- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

PCR-mediated One-day Synthesis of Guide RNA for the CRISPR/Cas9 System

(*contributed equally to this work) Published: Vol 11, Iss 13, Jul 5, 2021 DOI: 10.21769/BioProtoc.4082 Views: 4851
Reviewed by: Priyanka DasLifeng LiuAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Nowadays, CRISPR (clustered regularly interspaced short palindromic repeats) and the CRISPR-associated protein (Cas9) system play a major role in genome editing. To target the desired sequence of the genome successfully, guide RNA (gRNA) is indispensable for the CRISPR/Cas9 system. To express gRNA, a plasmid expressing the gRNA sequence is typically constructed; however, construction of plasmids involves much time and labor. In this study, we propose a novel procedure to express gRNA via a much simpler method that we call gRNA-TES (gRNA-transient expression system). This method employs only PCR, and all the steps including PCR and yeast transformation can be completed within 1 day. In comparison with the plasmid-based gRNA delivery system, the performance of gRNA-TES is more effective, and its total time and cost are significantly reduced.
Keywords: CRISPR/Cas9Background
Developing genome editing techniques is one of the central issues of genome science. For the past decade, the CRISPR/Cas9 system has contributed to easier and more precise genome editing as compared with previously developed techniques such as ZFN (zinc finger nuclease) and TALEN (transcription activator-like effector nuclease). For successful CRISPR/Cas9 engineering, design, expression, and delivery of the guide RNA (gRNA) components are the key factors (Stovicek et al., 2017). For prokaryotes like Escherichia coli and eukaryotes such as Saccharomyces cerevisiae, the most commonly employed method for expressing gRNA is to use a plasmid (Jiang et al., 2013; Li et al., 2015; DiCarlo et al., 2013; Bao et al., 2015; Jakočiunas et al., 2015a and 2015b); however, plasmid construction, including cloning steps for necessary components, is laborious, costly, and time-consuming. To express gRNA more simply, in this study we developed a method that we call gRNA-transient expression system (gRNA-TES), where gRNA is expressed from the PCR product. gRNA-TES is very fast and effective: it takes only 5-6 h to complete the whole process, including preparation of PCR products for expression of gRNA in yeast cells and yeast transformation. By contrast, it takes at least 3-4 days to construct a plasmid expressing gRNA including verification. As expected, when applied to replacement of desired chromosome regions in yeast, gRNA-TES effectively replaces single and multiple chromosomal regions (Easmin et al., 2019 and 2020). Therefore, we believe that gRNA-TES will be useful for other types of genome editing including segmental deletion, duplication, and splitting of chromosomes. Lastly, gRNA-TES is effective in yeast; therefore, it should be emphasized that gRNA-TES may also be efficacious in other organisms if suitable gene promoters are incorporated.
Materials and Reagents
0.1-10 μl pipette tips (BMBio, catalog number: BMT-10GXLR)
20-200 μl pipette tips (BMBio, catalog number: BMT-200R)
PCR tubes (Axygen, catalog number: SKPCRF)
p426-SNR52p-gRNA.CAN1.Y-SUP4t (Addgene, catalog number: 43803)
p414-TEF1p-Cas9-CYC1t (Addgene, catalog number: 43802)
Escherichia coli DH5α competent cells (NIPPON GENE, catalog number: 316-06233)
DNA, MB-grade from fish sperm (Roche Diagnostics, catalog number: 11467140001)
KOD plus neo (TOYOBO, catalog number: KOD-401)
2 mM dNTP solution (included with KOD plus neo)
25 mM magnesium sulfate (MgSO4) (included with KOD plus neo)
Oligonucleotide Primer Fw A for construction of fragment A (5’-GTTCGAAACTTCTCCGCAGT GAAAGATAAATGATCN20GTTTTAGAGCTAGAAATAGCAAG-3’) (synthesized by Hokkaido System Science, Japan) (N20 represents the 20-nt upstream sequence of the target PAM sequence)
Oligonucleotide primer Rv A for construction of fragment A (5’- ACTCACAAATTAGAGCTTCA -3’) (synthesized by Hokkaido System Science, Japan)
Oligonucleotide primer Fw B for construction of fragment B (5’- CGAACGACCGAGCGCAGCGA-3’) (synthesized by Hokkaido System Science, Japan)
Oligonucleotide primer Rv B for construction of fragment B (5’- TTTATCTTTCACTGCGGAGAAGTTTCGAAC-3’) (synthesized by Hokkaido System Science, Japan)
Ex Taq® DNA polymerase (TaKaRa, catalog number: RR001A)
10× Ex Taq buffer (Mg2+ plus) (included with Ex Taq® DNA Polymerase)
2.5 mM each dNTP mix (included with Ex Taq® DNA Polymerase)
Ampicillin (Nacalai tesque, catalog number: 02739-74)
Lithium acetate dihydrate (Sigma-Aldrich, catalog number: L6883-250G)
Polyethylene glycol 3,350 (Sigma-Aldrich, catalog number: P4338-500G)
Sodium hydroxide (NaOH) (FUJIFILM Wako Pure Chemical Corporation, catalog number: 192-15985)
Control primer 1 for amplifying the CNE1 region (5’-TCACAGGGTCGATTGCAAGG-3’) (synthesized in Hokkaido System Science, Japan)
Control primer 2 for amplifying the CNE1 region (5’-CTGGTGGTTCAGTGCCATCT-3’) (synthesized in Hokkaido System Science, Japan)
Oligonucleotide primer 1 for checking replacement (Use the 200-176 nt upstream sequence of the target region)
Oligonucleotide primer 2 for checking replacement (Use the 66-90 nt downstream reverse sequence of the target region)
Prime Star Max Premix 2× (TaKaRa, catalog number: R045A)
Agar (FUJIFILM Wako Pure Chemical Corporation, catalog number: 010-08725)
Gene Ladder Wide 2 (Nippon Gene, catalog number: 310-06971)
Glucose (FUJIFILM Wako Pure Chemical Corporation, catalog number: 043-31163)
Yeast nitrogen base without amino acids (BD, Difco, catalog number: DF0919-15-3)
Peptone (BD, BactoTM, catalog number: 211677)
Yeast extract (BD, BactoTM, catalog number: 288620)
Adenine HCL (FUJIFILM Wako Pure Chemical Corporation, product code: 016-00802)
Synthetic Complete (SC) medium (see Recipes)
YPDA medium (see Recipes)
Procedure
Designing the 20-nt gRNA target sequence
To design the gRNA target sequence, it is essential to have an appropriate PAM sequence. To design a target sequence with a PAM sequence for Cas9 cleavage (5’-NGG-3’ or 5’-CCN-3’ for opposite strand) close to the target region, we use the free software CRISPRdirect (https://crispr.dbcls.jp/). For S. cerevisiae, we select the S. cerevisiae representative strain S288C and paste 50-100 nt of the sequence located near the target site into the software. Then, CRISPRdirect quickly outputs the appropriate 20-nt gRNA target sequence with a PAM sequence.
Preparation of PCR fragments A and B
We use p426-SNR52p-gRNA.CAN1.Y-SUP4t as a common template plasmid to construct a PCR fragment (fragment C) expressing gRNA harboring the designed 20-nt target sequence. It contains the yeast promoter SNR52, which is responsible for expressing gRNA in yeast cells. If you want to express gRNA in another organism, you will need to use a template plasmid harboring a suitable promoter sequence obtained from that respective organism to prepare fragment C. Here, we explain the case in which gRNA is expressed from fragment C in a S. cerevisiae host. Before preparing fragment C, fragments A and B must be synthesized in two separate steps by first-round PCR.
Fragment A: For preparation of fragment A, design the forward primer (Fw A) to include 35 nt of the sequence from part of the SNR52 promoter region (3,855-3,889 nt) of the template plasmid p426-SNR52p-gRNA.CAN1.Y-SUP4t (full sequence is available at Addgene repository, https://www.addgene.org/43803/sequences/), followed by the 20-nt gRNA target sequence, and a further 23 nt of the sequence encoding the 5’ part of the gRNA scaffold from the template plasmid (3,910-3,932 nt) (Figure 1A). Design the reverse primer sequence (Rv A) based on 5,001-5,020 nt of the reverse sequence (5’-ACTCACAAATTAGAGCTTCA-3’) of the template plasmid.
Fragment B: Since fragment A does not contain the full sequence of the SNR52 promoter and it is essential to have this complete region for proper expression of gRNA, we need to incorporate the whole SNR52 promoter region with fragment A via fragment B. Therefore, we use a forward primer (Fw B) consisting of a 20-nt sequence (5’-CGAACGACCGAGCGCAGCGA-3’) and a reverse primer (Rv B) consisting of a 30-nt sequence (5’-TTTATCTTTCACTGCGGAGAAGTTTCGAAC-3’) of the template plasmid (3,301-3,320 nt and 3,855-3,884 nt in reverse sequence, respectively) to synthesize the whole SNR52 promoter sequence as fragment B (Figure 1A). The complementary sequence of the last 30 nt in fragment B is the same as the first 30 nt of fragment A in order to allow fragments A and B to be annealed in the second-round (overlap) PCR.
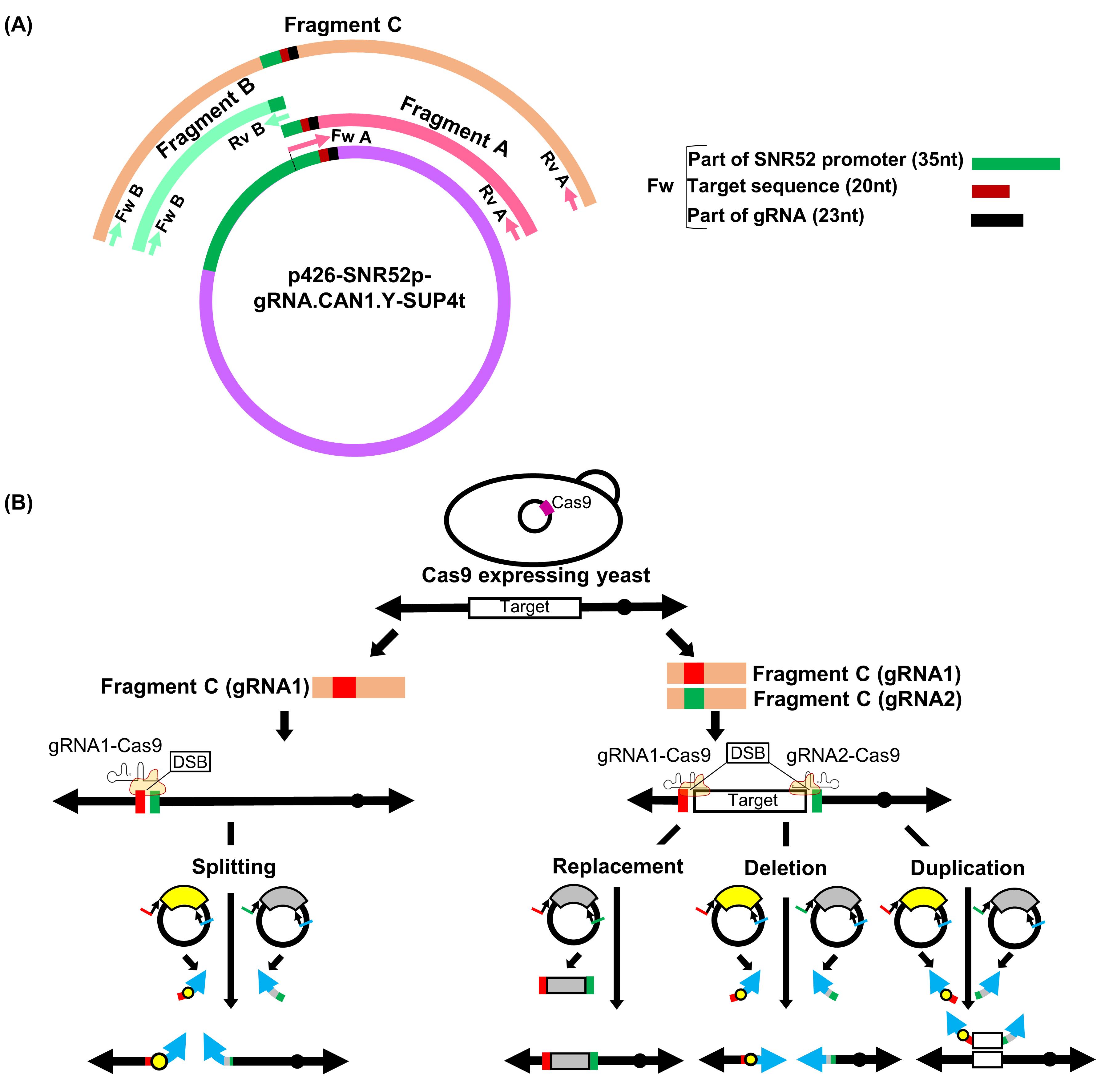
Figure 1. Overview of gRNA-TES. A. In gRNA-TES, we prepare three fragments, namely fragments A, B, and C, by PCR. Fragments A and B are prepared by a first PCR using plasmid p426-SNR52p-gRNA.CAN1.Y-SUP4t (DiCarlo et al., 2013) as a common template. Next, fragment C, for expressing the gRNA sequence, is synthesized by a second PCR (overlap PCR) using fragments A and B as templates. In gRNA-TES, a new fragment A is always needed because it contains the 20-nt unique gRNA target sequence. The 20-nt unique gRNA target sequence is changed depending on the target region. By contrast, fragment B is solely the SNR52 promoter region; therefore, it is not necessary to synthesize a new fragment B with a different sequence every time. Using this protocol, it is not possible to design a primer sequence that can amplify the target sequence along with the whole SNR52 promoter region since it is difficult to chemically synthesize a good-quality oligonucleotide with more than 100 nucleotides. Thus, we cannot amplify fragment C by a single round of PCR. B. gRNA-TES can be used for various genome manipulations such as chromosome splitting, replacement, deletion, and duplication. Only one fragment C, which delivers one gRNA, is necessary for splitting. Double-strand break (DSB) significantly increases the frequency of homologous recombination between the target sequence (red and green box) on the chromosome and its corresponding DNA modules synthesized from plasmids p3121 (Sugiyama et al., 2005) and pSJ69 (Easmin et al., 2019). As a consequence, a high frequency of splitting is thought to occur. Two kinds of fragment C, which deliver two independent gRNAs, are necessary for replacement, deletion, and duplication. For replacement, we prepare one DNA module harboring an appropriate marker gene (here Candida glabrata LEU2, CgLEU2) flanked with the target homology sequences using plasmid pSJ69 (Easmin et al., 2019). For segmental deletion and duplication, we need two DNA modules harboring the target homology sequences: one should have a centromere, and the other should contain the marker gene. In our experiments, we use p3121 (Sugiyama et al., 2005) harboring CEN4 and pSJ69 harboring CgLEU2 as templates to synthesize the DNA modules. After successful double-strand break, a high frequency of replacement, deletion, and duplication is expected. In panel B, the black circle on the chromosome denotes the native centromere; the yellow circle on the DNA modules denotes artificially supplied CEN4. The blue arrow indicates the artificially supplied telomere; the gray box represents CgLEU2. The plasmids containing yellow- and gray-colored curved boxes are p3121 and pSJ69, respectively.First-round PCR reaction

Preparation of fragment C by overlap PCR
To prepare fragment C, a 100-fold dilution (×1/100) of fragment A is mixed with the same dilution of fragment B and used as a template for the second-round PCR (overlap PCR) to generate fragment C with forward primer (Fw B) 5’-CGAACGACCGAGCGCAGCGA-3’ and reverse primer (Rv A) 5’-ACTCACAAATTAGAGCTTCA-3’.
Second-round PCR reaction

Note: We recommend using Ex Taq® DNA polymerase for gRNA-TES, especially for the second-round PCR (overlap PCR). Although we tested various DNA polymerases, we observed that those DNA polymerases frequently produced multiple unexpected bands, especially during the overlap PCR used to prepare fragment C.
Application of gRNA-TES to various genome manipulations
gRNA-TES has a variety of applications from splitting to segmental replacement, deletion, and duplication of chromosomes (Figure 1B). Depending on the genome engineering technique, up to two kinds of fragment C delivering two types of gRNA are necessary. After DSB, the frequency of splitting, replacement, deletion, and duplication is expected to be increased.
Preparation of DNA modules
To split, delete, replace, or duplicate target chromosomal regions, it is necessary to incorporate DNA modules into gRNA-TES. The type of DNA module that is constructed depends upon the purpose (splitting, deletion, replacement, or duplication) of genome editing. For replacement, only one DNA module is sufficient, while for splitting, deletion, or duplication, two DNA modules are needed (Figure 1B). For replacement, design and purchase oligonucleotide primers to amplify any genetic marker flanked with the homology sequence corresponding to the first and last 30 bp of the target region. For splitting and deletion, design forward primers to amplify the centromere or marker gene flanked with 50 bp of the homology sequence corresponding to the upstream or downstream sequence of the target splitting and deletion point. For duplication, design forward primers to amplify the marker gene or centromere flanked with the homology sequence corresponding to the first or last 50 bp of the target region. To include a telomere seed sequence in all DNA modules for splitting, deletion, and duplication, we use a common reverse primer including the 5’-CCCCAACCCCAACCCCAACCCCAACCCCAACCCCAA-3’ sequence. You can use any template plasmid depending on the purpose of genome editing. We use pSJ69 (Easmin et al., 2019) and p3121 (Sugiyama et al., 2005) to synthesize DNA modules. These plasmids have the same background because they were constructed from the same plasmid pUG6 (Güldener et al., 1996) and are available upon request. For splitting, deletion, or duplication, it is necessary to select an appropriate plasmid as a template for PCR so that the newly generated chromosomes contain only one centromere.
PCR reaction
10× KOD plus neo buffer 5 μl 2 mM dNTP solution 5 μl 25 mM MgSO4 3 μl Template plasmid 1 μl Primers for Replacement Oligonucleotide primer 1 (15 pmol)
5’-N30FGGCCGCCAGCTGAAGCTTCG-3’
1.5 μl (N30F represents the first 30-bp sequence of the target region) Oligonucleotide primer 2 (15 pmol)
5’-N30LAGGCCACTAGTGGATCTGAT-3’
1.5 μl (N30L represents the last 30-bp reverse sequence of the target region) Primers for Splitting and Deletion Forward primer (15 pmol)
5’-N50U/50DGGCCGCCAGCTGAAGCTTCG-3’
1.5 μl (N50U/50D represents the 50-bp upstream or downstream (reverse) sequence of the target region) Reverse primer (15 pmol)
5’-CCCCAACCCCAACCCCAACCCCAACCC CAACCCCAAAGGCCACTAGTGGATCTGAT-3’
1.5 μl Primers for Duplication Forward primer (15 pmol)
5’-N50F/50LGGCCGCCAGCTGAAGCTTCG-3’
1.5 μl (N50F/50L represents the first (reverse) or last 50-bp sequence of the target region) Reverse primer (15 pmol)
5’-CCCCAACCCCAACCCCAACCCCAACCC CAACCCCAAAGGCCACTAGTGGATCTGAT-3’
1.5 μl KOD plus neo 1 μl Water 32 μl Total 50 μl Pre-denaturation: 94°C, 2min Denature: 98°C, 10 s Annealing: 55°C, 30 s 30 cycles Extension: 68°C, 2 min Yeast transformation
Prepare in advance a yeast strain, for example, SJY30 (MATα ura3-52 his3-Δ200 leu2Δ1 lys2Δ202 trp1Δ63 harboring plasmid p414-TEF1p-Cas9-CYC1t [DiCarlo et al., 2013]), that expresses codon-optimized Cas9 by the introduction of p414-TEF1p-Cas9-CYC1t.
Cultivate the strain overnight in YPDA liquid medium.
Inoculate a fresh 5-ml aliquot of YPDA liquid medium with yeast cell pre-culture to an initial OD600 of approximately 0.2-0.3. Then incubate with a shaking speed of 140 rpm at 30°C until the OD600 reaches 0.8-1.0 (about 4-6 h).
Mix the appropriate DNA modules and gRNA-expressing fragment C (e.g., we need two kinds of fragment C [Figure 1B] to target both edges of the target region and one DNA module to replace chromosomal regions) and perform transformation using the conventional LiAc/PEG method (Gietz and Schiestl, 2007).
The aim of this protocol is to provide users with a procedure that is as easy as possible at every step, including colony PCR described in the next section. Therefore, the protocol was developed without measuring the DNA concentration of PCR products. For routinely performed yeast transformation, we consistently use 12 μl DNA module and 11 μl each fragment C PCR reaction mixture to replace chromosomal regions, since we have obtained sufficient transformants using such amounts (i.e., 12 μl + 11 μl + 11 μl = 34 μl) of PCR reaction mixture. Furthermore, the whole PCR reaction mixture can be directly used for yeast transformation without purification.
After yeast transformation, suspend the cells in 100 μl sterilized water, spread the whole suspension onto one or two selection plates, and incubate at 30°C for 2-3 days.
Confirmation of the expected chromosomal change by colony PCR and subsequent agarose gel electrophoresis
To make PCR-grade genomic DNA, take a small amount of cells from each colony and suspend in 10 μl 0.02 M NaOH solution. Heat at 98°C for 10 min in a heat block and then transfer to ice.
As an example, we describe how to confirm transformants obtained from a replacement experiment here. Design and purchase oligonucleotide primers for colony PCR. You can use any sequence from the upstream and downstream sequences of the target region, but we recommend designing the primer in such a way that the final PCR product will be <2 kb, since generating a larger PCR product may be problematic during colony PCR. For oligonucleotide primer 1, use a 200-176 nt upstream sequence of the target region and for oligonucleotide primer 2, use a 66-90 nt downstream reverse sequence of the target region. Since we use the CgLEU2 marker gene for replacement and the size of CgLEU2 is 1685 bp, after successful replacement of the target region, the size of the PCR product will be 200 bp + 1685 bp + 90 bp = 1975 bp (Figure 2). For example, to check the replacement of a 500-kb region in Chromosome 4 (coordinate number 494271-994270), design oligonucleotide primer 1 based on the 494071-494095 nt sequence (5’-CATATCAGTGTCTTCATCTTCATGA-3’) and oligonucleotide primer 2 based on the 994,336-994,360 nt reverse sequence (5’-TAGTGGATACGCAGGACGTGTTATC-3’) of Chromosome 4. In addition, we recommend designing control primers to check whether the PCR reaction is proceeding well. Any genomic region may be amplified as a control; we amplify the CNE1 region of Chromosome 1 as an internal control. Control primer 1 is based on the 211-230 nt sequence (5’-TCACAGGGTCGATTGCAAGG-3’) and control primer 2 on the 861-880 nt reverse sequence (5’-CTGGTGGTTCAGTGCCATCT-3’) of the CNE1 region. If the CNE1 gene is properly amplified, you will obtain a 670-bp PCR product.
PCR reaction
Prime Star Max Premix 2× 12.5 μl Template genomic DNA 0.5 μl Control primer 1 (7.5 pmol)
5’-TCACAGGGTCGATTGCAAGG-3’
0.75 μl Control primer 2 (7.5 pmol)
5’-CTGGTGGTTCAGTGCCATCT-3’
0.75 μl Oligonucleotide primer 1 (7.5 pmol)
(Use the 200-176 nt upstream sequence of the target region)
0.75 μl Oligonucleotide primer 2 (7.5 pmol)
(Use the 66-90 nt downstream (reverse) sequence of the target region)
0.75 μl Water 9 μl Total 25 μl Denature: 98°C, 10 s
Annealing: 55°C, 5 s 30 cycles Extension: 72°C, 10 s
Note: You can use any DNA polymerase for colony PCR. However, since our intention is to make this protocol as fast as possible, we use Prime Star Max Premix 2×, which requires an extension time of only 5 s/kb.
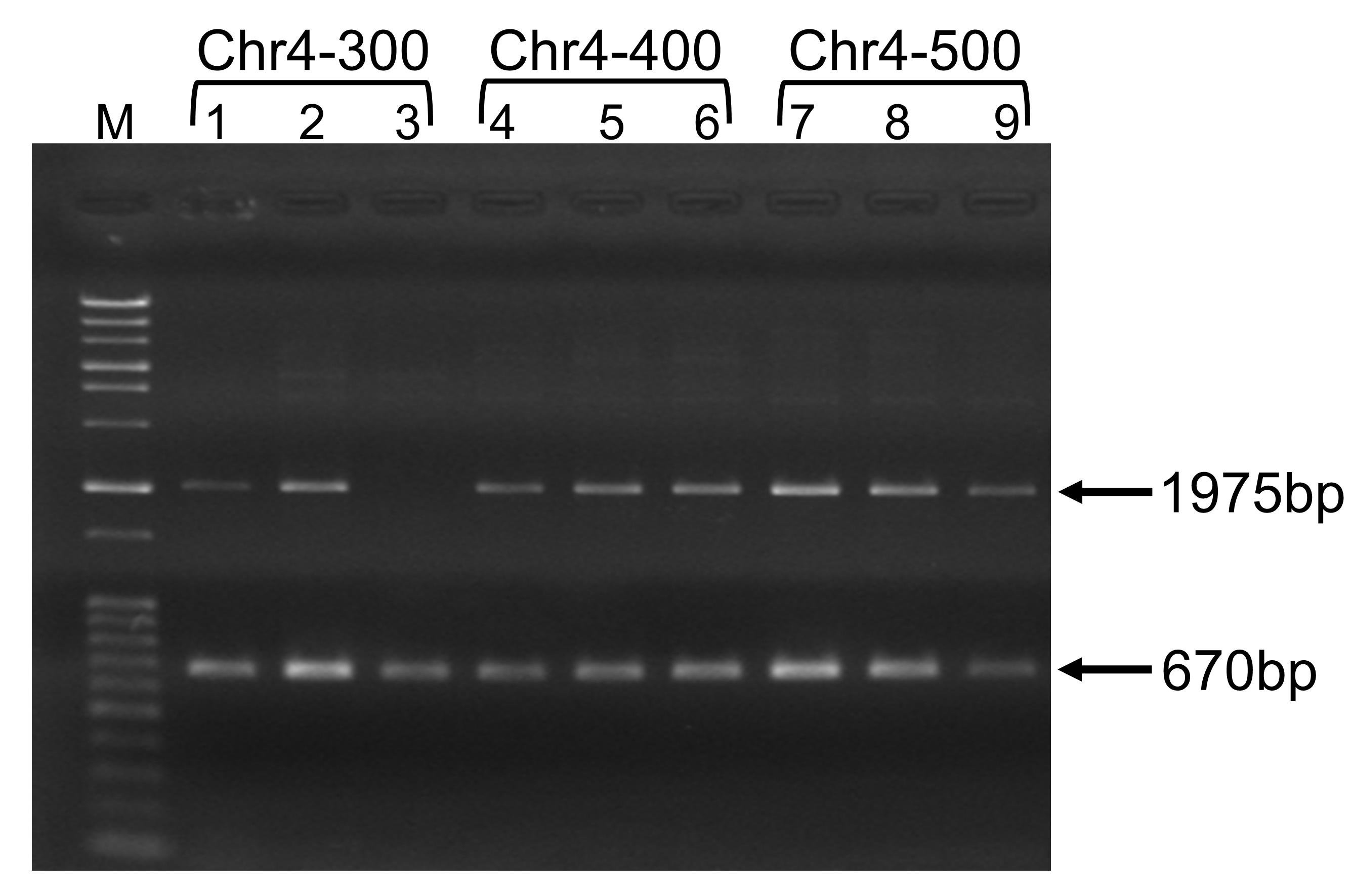
Figure 2. Typical agarose gel electrophoresis results for chromosome replacement analysis by gRNA-TES. Lanes 1 to 9 represent independent transformants. M represents size markers (Gene Ladder Wide 2, Nippon Gene, Toyama, Japan). Lanes 1 to 3 represent transformants obtained from replacement of a 300-kb region; lanes 4 to 6 represent transformants obtained from replacement of a 400-kb region; and lanes 7 to 9 represent transformants obtained from replacement of a 500-kb region of Chromosome 4. Almost all transformants (except those in lane 3) showed the expected replacement of the respective chromosomal region and yielded the 1975-bp band. The internal control band (670 bp) was also observed in all transformants.
Data analysis
The number of transformants varies in each experiment. For example, when we targeted the replacement of smaller chromosomal regions of, for example, 150 kb and 200 kb, we obtained 263 and 287 transformants, respectively, in a single transformation (Easmin et al., 2019). When we targeted 300-kb, 400-kb, and 500-kb regions, we obtained, respectively, 51, 43, and 103 transformants in a single transformation. We tested six transformants for replacement of each of the 150-kb and 200-kb regions, and 5 transformants for each of the 300-kb, 400-kb, and 500-kb regions. The frequencies of expected replacement of the 150-kb, 200-kb, 300-kb, 400-kb, and 500-kb regions were 100%, 66.6%, 80%, 100%, and 100%, respectively (Easmin et al., 2019). By contrast, a maximum of 16.6% expected frequency was observed for replacement of the 150-kb region when gRNA-TES was not applied; for replacement of the other chromosomal regions, no correct transformants were obtained when gRNA-TES was not employed.
Recipes
Synthetic complete (SC) medium
2% glucose
0.67% yeast nitrogen base without amino acids (e.g., BD Difco)
Note: 0.2% dropout mix containing all amino acids and nucleic acid bases lacking specific amino acids can be used for auxotrophic selection.
For the plate assay, 2% agar is added
YPDA medium
2% glucose
2% peptone (e.g., BD BactoTM)
1% yeast extract (e.g., BD BactoTM)
0.004% adenine HCl (FUJIFILM Wako Pure Chemical Corporation)
Acknowledgments
This work was supported by the Japan Society for the Promotion of Science (JSPS)-KAKENHI, Grant-in-Aid for Scientific Research (B), Grant numbers [JP 15H04475] to S.H. This protocol was adapted from our previously published work (Easmin et al., 2019).
Competing interests
The authors declare no competing interests.
References
- Bao, Z., Xiao, H., Liang, J., Zhang, L., Xiong, X., Sun, N., Si, T. and Zhao, H. (2015). Homology -Integrated CRISPR–Cas (HI-CRISPR) system for one-step multi-gene disruptions in Saccharomyces cerevisiae. ACS Synth Biol 4: 585-594.
- DiCarlo, J. E., Norville, J. E., Mali, P., Rios, X., Aach, J. and Church, G. M. (2013). Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res 41(7): 4336-4343.
- Easmin, F., Hassan, N., Sasano, Y., Ekino, K., Taguchi, H. and Harashima, S. (2019). gRNA-transient expression system for simplified gRNA delivery in CRISPR/Cas9 genome editing. J Biosci Bioeng 128(3): 373-378.
- Easmin, F., Sasano, Y., Kimura, S., Hassan, N., Ekino, K., Taguchi, H. and Harashima, S. (2020). CRISPR-PCD and CRISPR-PCRep: Two novel technologies for simultaneous multiple segmental chromosomal deletion/replacement in Saccharomyces cerevisiae. J Biosci Bioeng 129(2): 129-139.
- Gietz, R. D. and Schiestl, R. H. (2007). High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2(1): 31-34.
- Güldener, U., Heck, S., Fielder, T., Beinhauer, J. and Hegemann, J. (1996). A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res 24: 2519-2524.
- Jakočiunas, T., Bonde, I., Herrgård, M., Harrison, S. J., Kristensen, M., Pedersen, L. E., Jensen, M. K. and Keasling, J. D. (2015a). Multiplex metabolic pathway engineering using CRISPR/Cas9 in Saccharomyces cerevisiae. Metab Eng 28: 213-222.
- Jakočiunas, T., Rajkumar, A. S., Zhang, J., Arsovska, D., Rodriguez, A., Jendresen, C. B., Skjødt, M. L., Nielsen, A. T., Borodina, I., Jensen, M. K. and Keasling, J. D. (2015b). CasEMBLR: Cas9-Facilitated Multiloci Genomic Integration of in Vivo Assembled DNA Parts in Saccharomyces cerevisiae. ACS Synth Biol 4(11): 1226-1234.
- Jiang, W., Bikard, D., Cox, D., Zhang, F. and Marraffini, L. A. (2013). RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31(3): 233-239.
- Li, Y., Lin, Z., Huang, C., Zhang, Y., Wang, Z., Tang, Y. J., Chen, T. and Zhao, X. (2015). Metabolic engineering of Escherichia coli using CRISPR-Cas9 meditated genome editing. Metab Eng 31: 13-21.
- Stovicek, V., Holkenbrink, C. and Borodina, I. (2017). CRISPR/Cas system for yeast genome engineering: advances and applications. FEMS Yeast Res 17(5).
- Sugiyama, M., Ikushima, S., Nakazawa, T., Kaneko, Y. and Harashima, S. (2005). PCR-mediated repeated chromosome splitting in Saccharomyces cerevisiae. Biotechniques 38(6): 909-914.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Hassan, N., Easmin, F., Ekino, K. and Harashima, S. (2021). PCR-mediated One-day Synthesis of Guide RNA for the CRISPR/Cas9 System. Bio-protocol 11(13): e4082. DOI: 10.21769/BioProtoc.4082.
Category
Molecular Biology > DNA > Gene expression
Microbiology > Microbial genetics > CRISPR-Cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.