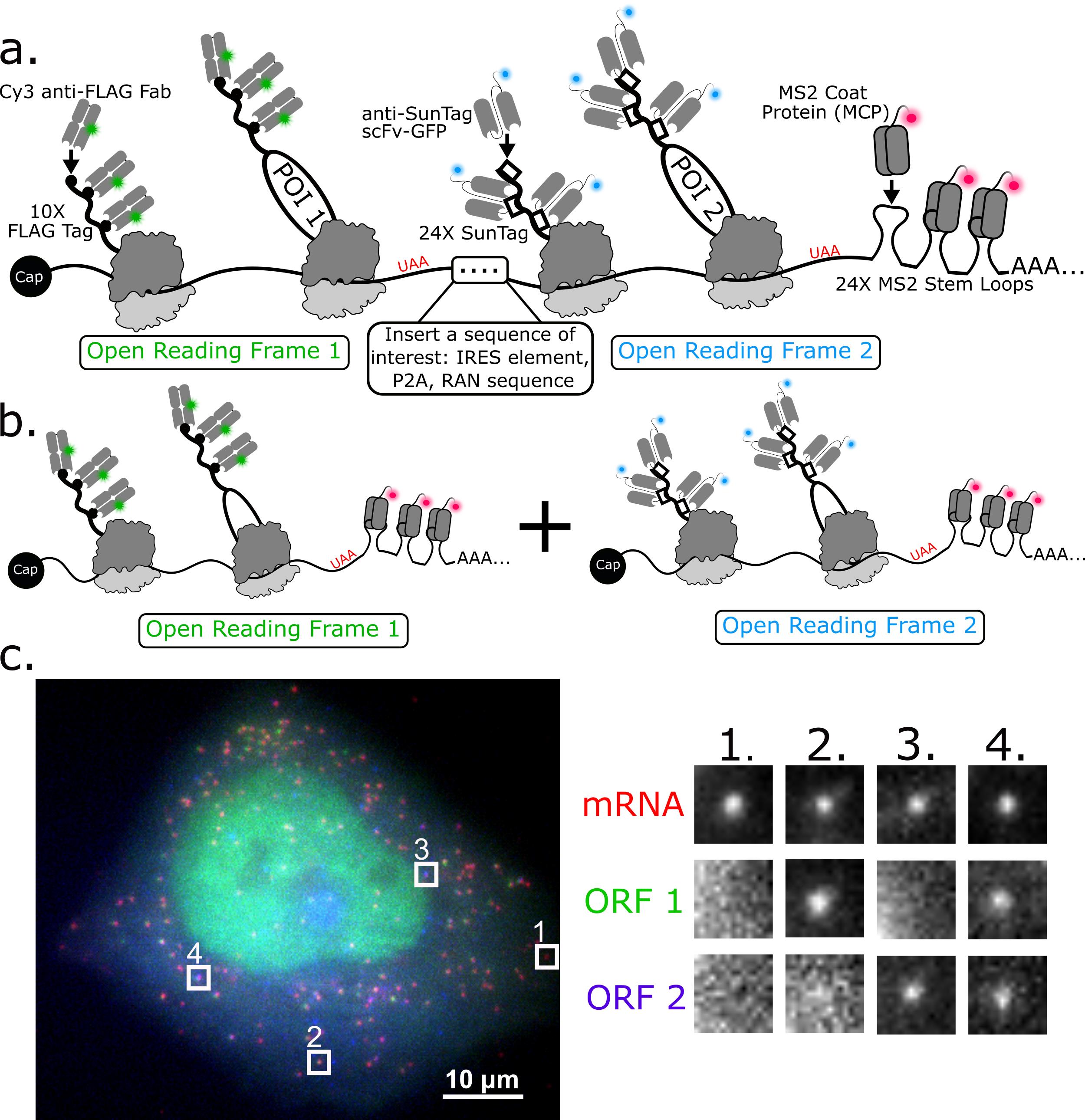
Figure 1. 3-color imaging of the translation of a single bicistronic or pair of monocistronic biosensors in live cells. a) Schematic illustrating a bicistronic 3-color single-molecule translation reporter. b) Schematic illustrating a 2-mRNA single-molecule translation reporter system. c) A 3-color bicistronic reporter (representative cell). Crops showing all types of translation are on the right panel.
Materials and Reagents
Anti-FLAG Fab generation and dye-conjugation
Pierce Fab preparation kit (Thermo Fisher Scientific, catalog number: 44985)
0.6 ml or 1.7 ml low retention tubes (Thomas Scientific, catalog numbers: 1149K01 and 1159M35PK, respectively)
PD-mini G-25 desalting column (GE Healthcare, catalog number: 95055-984)
Amicon Ultracel-50 (50 kDa-cutoff) 15-ml centrifugal filter unit (Millipore Sigma, catalog number: UFC9050)
Amicon Ultracel-10 (10 kDa-cutoff) 15-ml centrifugal filter unit (Millipore Sigma, catalog number: UFC9010)
Amicon Ultracel-10 (10 kDa-cutoff) 0.5-ml centrifugal filter unit (Millipore Sigma, catalog number: UFC5010)
FLAG antibodies (FujiFilm Wako Pure Chemical Corporation, catalog number: 012-22384 (Anti-DYKDDDDK mouse monoclonal IgG2b antibodies)
Cy3 N-hydroxysuccinimide ester mono-reactive dye pack (VWR, catalog number: 95017-373 PK)
Dimethyl sulfoxide (DMSO) (Millipore Sigma, catalog number: D8418)
Sodium bicarbonate (NaHCO3) (Millipore Sigma, catalog number: S5761-500G)
Phosphate-buffered saline (PBS) (Thermo Fisher Scientific, catalog number: AM9625)
Halo-MCP and anti-SunTag scFv-GFP purification
BL21 E. coli (DE3) pLysS competent cells (Novagen, EMD millipore, catalog number: 69451-3)
2× YT growth media (Research Products International, catalog number: X15600-5000.0)
Ampicilllin (sodium) USP grade (GoldBio, catalog number: A-301-100)
Chloramphenicol USP grade (GoldBio, catalog number: C-105-100)
Isopropyl-β-D-thiogalactoside (IPTG) (GoldBio, catalog number: I2481C100)
Phosphate-buffered saline (PBS) pH 7.4
Sodium chloride (NaCl) ACS grade
Protease inhibitor cocktail tablets (PierceTM EDTA-free Protease Inhibitor Mini Tablets, Thermo Scientific, catalog number: A32955)
4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF serine protease inhibitor) (GoldBio, catalog number: A-540-10)
Imidazole ACS reagent, ≥ 99% titration (Sigma-Aldrich)
Amicon Ultracel-30 (30 kDa-cutoff) 15-ml centrifugal filter unit (EMD Millipore, catalog number: UFC903024)
HEPES-based buffer (see Recipes)
HisTrap buffer A (see Recipes)
HisTrap buffer B (see Recipes)
Superdex buffer (see Recipes)
U-2 OS cell culture
U-2 OS cells (ATCC, catalog number: ATCC HTB-96)
DMEM (+/-), high glucose, no glutamine (See Recipes for DMEM (+) media) (Thermo Fisher Scientific, catalog number: 11960069)
Fetal bovine serum (Atlas Biologics, catalog number: F-0050-A)
Penicillin-streptomycin (Thermo Fisher Scientific, catalog number: 15140-122)
L-glutamine, 200 mM (100×) (Thermo Fisher Scientific, catalog number: 25030081)
Trypsin (Thermo Fisher Scientific, catalog number: 25300062)
100-mm cell culture dishes (VWR, catalog number: 82050-916)
Beadloading and staining Halo-MCP with JF646 ligand
106-µm glass beads (Millipore Sigma, catalog number: G4649)
Spectramesh Woven Filters Polypropylene Opening: 105-µm (Spectrum Labs, catalog number: 148496)
Plasmid DNA of interest
Cy3 anti-FLAG Fab (prepared as described in Step B8)
Purified anti-SunTag scFv-GFP (prepared as described in Step C19)
Purified Halo-MCP (prepared as described in Step C19)
Phenol-free DMEM (Thermo Fisher Scientific, catalog number: 31053036)
Janelia Fluor 646 HaloTag ligands (Promega, catalog number: GA1120)
0.6-ml low retention tubes (Thomas Scientific, catalog number: 1149K01)
Phosphate-buffered saline (Thermo Fisher Scientific, catalog number: AM9625)
Imaging
Glass-bottomed dishes, 35-mm, #1.5, 14-mm glass, uncoated (MatTek Corporation, catalog number: P35G-1.5-14-C)
Opti-MEM, Reduced Serum Medium (Thermo Fisher Scientific, catalog number: 31985070)
Puromycin (Thermo Fisher Scientific, catalog number: A1113803)
Harringtonine (Cayman Chemical Company, catalog number: 26833-85-2)
Sodium arsenite (NaAs) (Millipore Sigma, catalog number: S7400-100G)
Dithiothreitol (DTT) (Thermo Fisher, catalog number: R0861)
Equipment
UV-Vis spectrophotometer (Thermo Fisher Scientific, model: NanoDrop OneC)
Table top centrifuge (Beckman Coulter, model: Microfuge 20)
Cooled centrifuge (Thermo Fisher Scientific, model: Sorvall Legend XFR with F14 6x250LE)
Sonicator
HisTrap HP, 5-ml (Cytiva, formerly GE Healthcare catalog number: 17524801)
HiLoad 16/600 Superdex 200 pg column (Cytiva catalog number 28989335)
Fluorescence microscope (capable of thin sectioning; confocal, light-sheet, and HILO microscopes recommended) with a stage-top incubator
Software
Software for single-molecule tracking, e.g., TrackMate (Tinevez et al., 2017) or our custom Mathematica code found at:
https://github.com/Colorado-State-University-Stasevich-Lab/mRNA_Tracking_BioProtocol
Procedure
Concentrating anti-FLAG antibody and preparing anti-FLAG Fabs
General comment: 5 mg anti-FLAG antibodies are shipped at a concentration of ~0.4-0.6 mg/ml. In this step, 4 mg antibody is concentrated to 4-8 mg/ml to create the Fab. Note: the Ultracel-50 filter allows anything smaller than 50 kDa to pass through. Full-length antibody is ~150 kDa and thus does not pass through, but Fab does. Therefore, this filter is not suitable for concentrating Fab in Procedure B below.
Cool centrifuge to 4°C.
Pipette 4 mg anti-FLAG AB from the shipping container into an Ultracel-50 centrifugal filter unit (up to 15 ml sample volume). Add 1× PBS to fill the filter unit to 15 ml.
Centrifuge at 5,000 × g for 10-20 min at 4°C.
Discard flowthrough and add 1× PBS to fill the filter unit to 15 ml.
Repeat 4× to fully replace the buffer to 1× PBS and sufficiently concentrate the antibodies.
After centrifugation, around 0.5-1 ml antibody should remain at the top of the Ultracell chamber. Transfer this to a low retention tube.
After transfer, wash the sides of the Ultracel-50 filter by pipetting 50 μl 1× PBS. Pipette up and down a few times and let the PBS wash over the filter surface so that all residual antibody is washed to the bottom of the filter unit, where it can be easily pipetted and added to the concentrated antibody in the low retention tube (from step above).
Measure the concentration using a NanoDrop.
The final concentration should be around 4-8 mg/ml.
Use the Pierce Fab preparation kit to generate the Fab following the manufacturer’s instructions.
Tip: We recommend performing Fab preparation just once with 4 mg concentrated anti-FLAG antibody (keeping 1 mg full-length antibody as a backup). This is not only cost- and time-effective, but in our hands also leads to a minimal amount of antibody loss during the Fab preparation process.
The Pierce Fab preparation kit instructions can be found at:
https://assets.fishersci.com/TFS-Assets/LSG/manuals/MAN0011651_Pierce_Fab_Prep_UG.pdf.
Note that two Fabs are generated for each antibody, with each Fab roughly 50 kDa (or one-third of the full-length antibody). Thus, 4 mg antibody starting material will produce 2.67 mg Fab. Generally, we lose some antibody material in the process of making the Fab, so we typically end up with 1.5-2 mg Fab.
Concentrate the anti-FLAG Fab using a 15-ml Ultracel-10 centrifugal filter unit as in Procedure A above.
Notes:
Do not accidentally use the Ultracel-50 centrifugal filter unit! The Ultracel-10 will allow everything less than 10 kDa to pass through. Thus, Fab, which is ~50 kDa, will not pass through and will become concentrated at the top of the filter unit.
You can also use 0.5-ml Ultracel-10 centrifugal filter units for this step; however, this will require more time since only 0.5 ml can be concentrated at a time.
Measure the concentration using a NanoDrop.
The final concentration should ideally be around 2 mg/ml, such that 0.5 ml will be 1 mg Fab.
Store the concentrated anti-FLAG Fab at 4°C.
Tip: We find that both anti-FLAG full-length antibody and Fab can be stored in a standard 4°C refrigerator for years without degrading. Repeated freeze-thaw cycles will be detrimental to Fab function and are therefore not recommended.
Anti-FLAG Fab dye-conjugation (Cy3 anti-FLAG Fab)
General comment: The starting component is purified anti-FLAG Fab prepared in Procedure A.
Take a vial from a 5 pack of Cy3 mono-reactive NHS Ester.
Dissolve the Cy3-NHS Ester reagent in a vial in 100 μl DMSO and store at -20°C.
Pipette 100 μg purified Fab (~50 μl) from Step A into a low retention tube.
Add sterilized 1× PBS to reach a total volume of 90 μl.
Add 10 μl 1 M sodium bicarbonate to reach a total volume of 100 μl.
Tip: It is recommended to prepare fresh 1 M sodium bicarbonate before the reaction.
Add the correct amount of dye (this amount is found empirically, see Hayashi-Takanaka et al., 2014).
For Cy3 diluted in 100 μl DMSO, add 1.33 μl.
Tap the tube lightly to evenly distribute.
Place the tube in a light-proof bag and attach to a rotator or any equipment that can rotate the tube effectively.
Incubate for 1 hour at room temperature while rotating.
Purify Cy3 anti-FLAG Fab with a G-25 minitrap.
Cool the centrifuge to 4°C.
Discard the buffer from the G-25 minitrap and equilibrate by allowing 500 μl 1× PBS to fully run through by gravity flow.
Notes:
Repeat three times to fully equilibrate.
Be careful not to let the G-25 minitrap column dry out.
Briefly spin down the tube holding the incubated Fab to remove conjugated Fab from the lid and sides.
Pipette all 100 μl incubated Fab onto the top of the G-25 minitrap.
Note: Make sure the column is straight and pipette the mixture directly into the center.
Pipette 450 μl 1× PBS on top of the column for circulation.
Note: You can pipette an additional 50-100 μl to force the conjugated Fab (lower pinkish band) to move closer to the bottom of the column.
When the conjugated Fab band is close to the bottom of the column, obtain a new low retention tube to capture the conjugated Fab.
Pipette 500 μl 1× PBS into the center of the column. The lower Fab band will flow out and should be captured in the low retention tube.
Note: The solution containing the conjugated Fab will appear pinkish.
Concentrate the captured conjugated Fab protein by transferring it to an Ultra-10 0.5-ml centrifugal filter unit.
Align the filter perpendicular to the centrifuge.
Centrifuge at 12,000 × g for 5 min.
Discard the flowthrough and add up to 500 μl 1× PBS to the filter.
Usually, the flowthrough is clear, indicating that most of the dye is conjugated to the Fab.
Centrifuge at 4°C, 12,000 × g for 5 min.
Discard the flowthrough.
Add 500 μl 1× PBS.
Centrifuge at 12,000 × g for 10 min.
Pipette out the conjugated Fab that remains at the top of the Amicon Ultra filter and transfer to a new 0.6-ml low retention tube (Typically this volume is around 50 μl).
After transfer, wash the sides of the Ultracel-10 0.5-ml filter by pipetting 20-30 μl 1× PBS. Pipette up and down a few times and let the PBS wash over the filter surface so that all residual conjugated Fab is washed to the bottom of the filter unit, where it can be easily pipetted and added to the concentrated conjugated Fab in the 0.6-ml low retention tube (from step above).
Measure the concentration and absorbance spectrum with a NanoDrop.
Firstly, choose Protein and Labels; then choose the sample type as IgG and the appropriate Cy3 dye. The measured concentration should be close to 1 mg/ml with an absorbance ratio of ~1-2.
Calculate the degree of labeling:
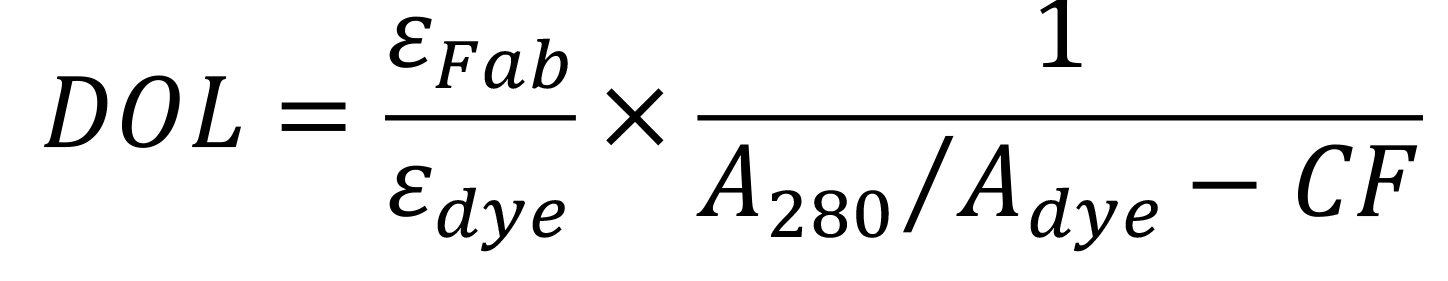
where εFab is the extinction coefficient of Fab (70,000 M-1cm-1), εdye is the extinction coefficient of the dye used for conjugation (150,000 M-1cm-1 for Cy3), A280 and Adye are the measured absorbances of dye-conjugated Fab fragments at 280 nm and at the peak of the emission spectrum of the dye (570 nm for Cy3), respectively, and CF is the correction factor of the dye (the ratio of the absorbances of the dye alone at 280 nm to at the peak; 0.08 for Cy3).
Ideally, the DOL should be close to 1, corresponding to ~1 dye per Fab. If the DOL is too low, the entire process can be repeated.
For working stocks, dilute Cy3 anti-FLAG Fab to a concentration of 0.5 mg/ml in 1× PBS.
Note: Store at 4°C.
Purification of MCP fused to HaloTag (Halo-MCP) and anti-SunTag scFv fused to eGFP (anti-SunTag scFv-GFP)
Transform E. coli BL21 (DE3) pLysS competent cells with the expression plasmid (pHis-Halo-2MCP or pHis-scFv-GFP – plasmids are available upon request).
Grow and shake transformed cells at 37°C in 2× YT broth containing 100 mg/L ampicillin and 25 mg/L chloramphenicol until the cells reach OD600.
Lower the temperature to 30°C (Halo-MCP) or 18°C (anti-SunTag scFv-GFP) when this density is reached.
Add 0.4 mM IPTG.
Incubate for 3 or 16 h for Halo-MCP or anti-SunTag scFv-GFP, respectively.
Cool the centrifuge to 4°C.
Pellet the cells by centrifugation at 2,041 × g for 30 min at 4°C.
Resuspend the pellet in HisTrap buffer A (20 ml per 1 L culture) and add protease inhibitor cocktail (manufacturer’s suggestion).
Lyse the cells by sonication at 50% output; 30 s on and 90 s off.
Pellet the lysate by centrifugation at 30,186 × g, 4°C for 30 min.
Connect two HisTrap HP 5-ml columns in series and pre-equilibrate columns with HisTrap buffer A.
Load the supernatant onto the columns.
Wash columns with 5 column volumes (CV, 50 ml) of HisTrap buffer A.
Elute with 10 CV (100 ml) of a linear gradient of 0-500 mM imidazole (HisTrap buffer B).
Concentrate the purified protein (Halo-MCP or anti-SunTag scFv-GFP) fraction using an Amicon Ultracel-30 (30 kDa-cutoff) 15-ml centrifugal filter unit to a final volume of approximately 2 ml.
Equilibrate a HiLoad 16/600 Superdex 200 pg column with Superdex buffer.
Load concentrated sample onto the equilibrated HiLoad 16/600 Superdex 200 pg column.
Check the purity of the fractions by 15% SDS-PAGE. Concentrate the fractions containing purified protein using an Amicon Ultracel-30 15-ml centrifugal filter unit.
Determine the protein concentration using a NanoDrop.
Aliquot into small volumes, flash freeze with liquid nitrogen, and store at -80°C.
For working stocks, dilute purified Halo-MCP to a concentration of 0.13 mg/ml in 1× PBS and anti-SunTag scFv-GFP to 0.5 mg/ml in 1× PBS.
Store at 4°C.
Tip: Try to make a small working stock volume of 10-20 μl. Diluted purified protein lasts for ~2 months at 4°C.
Preparing glass beads by acid washing
Note: Be careful not to inhale the glass beads; they are toxic to the lungs.
Sterilize 5 ml glass beads in 25 ml 2 M NaOH for 2 h in a 50-ml conical tube with gentle mixing, preferably using a rotator.
Decant the NaOH, retaining as many beads as possible. If the beads are in suspension, they can be briefly spun in a centrifuge.
Wash the beads thoroughly with cell culture grade water until the pH is neutral (use a pH test strip on the eluate). Decant the wash water each time, as before.
Rinse the beads thoroughly with 100% ethanol 2-3 times. Decant the ethanol each time, as before.
Sprinkle the beads into a sterile container (such as a 100-mm Petri dish) and allow to air dry in a biosafety cabinet overnight.
Note: When the beads are completely dry, they will look sandy, i.e., they will not clump or flake when the container is tapped.
UV-sterilize the beads for 30 min, if possible.
Store in a desiccator to keep the beads dry.
U-2 OS cell culture
Make DMEM (+). See recipe below.
Store at 4°C.
Maintain U-2 OS cells at a confluence no greater than 90%.
The day before imaging, split cells into MatTek chambers as follows:
Warm up the Trypsin and DMEM (+).
Remove the cells from the 37°C, 5% CO2 incubator.
Remove all media.
Wash 2-3 times with 1× PBS.
Add enough Trypsin to cover the bottom of the dish.
Incubate at room temperature for 30 s.
Aspirate or pipette out the Trypsin. This will leave a thin layer on top that is sufficient for cell detachment. Tilt the dish back and forth a few times to make sure the thin layer of Trypsin is evenly spread.
Wait 1-5 min for the cells to detach. This can be facilitated by shaking/tapping the dish or placing the dish in the incubator at 37°C briefly.
Add DMEM (+) (10 ml for a 100-mm dish).
Tilt the dish at a 45° angle.
To ensure that most cells are removed, pipette the media at the top of the dish so that it runs down and washes the cells toward the bottom, where they can be pipetted. Repeated pipetting can also help to remove cell clumps.
Transfer the cells and media into a 15-ml conical tube. This is optional but will help to prevent cells from re-adhering to the dish if there is some time before plating onto MatTek chambers.
Plate cells onto MatTek chambers at 75% confluence (approximately 9.0 × 105 cells).
1-2 hours before beadloading, change media from DMEM (+) to Opti-MEM supplemented with 10% FBS (v/v):
Wash the cells twice with 1× PBS.
Add Opti-MEM + 10% FBS (v/v).
Incubate at 37°C.
Note: This step is optional. Cells can be left in DMEM (+) before beadloading. Our lab has empirically found that cells in OPTI-MEM + FBS are more likely to express the plasmid of interest and are generally healthier.
Beadloading
Note: Applying the glass beads to the top of the cells can be achieved in multiple ways. We recommend using a hand-made beadloader that will effectively sprinkle an evenly distributed monolayer of beads across the cells. See also Cialek et al. (2021).
Create a custom beadloader (Figure 2).
Remove the glass bottom from a MatTek chamber.
Place a ~100-µm nylon mesh over the hole. Secure with tape.
Add the glass beads.
Cover the top with a MatTek lid. Secure with parafilm.
Cover the bottom of the dish (side with the mesh) with another MatTek lid.
Store in a desiccator upside down to keep the beads dry and within the chamber.
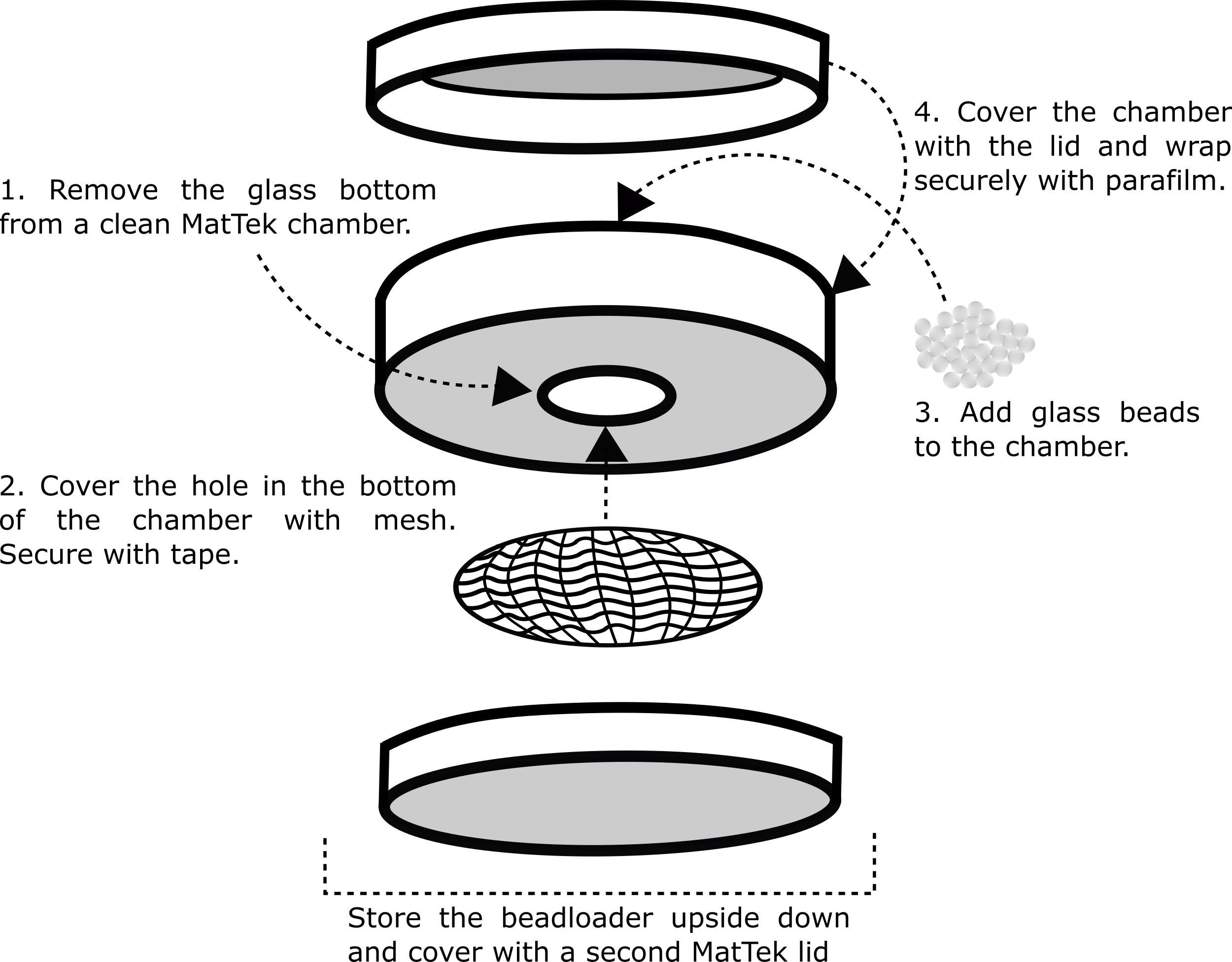
Figure 2. Steps depicting how to make a custom beadloaderOn the bench, add the following to a low retention 0.6-ml tube:
~0.5 µg Cy3 anti-FLAG Fab (~1 µl working stock prepared in Step B8).
~0.5 µg anti-SunTag scFv-GFP (~1 µl working stock prepared in Step C19).
~0.13 µg purified Halo-MCP (~1 µl working stock prepared in Step C19).
750 ng plasmid DNA of interest.
1× PBS to a total volume of 4 μl.
Mix well by pipetting and flicking the tube; do not vortex.
Spin down rapidly.
Take the MatTek chamber containing the cells out of the incubator and into a sterile hood.
Remove the medium (Opti-MEM + FBS) and place in a 15-ml conical tube.
Tip: Make sure to remove all medium to avoid diluting the sample solution, especially from the coverslip area that can retain some medium because of its concave shape. If medium is left on the coverslip, glass beads sprinkled on top in step 8 below will sometimes float and not actually contact the cells. Be careful not to allow cells to dry out by proceeding through the following steps promptly.
Add the 4 µl solution to the top of the cells.
Sprinkle glass beads onto the cells by gently tapping the homemade beadloader against the chamber.
Tips:
The beadloader helps to sprinkle the beads in a uniform fashion, so that it is easy to obtain a monolayer across the entire coverslip. However, this can be achieved without a beadloader by simply pipetting the beads using a 1,000-µl pipette tip and briefly shaking the tip over the cells to sprinkle the beads on top. While this method works, sometimes the beads will fall in clumps on the cells, which tends to cause the cells to peel more readily.
The easiest cells to beadload are adherent, such as U-2 OS, HeLa, RPE1, or fibroblast cells. Other cells that easily detach (for example, HEK293 cells) are more difficult to beadload because they tend to peel. In this case, we recommend polylysine to facilitate adherence to coverslips and prevent peeling after beadloading.
If the cells are too confluent, they can peel in one massive layer after beadloading. The ideal confluence is 70-90%.
Remove the beadloader.
Tap the chamber against the hood bench 5-12 times.
Note: This action causes the glass beads to roll over and bang against the cells. Similar to the action of electroporation, the rolling beads induce tiny tears in the cell membranes. Purified protein/DNA pipetted on top of the cells can then diffuse through these tiny tears (whilst the beads are much too large to enter). Although these tears do physically damage cells, we find the the cells recover pretty quickly. In our experience, in 2-3 h, cells are fine, display no stress granules, and are ready for imaging.
Tip the chamber at a 45° angle.
Pour the Opti-MEM + FBS medium that was removed in step 5 back onto the cells.
Tip: When pouring medium back onto cells, be careful not to disturb the cells too much. We recommend pouring the medium into the corner of the MatTek chamber and allow it to gently wash over the coverslip. By tipping the chamber, this step also helps the beads to float off to the edge, where they can easily be removed using a pipette or aspirator.
Incubate at 37°C, 5% CO2 for 1 h.
During the incubation time, warm phenol-free (white) DMEM +/- media to 37°C.
Staining Halo-MCP with Janelia Fluor 646 HaloTag ligands (JF646-Halo ligand)
Dissolve the JF646-Halo ligand to a concentration of 10 mM in DMSO as the stock solution. Dilute the stock solution to a concentration of 0.2 mM in DMSO as the working stock.
Store at -20°C.
Make aliquots of 10-15 μl to avoid excessive freeze-thaw cycles.
Remove an aliquot of JF646-Halo ligand from -20°C.
Bring the JF646-Halo ligand and warmed white DMEM (+/-) medium into the hood.
Prepare a 1:1,000 dilution of the JF646-Halo ligand in white DMEM (+) medium.
The final volume of the dilution should be sufficient to cover the surface of the cells.
For 35-mm MatTek chambers, 500 μl is sufficient.
Remove cells from the incubator and bring them into the hood.
Tilt the chamber at a 45° angle.
Remove all medium.
Wash cells gently with white DMEM (-) until most of the beads have been removed.
Add the JF646-Halo ligand dilution to the top of the cells.
Incubate at 37°C, 5% CO2 for 20 min.
Wash cells twice with white DMEM (-) medium.
Add 2 ml white DMEM (+) to the cells.
Cells can be imaged immediately after washing.
Keep cells at 37°C, 5% CO2 until ready to image.
Imaging [Video 1 - Example of a 3-color movie]
Note: Imaging can be performed on any microscope equipped with ~488, ~563, and ~647 nm lasers, appropriate filters, a stage-top incubator, and sensitive EMCCD or sCMOS cameras. We recommend using a HILO microscope (Tokunaga et al., 2008), a light-sheet microscope, or a confocal microscope to reduce the background signal. TIRF microscopy can be used to examine translation sites near the coverslip. Widefield can also be used, but signal-to-noise for single RNA translation sites will be somewhat diminished because there is no sectioning.
Tip: Autofluorescence can look like translating spots because it fluoresces over a wide range of wavelengths and therefore appears in multiple channels simultaneously. Thus, it is Important to add puromycin to confirm that the spots are translation sites.
Place cells onto a stage-top incubator (37°C, 5% CO2)
Add puromycin to test for active translation
Set up your imaging experiment to capture the entire cell with a 1-min interval between each volume capture for 15 total timepoints.
Notes:
The interval between each volume capture can be decreased to more precisely measure the dynamics of translation shut-off in a single cell.
Volume indicates the z-stacks needed to image the entire cell from top to bottom. For our purpose, we image 13 z-stacks, where each stack is 0.5 μm. The stack number and size can be adjusted to fit specific needs.
Acquire five timepoints.
After the first five timepoints, add puromycin at a final concentration of 50-100 μg/ml directly to the top of cells.
Note: Avoid touching the chamber directly, as this could move the chamber and change your imaging field.
Continue imaging cells for 10 min (10 frames). If translation sites are real, they will disappear quickly, typically within a minute or two.
Note: As an initial test for cell stressors and Harringtonine-induced ribosomal run-off assays, we recommend conducting imaging experiments with a larger interval (3-5 min) for several hours. After determining the time of response, intervals and imaging time can be adjusted accordingly. The following describes the imaging conditions in U-2 OS cells that we found fruitful for stressors and Harringtonine experiments.
Add Harringtonine to test for ribosomal run-off times
Set up your imaging experiment to capture the entire cell with a 1-min interval between each volume capture for 50 total timepoints.
Acquire five timepoints.
After the first five timepoints, add Harringtonine at a final concentration of 3-5 μg/ml directly to the top of cells.
Note: Avoid touching the chamber directly, as this could move the chamber and change your imaging field.
Continue imaging cells for the remaining 45 frames (45 min).
Adding stressors such as sodium arsenite (NaAs) and dithiothreitol (DTT)
Set up your imaging experiment to capture the entire cell with intervals of 3 min for NaAs and 2 min for DTT between each volume capture for 35 total timepoints.
Acquire five timepoints.
After the first five timepoints, add NaAs or DTT at a final concentration of 0.5 mM or 0.75 mM, respectively.
Note: Avoid touching the chamber directly, as this could move the chamber and change your imaging field.
Continue imaging cells for the remaining 30 frames.
 Video 1. Representative movie of a cell after the image pre-processing step (Step I1). The movie was acquired at one frame per 6 s. The field of view is 512 pixels × 512 pixels = 66.56 μm × 66.56 μm.
Video 1. Representative movie of a cell after the image pre-processing step (Step I1). The movie was acquired at one frame per 6 s. The field of view is 512 pixels × 512 pixels = 66.56 μm × 66.56 μm.
Data analysis (Figure 3)
Preprocessing movies (Video 1)
Create maximum intensity projection through z.
Subtract the background intensity from each channel.
Note: Background levels were determined by measuring the inherent noise of the cameras used. This can be achieved by taking images with the camera shutters closed and then calculating the average pixel intensity of those images.
Make a mask that contains the entire cell. This masking step will avoid including any molecules from adjacent cells.
Create a running average projection over time (using a few frames). This is necessary if the signal-to-noise ratio is poor.
Note: We use the ImageJ software for Steps I1a-I1b and Mathematica for Steps I1c-I1d.
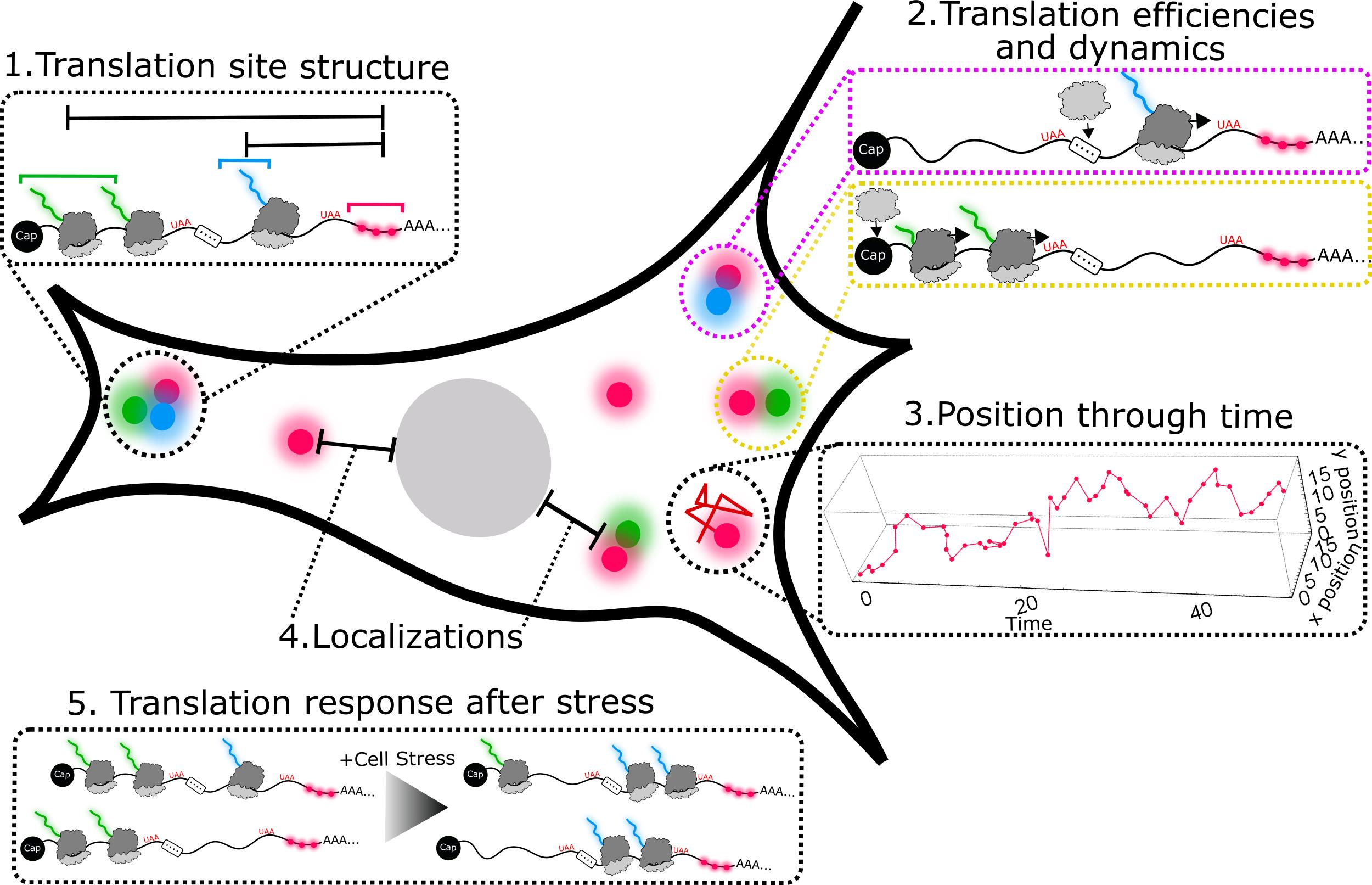
Figure 3. Schematic illustration of example biophysical parameters that analysis of the acquired data can provideTrack mRNAs. Any single-molecule tracking package can be used. We recommend TrackMate for its ease of use (Tinevez et al., 2017). We also have custom Mathematica code available on Github at:
https://github.com/Colorado-State-University-Stasevich-Lab/mRNA_Tracking_BioProtocol
Detect mRNA molecules in each time frame.
Apply a difference-of-Gaussian filter to enhance the signals of single molecules.
Set a threshold to binarize the image such that all mRNAs are recognized.
Detect objects and their intensity centroids.
Filter objects out that are either too large or too small. This will exclude aggregations and noise, respectively.
If analyzing Harringtonine or stress experiments, skip to Step I5.
Link detected mRNAs over time.
A nearest-neighbor tracking algorithm can be used. We allow a maximum mRNA jump size of 5 pixels (650 nm) between subsequent frames (assuming <10 sec intervals).
Hand-curate tracks to merge tracks that are split accidentally because of dimmer mRNA signals for a few frames and/or to omit tracks that are indistinguishable among other mRNAs due to dense environments.
Categorize mRNA into four different types of translation status (no translation, translation of ORF 1, translation of ORF 2, and translation of ORF 1 and ORF 2 simultaneously).
Make crops in all channels by centering around tracked mRNAs.
In the case that multiple cameras are used, a correction factor must be applied to adjust for potential camera misalignment. For this, a calculated transformation function is applied to other channels based on the centroid position of the tracked mRNA signals. The transformation function can be determined by imaging fixed beads that fluoresce in all channels simultaneously (for example, TetraSpeck Microspheres, 0.1 μm, Thermo Fisher Scientific, T7279).
Average the cropped images for each track in all channels.
Detect signals in the other channels (i.e., non-mRNA channels) and categorize into four groups based on the presence or absence of signals in the other channels.
Hand-curate the categorization. This is a check to ensure that the generated categorization was correct.
Use these results to analyze the population of different mRNAs within cells.
Fit a 2D Gaussian to the cropped data; this will calculate intensities in all channels and super-resolve the localization of mRNAs.
Use these results to analyze ribosome density and mean square displacement.
Stress and Harringtonine analysis
Use detected RNA molecules found in Step I2a.
Note: Linking tracks over time is not possible when using a large time interval between each frame. Since Harringtonine and stress-induced ribosomal run-offs can take tens of mins to an hour, we typically image infrequently (once per minute or longer) so individual translation sites cannot all be tracked (although there usually are a few that can be tracked by chance because they are relatively immobile). The analysis below therefore does not require the tracking of individual translation sites.
Make crops in all channels by centering around detected mRNA signals.
Detect signals in other channels and categorize into four groups based on the presence or absence of signals in the other (non-mRNA) channels.
Fit a 2D Gaussian to the cropped data; this will calculate the intensities in all channels.
Combine the translation site intensity data from all cells and all experiments.
Plot as intensity or translation site number over time.
Recipes
1 M HEPES buffer
25 mM HEPES pH 7.9
12.5 mM MgCl2
100 mM KCl
0.1 mM EDTA
0.01% NP40
10% glycerol
1 mM DTTHisTrap buffer A
1× PBS pH 7.4
300 mM NaCl
0.2 mM AEBSF
5 mM b-mercaptoethanolHisTrap buffer B
1× PBS pH 7.4
300 mM NaCl
500 mM immidazole
0.2 mM AEBSF
5 mM b-mercaptoethanolSuperdex buffer
25 mM HEPES pH 7.9
12.5 mM MgCl2
100 mM KCl
0.1 mM EDTA
0.01% NP40
10% glycerol
1 mM DTTDMEM (+) media (for phenol-free as well)
To 500 ml DMEM, add 50 ml FBS, 5 ml Pen/strep, 5 ml L-Glu
Acknowledgments
We sincerely thank Dr. Hataichanok (Mam) Scherman for the protocol to purify Halo-MCP and anti-SunTag scFv-GFP. We thank Dr. Luke Lavis for kindly providing the JF646-labeled HaloTag ligand. We thank all members of the Stasevich lab for their support and helpful discussion. TJS, ALK, and TM were supported by the NIH (grant no. R35GM119728). TJS was also supported by the NSF (grant no. MCB-1845761). This protocol was derived from the work published in Koch et al. (2020). The beadloading protocol was first described in Hayashi-Takanaka et al. (2011) and the single-molecule translation imaging assay was first described in Morisaki et al. (2016).
Competing interests
The authors declare no competing interests.
Ethics
No human and/or animal subjects are used in this protocol.
References
- Boersma, S., Khuperkar, D., Verhagen, B. M. P., Sonneveld, S., Grimm, J. B., Lavis, L. D. and Tanenbaum, M. E. (2019). Multi-Color Single-Molecule Imaging Uncovers Extensive Heterogeneity in mRNA Decoding. Cell 178: 458-472.e19.
- Bornes Stéphanie, Prado-Lourenco Leonel, Bastide Amandine, Zanibellato Catherine, Iacovoni Jason S., Lacazette Eric, Prats Anne-Catherine, Touriol Christian, and Prats Hervé (2007). Translational Induction of VEGF Internal Ribosome Entry Site Elements During the Early Response to Ischemic Stress. Circ Res 100: 305-308.
- Cialek, C. A., Koch, A. L., Galindo, G., and Stasevich, T. J. (2020). Lighting up single-mRNA translation dynamics in living cells. Curr Opin Genet Dev 61: 75-82.
- Cialek, C. A., Galindo, G., Koch, A. L., Saxton, M. N. and Stasevich, T. J. (2021). Bead Loading Proteins and Nucleic Acids into Adherent Human Cells. J Vis Exp e62559.
- Coulon, A., Chow, C. C., Singer, R. H., and Larson, D. R. (2013). Eukaryotic transcriptional dynamics: from single molecules to cell populations. Nat Rev Genet 14(8): 572-584.
- Firth, A. E. and Brierley, I. (2012). Non-canonical translation in RNA viruses. J Gen Virol 93: 1385-1409.
- Hayashi-Takanaka, Y., Stasevich, T.J., Kurumizaka, H., Nozaki, N. and Kimura, H. (2014). Evaluation of chemical fluorescent dyes as a protein conjugation partner for live cell imaging. PloS One 9: e106271.
- Hayashi-Takanaka, Y., Yamagata, K., Wakayama, T., Stasevich, T.J., Kainuma, T., Tsurimoto, T., Tachibana, M., Shinkai, Y., Kurumizaka, H., Nozaki, N., et al. (2011). Tracking epigenetic histone modifications in single cells using Fab-based live endogenous modification labeling. Nucleic Acids Res 39(15): 6475-6488.
- Koch, A., Aguilera, L., Morisaki, T., Munsky, B. and Stasevich, T. J. (2020). Quantifying the dynamics of IRES and cap translation with single-molecule resolution in live cells. Nat Struct Mol Biol 27: 1095-1104.
- McNeil, P. L., and Warder, E. (1987). Glass beads load macromolecules into living cells. J Cell Sci 88 (Pt 5): 669-678.
- Morisaki, T. and Stasevich, T. J. (2018). Quantifying Single mRNA Translation Kinetics in Living Cells. Cold Spring Harb PerspectBiol 10(11): a032078.
- Morisaki, T., Lyon, K., DeLuca, K. F., DeLuca, J. G., English, B. P., Zhang, Z., Lavis, L.D., Grimm, J. B., Viswanathan, S., Looger, L. L., et al. (2016). Real-time quantification of single RNA translation dynamics in living cells. Science 352(6292): 1425-1429.
- Pichon, X., Bastide, A., Safieddine, A., Chouaib, R., Samacoits, A., Basyuk, E., Peter, M., Mueller, F. and Bertrand, E. (2016). Visualization of single endogenous polysomes reveals the dynamics of translation in live human cells. J Cell Biol 214(6): 769-781.
- Pichon, X., Robert, M.-C., Bertrand, E., Singer, R.H., and Tutucci, E. (2020). New Generations of MS2 Variants and MCP Fusions to Detect Single mRNAs in Living Eukaryotic Cells. Methods Mol Biol 2166: 121-144.
- Tanenbaum, M. E., Gilbert, L. A., Qi, L. S., Weissman, J. S. and Vale, R. D. (2014). A Protein-Tagging System for Signal Amplification in Gene Expression and Fluorescence Imaging. Cell 159(3): 635-646.
- Tinevez, J. Y., Perry, N., Schindelin, J., Hoopes, G. M., Reynolds, G. D., Laplantine, E., Bednarek, S. Y., Shorte, S. L. and Eliceiri, K. W. (2017). TrackMate: An open and extensible platform for single-particle tracking. Methods 115: 80-90.
- Tokunaga, M., Imamoto, N., and Sakata-Sogawa, K. (2008). Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat Methods 5: 159-161.
- Wang, C., Han, B., Zhou, R. and Zhuang, X. (2016). Real-Time Imaging of Translation on Single mRNA Transcripts in Live Cells. Cell 165(4): 990-1001.
- Wu, B., Eliscovich, C., Yoon, Y. J. and Singer, R. H. (2016). Translation dynamics of single mRNAs in live cells and neurons. Science 352(6292): 1430-1435.
- Yan, X., Hoek, T. A., Vale, R. D. and Tanenbaum, M. E. (2016). Dynamics of Translation of Single mRNA Molecules In Vivo. Cell 165(4): 976-989.
- Zhao, N., Kamijo, K., Fox, P. D., Oda, H., Morisaki, T., Sato, Y., Kimura, H. and Stasevich, T. J. (2019). A genetically encoded probe for imaging nascent and mature HA-tagged proteins in vivo. Nat Commun 10(1): 2947.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Koch, A. L., Morisaki, T. and Stasevich, T. J. (2021). A Multi-color Bicistronic Biosensor to Compare the Translation Dynamics of Different Open Reading Frames at Single-molecule Resolution in Live Cells. Bio-protocol 11(14): e4096. DOI: 10.21769/BioProtoc.4096.
Category
Biophysics > Microscopy
Molecular Biology > RNA > mRNA translation
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.