- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Simple Method to Generate Super-sensitive AID (ssAID)-based Conditional Knockouts using CRISPR-based Gene Knockout in Various Vertebrate Cell Lines
Published: Vol 11, Iss 14, Jul 20, 2021 DOI: 10.21769/BioProtoc.4092 Views: 4162
Reviewed by: Alessandro DidonnaQin TangSabine Le Saux
Original research article
The authors used this protocol in:

Oct 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Inducing loss of function of a target protein using methods such as gene knockout is a powerful and useful strategy for analyzing protein function in cells. In recent years, the CRISPR/Cas-9-based gene knockout technology has been widely used across a variety of eukaryotes; however, this type of simple gene knockout strategy is not applicable to essential genes, which require a conditional knockout system. The auxin-inducible degron (AID) system enables rapid depletion of the target protein in an auxin-dependent manner and has been used to generate conditional mutants in various eukaryotic cell lines. One problem with the AID system is the use of high auxin concentrations for protein degradation, which can cause cytotoxicity. Recently, we established a super-sensitive AID (ssAID) system that allowed a reduction in the amount of auxin required by more than 1,000-fold. We also utilized a single-step method to generate AID-based conditional knockout cells with a ssAID system in various cell lines. In this protocol, we introduce our improved method, which provides a powerful tool for the investigation of the roles of essential genes.
Keywords: AuxinBackground
The auxin-inducible degron (AID) system enables the direct and conditional depletion of a target gene product in various eukaryotic cells (Nishimura et al., 2009; Holland et al., 2012). This system utilizes an auxin-dependent degradation system in plants. Transport inhibitor response 1 (TIR1) is a plant-specific F-box protein that forms an E3 ubiquitin ligase, namely the SCFTIR1 complex, which ubiquitinates and degrades proteins in the AUX/IAA family in an auxin-dependent manner in plant cells (Dharmasiri et al., 2005; Kepinski and Leyser, 2005). In non-plant cells, SCFTIR1 is formed via the expression of TIR1, with the target protein fused with an AID-tag, which is degraded in an auxin-dependent manner (Figure 1, AID). The AID system has been widely used to generate conditional knockout yeast and vertebrate cell lines (Holland et al., 2012; Maric et al., 2014; Nora et al., 2017; Gibcus et al., 2018).
In the conventional AID system, a natural auxin, indole-3-acetic acid (IAA), is used to induce degradation (Nishimura et al., 2009; Holland et al., 2012). However, one issue with this system is the amount of IAA required for protein degradation (Yesbolatova et al., 2019), as high IAA concentrations (500 μM) can cause cytotoxicity in some mammalian cell lines. To overcome this problem, we focused on a high-affinity binding pair, the synthetic auxin 5-Ad-IAA and its high-affinity partner Oryza sativa (Os) TIR1F74A, which was identified based on previous studies in A. thaliana (Uchida et al., 2018; Yamada et al., 2018). We succeeded in establishing a super-sensitive AID (ssAID) system that incorporates a high-affinity binding pair, 5-Ad-IAA and OsTIR1F74A (Figure 1, ssAID) (Nishimura et al., 2020). This ssAID system was shown to have a 1,000-fold higher sensitivity than the conventional AID system using OsTIR1WT and IAA. Another group developed a similar system called auxin-inducible degron 2, which uses OsTIR1F74G instead of OsTIR1F74A (Yesbolatova et al., 2020). Additionally, they suggested that OsTIR1F74A and OsTIR1F74G had similar effects on AID degradation; however, we found that OsTIR1F74A is 100-fold more sensitive to 5-Ad-IAA than is OsTIR1F74G.
Moreover, we also developed a single-step method to quickly generate AID-based conditional knockouts in various vertebrate cell lines (Nishimura and Fukagawa, 2017). In this method, we use CRISPR/Cas9-based gene targeting combined with integration of an AID plasmid that contains the expression cassettes for OsTIR1 and the target protein with an AID-tag. The combination of an ssAID system and a single-step method may provide a powerful tool for elucidating the roles of various gene products in vertebrate cells. Here, we introduce a protocol to generate AID-based conditional knockouts in various vertebrate cell lines using an ssAID system.
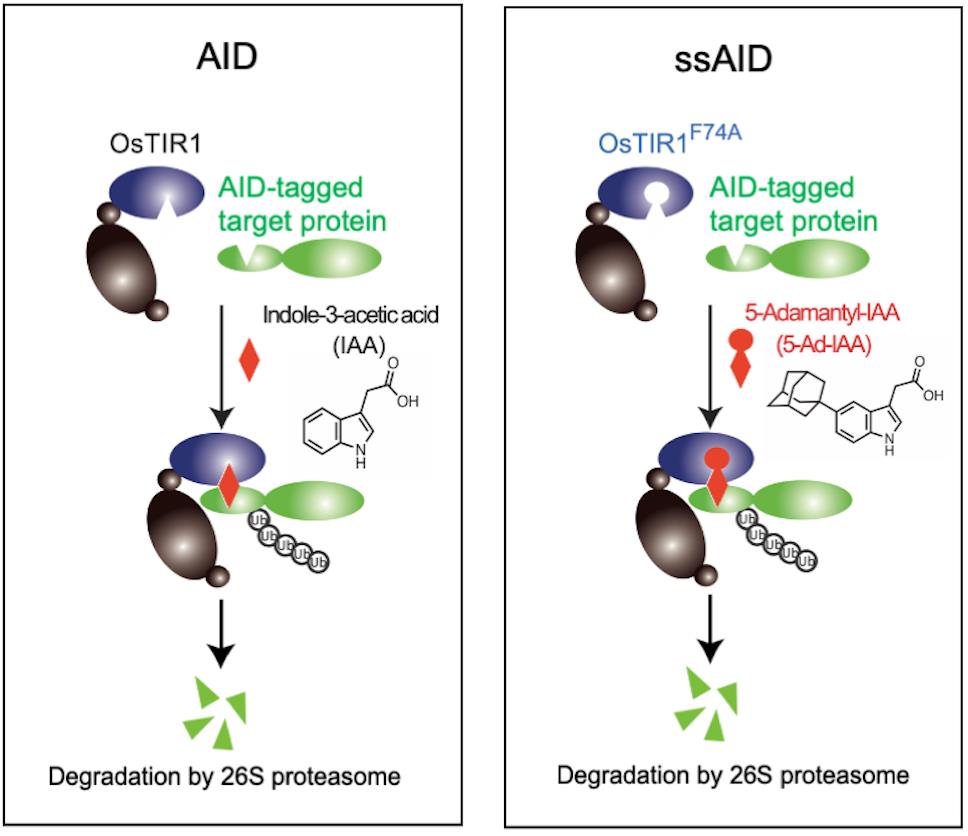
Figure 1. The concept of Auxin Inducible Degron (AID) for rapidly degrading AID-tagged target proteins in an auxin (IAA)-dependent manner. The protein of interest with an AID-tag is conditionally degraded by applying the auxin IAA. ssAID uses the IAA derivative 5-Ad-IAA and its high-affinity binding partner OsTIR1F74A.
Materials and Reagents
Laboratory disposables:
Pipettes (e.g., Thermo Fisher Scientific, catalog numbers: FN-4641010N, FN-4641060N, FN-4641080, FN-4641100)
15-ml tubes (e.g., Greiner, catalog number: 188261)
50-ml tubes (e.g., Greiner, catalog number: 227261)
1.5-ml tubes (e.g., Watson, catalog number: 9833)
PCR tubes (e.g., Greiner, catalog number: 673210)
96-well plates (Corning, catalog number: 3596)
24-well plates (Corning, catalog number: 3526)
6-well plates (Corning, catalog number: 3516)
10-cm culture dishes (Corning, catalog number: 430167)
E. coli (Dh5α) competent cells (Takara, catalog number: 9057)
pX330 plasmid (Addgene, catalog number: 42230)
pAID plasmids
pAIDFA-EF1a-NmScarlet-mAID (Addgene, catalog number: 140615)
pAIDFA-CMV-NmScarlet-mAID (Addgene, catalog number: 140616)
pX330 plasmids for linearization of the pAID plasmids
pAID-CMV linearizing in pX330 (Addgene, catalog number: 140609)
pAID-EF1a linearizing in pX330 (Addgene, catalog number: 140610)
EcoRV (NEB, catalog number: R0195S)
PrimeStar GXL PCR polymerase (Takara, catalog number: R050A)
Gibson Assembly Cloning kit (NEB, catalog number: E5510S)
InFusion HD Cloning Plus (Takara, catalog number: Z8909N)
1× Tris-acetate-EDTA (TAE) buffer (0.04 M Tris-acetate and 0.001 M EDTA) for electrophoresis
Agarose (SeaKem LE Agarose: 50001) for electrophoresis
Ethidium bromide solution for staining DNA gels (Nacalai, catalog number: 14631-51)
Gotaq Master Mix (Promega, catalog number: M7122)
Wide range DNA ladder (Takara, catalog number: 3415A)
Wizard SV Gel and PCR Clean-Up System (Promega, catalog number: A9281) for plasmid construction
NucleoBond Xtra Maxi kit (Macherey-Nagel, catalog number: 740414.100)
Neon transfection system 100 µl kit (Thermo Fisher Scientific, catalog number: MPK10096)
Polyethylenimine MW 25000, transfection grade (PolySciences, catalog number: 23866-1)
DMEM (Nacalai, catalog number: 09891-25)
PBS (Nacalai, catalog number: 14249-95)
FBS (Gibco, catalog number: 10270-106)
Chicken serum (Gibco, catalog number: 16110-082)
Trypsin solution (Nacarai, catalog number: 32777-15)
Penicillin-streptomycin (Nacalai, catalog number: 09357-34)
Knockout serum replacement (Thermo Fisher Scientific, catalog number: 10828010)
MEM non-essential amino acids (Nacalai, catalog number: 06344-56)
L-glutamate (Nacalai, catalog number: 16948-04)
2-mercaptoethanol (Sigma, catalog number: M6250)
1× PBS (Nacalai, catalog number: 14249-95)
Gelatin (Sigma, catalog number: G1890-100G)
Blasticidin S hydrochloride (Wako, catalog number: 029-18701)
L-glutamic acid (Wako, catalog number: 072-00501)
5-adamantyl-IAA (TCI, catalog number: A3390)
Dimethyl sulfoxide (DMSO) (Wako, catalog number: 043-07216)
1 M NaOH solution (Wako, catalog number: 193-02175)
Filter (Merck, catalog number: S2HVU11RE)
LB medium (Nacalai, catalog number: 20068-75)
Kanamycin (Nacalai, catalog number: 19860-31)
Ampicillin (Nacalai, catalog number: 02739-74)
Culture medium for chicken DT40 cells (see Recipes)
Culture medium for mammalian cells (see Recipes)
Culture medium for mouse ES cells (see Recipes)
0.1% gelatin solution (see Recipes)
1 mg/ml polyethylenimine solution (see Recipes)
5-Ad-IAA solution (see Recipes)
20 mM glutamic acid solution (pH 4.0) (see Recipes)
Equipment
Thermal cycler (e.g., Thermo Fisher Scientific: MiniAmp)
Mupid 2plus (Mupid: AD110)
CO2 incubator (at 5% CO2 and 37°C for mammalian cells, 38.5°C for chicken DT40 cells) (e.g., PHCBi: MCO-170AIC)
Countess II (Thermo Fisher Scientific: C10228)
Neon transfection system (Thermo Fisher Scientific: MPK5000)
Procedure
DNA preparation
pAID plasmid construction
Note: pAID plasmids contain the OsTIR1F74A and mScarlet-mAID expression cassette under the control of the CMV (Addgene #140615) or EF-1α promoter (Addgene #140616) (Figure 1A) (Nishimura et al., 2020). The CMV promoter is for the chicken DT40 cell line, while the EF1a promoter is for mammalian cell lines. The cDNA of the target protein can be inserted into the EcoRV site at the end of the mScarlet-mAID cassette using the InFusion (Takara) or Gibson assembly (NEB) cloning systems (Figure 2B).
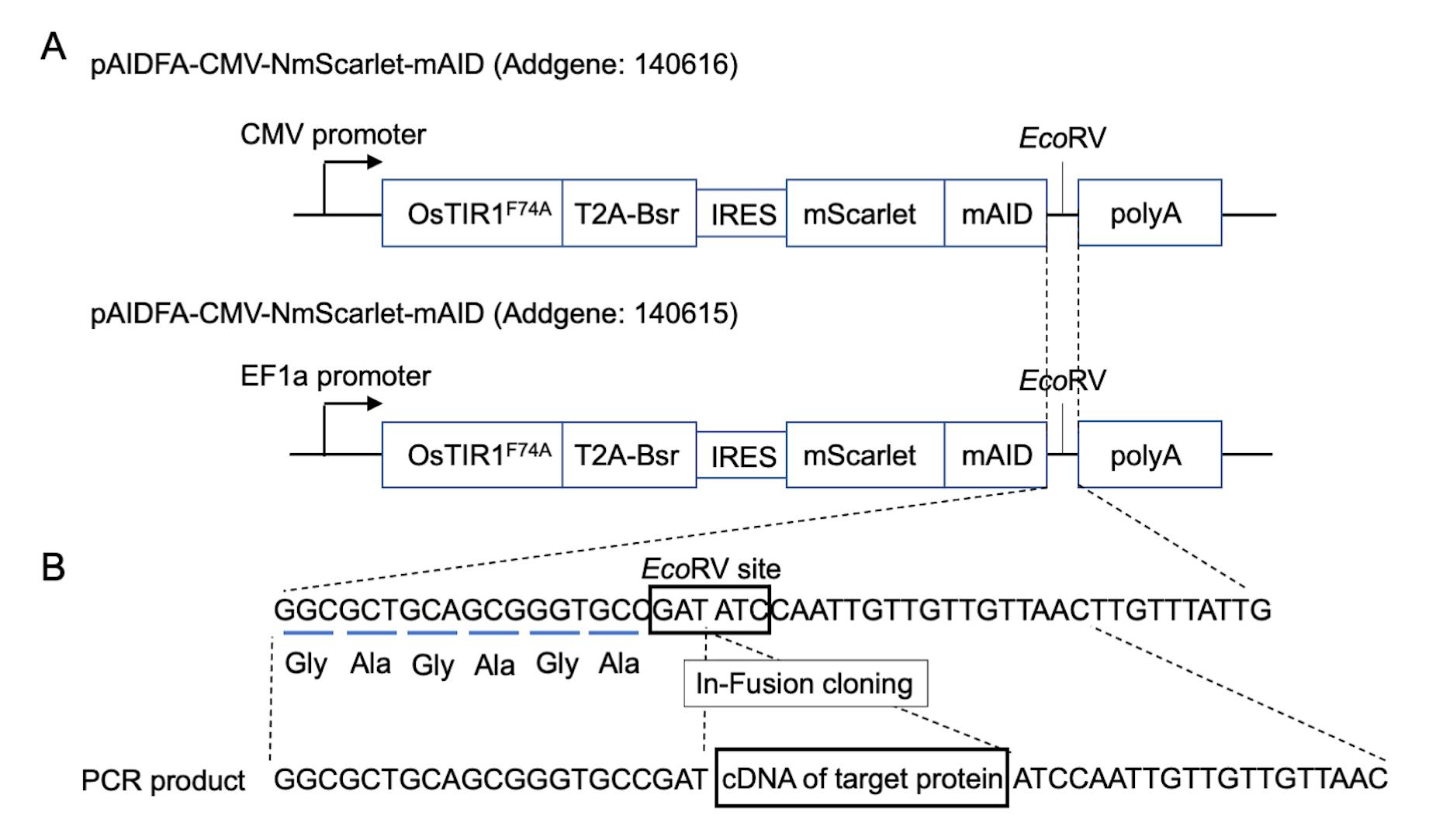
Figure 2. Construction of pAID plasmids. A. Maps of the pAID plasmids. pAID plasmids contain the OsTIR1F74A and mScarlet-mAID expression cassette under the control of the CMV or EF-1α promoter. B. Construction of the pAID plasmid using In-Fusion cloning. Purified PCR fragment is cloned into the EcoRV site of the pAID plasmid by In-Fusion cloning.Digest the pAID plasmid with EcoRV.
10× NEB buffer 2 3 µl
pAID plasmid 3 µg
EcoRV 1 µl
DW up to 30 µl
Incubate at 37°C for 2 h
Amplify the target protein cDNA by PCR using the following primer pair:
pAID-Ncloning1: 5-CTGGAGCGGGTGCCGAT[5 end of your target cDNA]-3
pAID-Ncloning2: 5-GTTAACAACAACAATTGGAT[3 end of your target cDNA]-3
5× PrimeStar GXL buffer 10 µl
dNTP Mixture (2.5 mM each) 4 µl
pAID-Ncloning 1 10 pmol
pAID-Ncloning 2 10 pmol
Template (cDNA) 25-750 ng (Plasmid DNA: 100 pg-10 ng)
PrimeStar GXL polymerase 1 µl
DW up to 50 µl
PCR steps (30 cycles)
98°C 10 s
60°C 16 s
68°C 1 min/kb
Purify the linearized pAID plasmid and PCR product using Wizard SV PCR and the Gel Purification Clean-UP system after 1% agarose gel electrophoresis.
Note: Digested pAID plasmid and PCR fragments can be estimated by comparing the fluorescent signals with DNA ladder marker after ethidium bromide staining.Assemble the PCR fragment and linearized pAID plasmid using the Gibson assembly (NEB) or In-Fusion (Takara) cloning systems. Please see the user manual of the In-Fusion HD cloning kit for details.
Purified PCR fragment 10-200 ng
Linearized pAID vector 50-200 ng
5× In-Fusion HD Enzyme Mix 2 µl
DW up to 10 µl
Incubate the reaction mixture at 50°C for 15 min.
Transform E. coli (DH5α) competent cells with the assembled DNA. Please see the user manual of the In-Fusion HD cloning kit for details.
Perform colony PCR to collect the transformed E. coli colonies.
Note: We use colony PCR to collect E. coli colonies by GoTaq (Promegega) before isolation of the corresponding plasmids. We usually check 8 colonies per sample using the following primer set:
pAID-Ncheck1: 5-CAGGCGGTGGAAGCGGGTGCCG-3
pAID-Ncheck2: 5-ACAAGTTAACAACAACAATTGG-3
Collect two of the E. coli colonies and isolate the corresponding plasmids.
Confirm the sequence of the cloned plasmid by Sanger sequencing.
For the transfection step, transfection grade pure plasmid DNA is necessary. We usually use the NucleoBond Xtra Maxi kit for plasmid isolation to obtain transfection grade plasmids; we typically obtain several mg/ml plasmid solution. If the concentration of plasmid DNA is lower than 1 mg/ml, the DNA should be concentrated by ethanol precipitation.
pX330 construction and design of sgRNA for target sequences
Note: We usually design sgRNA at the intron-exon boundary to avoid digesting the cDNA in the pAID plasmid (Figure 3). The pX330 plasmid was constructed as described in a previously published study (Pyzocha et al., 2014). Please see Step A9 regarding plasmid preparation.
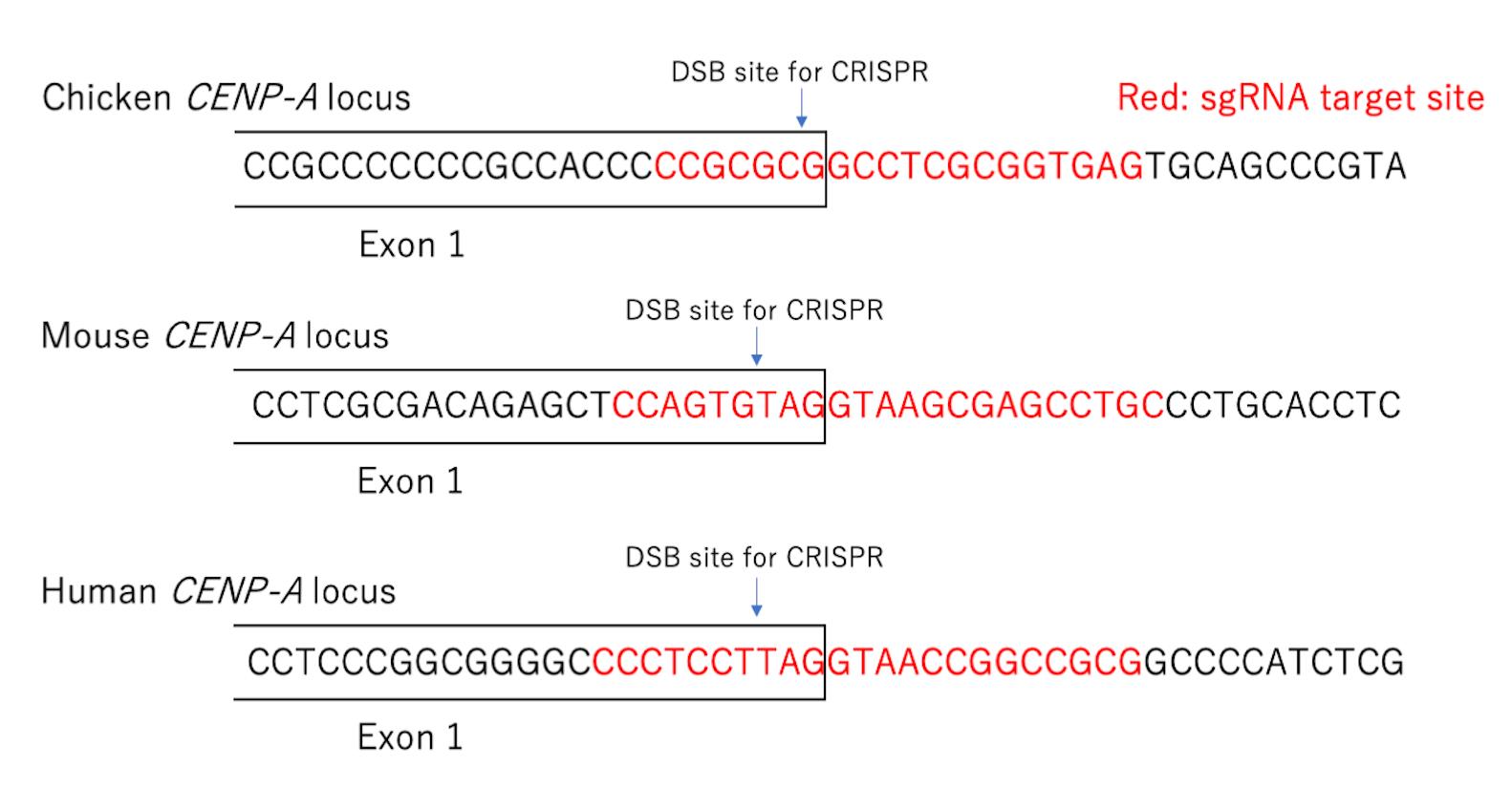
Figure 3. sgRNA design for target genes. SgRNA for gene targeting is designed at the intron-exon boundary. There are three examples for designing a target site for CRISPR/Cas9 at the CENP-A gene in chicken, mouse, or human cells. The boxed area shows the exon 1 sequence of CENP-A. The sequence shown in red is the target site for CRISPR/Cas9. The double-strand break site introduced by CRISPR/Cas9 is labeled as DSB site for CRISPR.Transfection
A single-step method enables the generation of AID-based conditional knockouts in various vertebrate cell lines. In this method, we used CRISPR/Cas9-based gene targeting combined with the integration of an AID plasmid, which contains the expression cassettes for OsTIR1 and the target protein with an AID-tag (Figure 4).
Note: Although we transfected cells using polyethylenimine or the Neon transfection system, other methods (such as lipofection) can be used for the transfection of mammalian cell lines. It is important to use transfection methods with low toxicity, which will not inhibit cell growth after transfection.
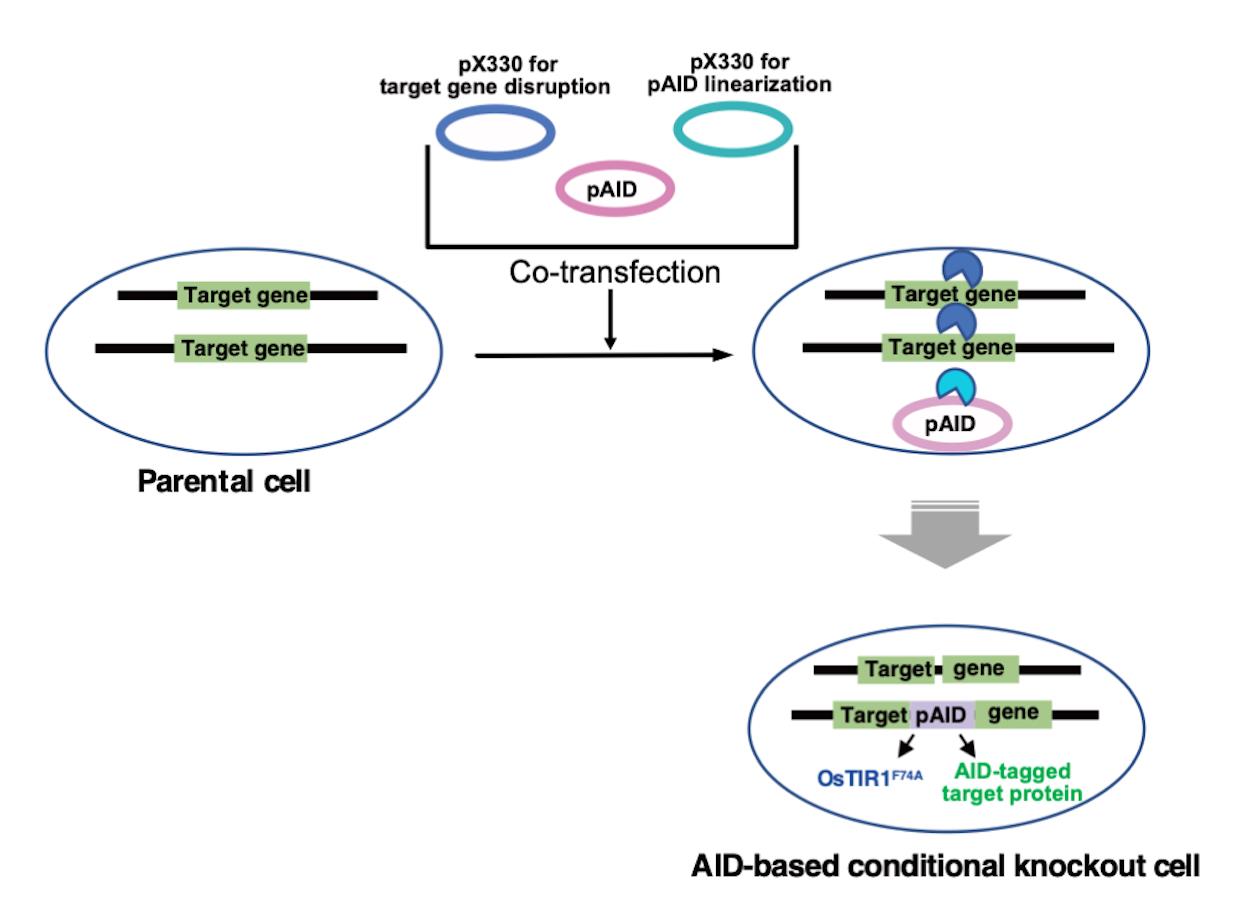
Figure 4. A single-step method to quickly generate an AID-based conditional knockout cell line. CRISPR/Cas9-based gene targeting is coupled to pAID-plasmid integration to express both OsTIR1 and an AID-tagged target protein. Parental cells are transfected simultaneously with three different plasmids: pAID, pX330 for pAID linearization, and pX330 for target gene disruption.After transfection, Cas9 protein induces DNA double-strand breaks in the target locus and pAID. Target genes are disrupted by pAID-plasmid integration and/or internal deletion/insertion.
Co-transfection induces pAID plasmid integration into the genome and target gene disruption. OsTIR1F74A and AID-tagged target protein is expressed from the pAID plasmid that is integrated into the genome.
Neon transfection of chicken DT40 cells
DNA preparation (a)
Prepare 5 µg pAID plasmid, 5 µg pX330 plasmid for pAID plasmid linearization, and 10 µg pX330 plasmid containing sgRNA sequences for gene targeting. Add all plasmids to 130 µl R buffer for Neon transfection.
Note: The volume of plasmid DNA solutions should not exceed 10 µl. If they exceed 10 µl, the DNA should be concentrated by ethanol precipitation.
Cell preparation (b-k)
Transfer cells to a 15-ml tube.
Centrifuge cells for 5 min at 200 × g at room temperature (RT).
Remove the media by aspiration. Resuspend the cells in 1 ml 1× PBS at RT.
Transfer 2 × 105 cells to a 15-ml tube.
Centrifuge cells for 5 min at 200 × g. Resuspend the cells in 130 µl R buffer containing the plasmids.
Transfect cells by electroporation using the Neon transfection system (1,400 V, 5 ms, 6 pulses).
Immediately after electroporation, resuspend the cells in 10 ml pre-warmed DT40 culture medium at 38.5°C (see Recipes).
Culture cells in a 10-cm dish for 48 h at 38.5°C in a CO2 incubator.
Carry out single-cell cloning by limiting dilution. Take 3 ml, 0.6 ml, and 120 µl cell culture suspension and dilute each in 40 ml DT40 culture media containing 25 µg/ml Blasticidin S. Split cells into two 96-well plates (200 µl/well) and culture them in a CO2 incubator for 1-2 weeks at 38.5°C.
Harvest surviving cells from each well and plate in a 24-well plate.
Neon transfection of human HeLa, HeLa-S3, HT1080, and RPE1 cells
DNA preparation (a)
Prepare 5 µg pAID plasmid, 5 µg pX330 plasmid for pAID plasmid linearization, and 10 µg pX330 plasmid containing sgRNA sequences for gene targeting. Add all plasmids to 130 µl R buffer for Neon transfection.
Note: The volume of plasmid DNA solutions should not exceed 10 µl. If they exceed 10 µl, the DNA should be concentrated by ethanol precipitation.
Cell preparation (b-i)
Wash culture cells with PBS and add 1 ml Trypsin solution. Harvest and collect cells into a 15-ml tube after incubation for 2 min at 37°C.
Centrifuge cells for 5 min at 200 × g.
Remove the media by aspiration. Resuspend the cells in 1 ml 1× PBS.
Transfer 2 × 105 cells to a 15-ml tube.
Centrifuge cells for 5 min at 200 × g. Aspirate the PBS and resuspend the cells in 130 µl R buffer containing the plasmids.
Transfect cells by electroporation using the Neon transfection system (1,400 V, 20 ms, 1 pulse).
Immediately after electroporation, resuspend the cells in 10 ml pre-warmed mammalian cell culture media (see Recipes).
Culture cells for 48 h at 37°C in a CO2 incubator.
Aspirate the media and add 10 ml fresh culture medium containing 4 µg/ml (for HeLa-S3), 8 µg/ml (for HeLa), or 10 µg/ml (for HT1080 and RPE1) Blasticidin S to the cells.
Note: The effective concentration of Blasticidin S depends on the cell type. It is preferable to determine the effective concentration of Blasticidin S for each cell line.
Culture cells for 1-2 weeks in a CO2 incubator at 37°C.
Note: Cells sometimes grow well in a plate, which makes it difficult to isolate a single colony (cell clone). In such cases, we usually use limiting dilution to make 1:3 and 1:9 dilution cultures for every transfection.
Isolate single-cell clones and transfer them into 24-well plates.
Polyethylenimine transfection of mouse ES (Feeder-free E14tg2a) cells
DNA preparation (a)
Prepare 1 µg pAID plasmid, 1 µg pX330 plasmid for pAID plasmid linearization, and 2 µg pX330 plasmid containing sgRNA sequences for gene targeting. Add all plasmids to a test tube containing 50 µl 20 mM glutamic acid (pH 4.0, see Recipes).
Cell preparation (b-u)
Add 18 µl 1 mg/ml polyethylenimine to another test tube containing 50 µl 20 mM glutamic acid (pH 4.0).
Vortex both tubes.
Mix and vortex the two tubes again.
Incubate the DNA and polyethylenimine mixture at room temperature for 10 min.
Note: During the incubation time, perform steps f-k.
Wash cultured cells with PBS and add 1 ml trypsin solution. Harvest and collect cells into a 15-ml tube after incubation for 2 min at 37°C.
Centrifuge cells for 5 min at 200 × g.
Remove the media by aspiration. Resuspend the cells in 1 ml 1× PBS.
Transfer 1 × 106 cells to a 15-ml tube.
Centrifuge cells for 5 min at 200 × g.
Aspirate the PBS and resuspend the cells in 300 µl serum-free DMEM media.
Add the DNA and polyethylenimine mixture to the cell suspension.
Vortex the sample.
Incubate the sample at room temperature for 30 min.
Add 1 ml 0.1% gelatin solution (see Recipes) to all wells of a 6-well plate.
After 5 min, aspirate the gelatin solution.
Carry out single-cell cloning by limiting dilution. Plate the cells with 2 ml mouse ES medium in a gelatin-coated 6-well plate at different dilutions (for example, 1:5, 1:25, 1:125, 1:625, or higher).
Culture the cells in the absence of Blastcidin S for 24 h.
Aspirate the medium and add fresh mouse ES culture medium containing 10 µg/ml Blastcidin S.
Culture the cells for 5-7 d in a CO2 incubator at 37°C.
Isolate single-cell clones and transfer them to 24-well plates.
- Troubleshooting
No colonies are found after blasticidin selection.
Sensitivities to Blasticidin depend on cell type and species. Optimization of the concentrations of Blasticidin S for drag selection of the desired cell line is recommended.
When no expression of the AID-tagged target protein is found after blasticidin selection, our recommendation is to confirm that the pAID plasmid sequence has no frameshift and/or that the pAID plasmid is stably integrated into the genome.
When the AID-tagged protein expression does not compensate for disruption of the target gene, our recommendation is to fuse an AID-tag at the opposite terminus of the target protein (C-terminus or N-terminus). The AID-tag fusion occasionally inhibits target protein function.
Data analysis
Each colony should be cultured in the presence of 5 µM 5-Ad-IAA or DMSO (control). It is preferable to use parental cells as a growth control. AID-based conditional knockout cells for essential genes exhibit cell death or slow growth in the presence of 5 µM 5-Ad-IAA. After checking 5-Ad-IAA sensitivity, target gene disruption must be confirmed by DNA sequencing and immunoblotting analysis with antibodies against the respective target protein, as described (Nishimura et al., 2020).
Recipes
Culture medium for chicken DT40 cells
DMEM 500 ml
FBS 50 ml
Chicken serum 5 ml
Penicillin-streptomycin 5 ml
10 mM 2-mercaptoethanol 500 µl
Culture medium for mammalian cells
DMEM 500 ml
FBS 50 ml
Penicillin-streptomycin 5 ml
Culture medium for mouse ES cells
DMEM 500 ml
FBS 50 ml
L-glutamate 5 ml
MEM NEAA 5 ml
Penicillin-streptomycin 5 ml
10 mM 2-mercaptoethanol 500 µl
LIF protein-containing media 1 ml
0.1% gelatin solution
0.1% gelatin is diluted with PBS. This solution is sterilized at 120°C for 20 min.
1 mg/ml polyethylenimine solution
Note: The polyethylenimine solution is used according to the protocol in PolyScience Inc.
5-Ad-IAA solution
5-Ad-IAA is dissolved in DMSO. We usually make 0.5 M or 5 mM stock solutions. These solutions are stored at -30°C in the dark. The stock solutions are stable for at least a year.
20 mM glutamic acid solution (pH 4.0)
Glutamic acid is diluted in water at a concentration of 20 mM. Adjust the pH to 4.0 with NaOH. The solution is purified via filter purification.
Acknowledgments
This work was supported by JSPS KAKENHI Grant Numbers, JP17K15041, JP19K06611, and JP20K21423 to K.N.; JP17H06167, JP16H06279, and JP15H05972 to T.F. from the Japan Society for the Promotion of Science (JSPS); and by the Hori Sciences and Arts Foundation to K.N.
Competing interests
No conflicting interests.
References
- Dharmasiri, N., Dharmasiri, S. and Estelle, M. (2005). The F-box protein TIR1 is an auxin receptor. Nature 435(7041): 441-445.
- Gibcus, J. H., Samejima, K., Goloborodko, A., Samejima, I., Naumova, N., Nuebler, J., Kanemaki, M. T., Xie, L., Paulson, J. R., Earnshaw, W. C., Mirny, L. A. and Dekker, J. (2018). A pathway for mitotic chromosome formation. Science 359(6376).
- Holland, A. J., Fachinetti, D., Han, J. S. and Cleveland, D. W. (2012). Inducible, reversible system for the rapid and complete degradation of proteins in mammalian cells. Proc Natl Acad Sci U S A 109(49): E3350-3357.
- Kepinski, S. and Leyser, O. (2005). The Arabidopsis F-box protein TIR1 is an auxin receptor. Nature 435(7041): 446-451.
- Maric, M., Maculins, T., De Piccoli, G. and Labib, K. (2014). Cdc48 and a ubiquitin ligase drive disassembly of the CMG helicase at the end of DNA replication. Science 346(6208): 1253596.
- Nishimura, K. and Fukagawa, T. (2017). An efficient method to generate conditional knockout cell lines for essential genes by combination of auxin-inducible degron tag and CRISPR/Cas9. Chromosome Res 25(3-4): 253-260.
- Nishimura, K., Fukagawa, T., Takisawa, H., Kakimoto, T. and Kanemaki, M. (2009). An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods 6(12): 917-922.
- Nishimura, K., Yamada, R., Hagihara, S., Iwasaki, R., Uchida, N., Kamura, T., Takahashi, K., Torii, K. U. and Fukagawa, T. (2020). A super-sensitive auxin-inducible degron system with an engineered auxin-TIR1 pair. Nucleic Acids Res 48(18): e108
- Nora, E. P., Goloborodko, A., Valton, A. L., Gibcus, J. H., Uebersohn, A., Abdennur, N., Dekker, J., Mirny, L. A. and Bruneau, B. G. (2017). Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 169(5): 930-944 e922.
- Pyzocha, N. K., Ran, F. A., Hsu, P. D. and Zhang, F. (2014). RNA-guided genome editing of mammalian cells. Methods Mol Biol 1114: 269-277.
- Uchida, N., Takahashi, K., Iwasaki, R., Yamada, R., Yoshimura, M., Endo, T. A., Kimura, S., Zhang, H., Nomoto, M., Tada, Y., Kinoshita, T., Itami, K., Hagihara, S. and Torii, K. U. (2018). Chemical hijacking of auxin signaling with an engineered auxin-TIR1 pair. Nat Chem Biol 14(3): 299-305.
- Yamada, R., Murai, K., Uchida, N., Takahashi, K., Iwasaki, R., Tada, Y., Kinoshita, T., Itami, K., Torii, K. U. and Hagihara, S. (2018). A Super Strong Engineered Auxin-TIR1 Pair. Plant Cell Physiol 59(8): 1538-1544.
- Yesbolatova, A., Saito, Y., Kitamoto, N., Makino-Itou, H., Ajima, R., Nakano, R., Nakaoka, H., Fukui, K., Gamo, K., Tominari, Y., Takeuchi, H., Saga, Y., Hayashi, K. I. and Kanemaki, M. T. (2020). The auxin-inducible degron 2 technology provides sharp degradation control in yeast, mammalian cells, and mice. Nat Commun 11(1): 5701.
- Yesbolatova, A., Tominari, Y. and Kanemaki, M. T. (2019). Ligand-induced genetic degradation as a tool for target validation. Drug Discov Today Technol 31: 91-98.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Nishimura, K. and Fukagawa, T. (2021). A Simple Method to Generate Super-sensitive AID (ssAID)-based Conditional Knockouts using CRISPR-based Gene Knockout in Various Vertebrate Cell Lines. Bio-protocol 11(14): e4092. DOI: 10.21769/BioProtoc.4092.
Category
Molecular Biology > Protein > Targeted degradation
Stem Cell > Embryonic stem cell
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.