- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Lipidomics Approach to Measure Phosphatidic Acid Species in Subcellular Membrane Fractions Obtained from Cultured Cells
Published: Vol 11, Iss 12, Jun 20, 2021 DOI: 10.21769/BioProtoc.4066 Views: 3796
Reviewed by: David F DelotterieSébastien Gillotin
Original research article
The authors used this protocol in:

Aug 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Over the last decade, lipids have emerged as possessing an ever-increasing number of key functions, especially in membrane trafficking. For instance, phosphatidic acid (PA) has been proposed to play a critical role in different steps along the secretory pathway or during phagocytosis. To further investigate in detail the precise nature of PA activities, we need to identify the organelles in which PA is synthesized and the PA subspecies involved in these biological functions. Indeed, PA, like all phospholipids, has a large variety based on its fatty acid composition. The recent development of PA sensors has helped us to follow intracellular PA dynamics but has failed to provide information on individual PA species. Here, we describe a method for the subcellular fractionation of RAW264.7 macrophages that allows us to obtain membrane fractions enriched in specific organelles based on their density. Lipids from these membrane fractions are precipitated and subsequently processed by advanced mass spectrometry-based lipidomics analysis to measure the levels of different PA species based on their fatty acyl chain composition. This approach revealed the presence of up to 50 different species of PA in cellular membranes, opening up the possibility that a single class of phospholipid could play multiple functions in any given organelle. This protocol can be adapted or modified and used for the evaluation of other intracellular membrane compartments or cell types of interest.
Keywords: LipidomicsBackground
The acyl chain diversity in glycerophospholipids has been studied for decades; however, technical and conceptual limitations have restricted our understanding of its contribution to cell function. Among the phospholipids, phophatidic acid (PA) plays an important role as an intermediate metabolite in glycerophospholipid metabolism and has also been suggested to display key signaling functions. Chemically, PA consists of a glycerol backbone, to which two fatty acyl chains and a phosphate are attached, through esterification, at positions sn-1, sn-2, and sn-3, respectively (Tanguy et al., 2019a). The unique feature of PA as compared with other glycerophospholipids is its phosphomonoester link to a small anionic phosphate headgroup (Tanguy et al., 2018). The small headgroup of PA is thought to adopt a cone-shaped structure in membranes, which has often served as an explanation for its contribution to diverse membrane trafficking events (Ammar et al., 2013; Tanguy et al., 2016). However, despite many implications, the precise functions of PA in these membrane trafficking events remain unclear. The diversity of the PA biosynthetic routes, together with the possible occurrence of many different PA species based on their fatty acyl chain composition, opens up the possibility for multiple roles in a given cellular function, which prompted us to develop a method to characterize the profile of the different PA species in different membrane organelles.
Macrophages are the crux of many innate immune responses to invading pathogens or danger signals for which the contribution of PA produced by phospholipase D (PLD) has been well established in phagocytosis (Corrotte et al., 2006; Tanguy et al., 2019b). However, despite evidence suggesting that PA levels increase at the plasma membrane during phagocytosis, there is little data available on PA dynamics in additional membrane compartments regarding the species involved. We took advantage of a frustrated phagocytosis assay that occurs when macrophages are exposed to immobilized immune complexes in culture and spread as if trying to engulf them, which is believed to mimic some key steps of the phagocytic process (Labrousse et al., 2011). Hence, we describe a protocol for subcellular fractionation, followed by lipid extraction and the quantitation of PA species by advanced mass spectrometry-based lipidomics analysis. These approaches allowed us to obtain quantitative information regarding PA species composition in specific organelles during frustrated phagocytosis.
Materials and Reagents
Calibrated pipetman and pipette tips of any brand
10 cm cell tissue culture dishes, Falcon (Fisher Scientific, catalog number: 353003)
15 ml Falcon tubes (Dutscher, catalog number: 352097)
50 ml Falcon tubes (Dutscher, catalog number: 352070)
Cell scrapers, Falcon (Fisher Scientific, catalog number: 353085)
Centrifuge tubes (thinwall, ultra-ClearTM, 14 ml, 14 × 95 mm) (Beckman Coulter, catalog number: 344060)
Cryovials (Nalgene, Dutscher, catalog number: 028001)
RAW264.7 cells (ATCC, catalog number: TIB-71)
RPMI-Glutamax medium (Gibco, catalogue number: 61870-044)
Fetal bovine serum (FBS) (Gibco, catalogue number:16030-074)
DMSO (Fisher Scientific, catalog number: BP231-100)
Trypsin/EDTA (Gibco, catalogue number: 25300-054)
Sucrose (Sigma-Aldrich, catalog number: S9378-500G)
Penicillin/streptomycin solution 100× (Pen/Strep) (Sigma, catalogue number: P4458-100ml)
Coomassie Brilliant Blue G-250 (Thermo Fisher Scientific, catalog number: 100-25)
Novex 4-12% Bis-Tris SDS-PAGE gels (Invitrogen, catalogue number: NP0322BOX)
Polyclonal rabbit anti-BSA IgG (Thermo Scientific, catalog number: A11133)
Polyclonal Sheep anti-BSA IgG (Euromedex, catalog number: GTX77113)
Chloroform (HPLC grade, Sigma, catalogue number: C2432)
Methanol (HPLC grade, Sigma, catalogue number: 34860)
Disposable culture tubes and their phenolic caps, PTFE liners (Corning, catalog number: 99447-13 and 9998-13)
Internal standard 17:0/17:0 PA that can be noted 34:0_17:0, with the first number reflecting the total carbons in both fatty acids and the second number corresponding to the number of carbons in the fatty acid at the sn1 position (Avanti Polar lipids, catalog number: 830856)
Formic acid, 98% (Sigma, catalog number: 33015)
Ammonium hydroxide solution (Sigma, catalog number: 221228)
Glycerol (Merck, catalog number: 104093)
SDS (Sigma, catalog number: L4509)
Tris-HCl (Sigma, catalog number: 25285-9)
β-mercaptoethanol (Sigma, catalog number: M3148)
HEPES free acid (Sigma, catalog number: H4034)
EGTA (Sigma, catalog number: E4378)
MgCl2 (Sigma, catalog number: M9272)
OptiPrep Solution (Sigma, catalog number: D1556)
Isopropanol (Sigma, catalog number: I9516)
Vials and caps for injection (Agilent, catalog number: 5183-2067 and 5190-1599)
Tissue culture freezing media (see Recipes)
2× Laemmli buffer (see Recipes)
Isolation medium (see Recipes)
Gradient media (see Recipes)
LC solvent (see Recipes)
Solvent A and solvent B
Equipment
Incubator at 37°C with 5% CO2
Centrifuge (Eppendorf, model: 5810)
Ultracentrifuge (Beckman Coulter, model: CO-LE80K)
Pre-chilled swing bucket rotor (Beckman Coulter, model: SW41)
NuPage Novex protein electrophoresis apparatus (Invitrogen)
Western blotting apparatus transblot turbo (Bio-Rad)
Ultrasonic bath sonicator (Liebisch)
Evaporator under N2 flow (Organomation)
Ultra-high-performance LC 1290 Infinity II LC System (Agilent)
Qtrap 6500 Mass spectrometer (Sciex)
Luna C8 150 × 1 mm column, with 100 Å pore size, 5 μm particles (Phenomenex)
Dounce homogenizer and pestle (Sigma, catalog number: P0735-1EA)
Software
Analyst software, version 1.7.1 (Sciex) and Analyst Device Driver for Agilent 1.3 (Sciex)
Multiquant software version 3.0.3 (Sciex)
Procedure
RAW264.7 cell culture
Maintain mouse macrophage RAW 264.7 cells in RPMI-Glutamax medium supplemented with 10% heat-inactivated FBS, 100 U/ml penicillin, and 100 µg/ml streptomycin in the incubator at 37°C under 5% CO2. Use cells for a maximum of 15 passages.
For each passage, rinse cells with 2 ml Trypsin-EDTA and subsequently incubate for 5 min at 37°C. Suspend cells in 8 ml RPMI-glutamax medium supplemented with 10% heat-inactivated FCS before centrifugation at 800 × g.
Recover cell pellets in 500 µl tissue culture freezing media (see Recipes) and transfer to cryovials for storage in liquid nitrogen, or in 10 ml culture medium for dispatch on 10-cm plates for further culture or frustrated phagocytosis.
Frustrated phagocytosis of RAW264.7 cells
Pre-coat 10-cm dishes with BSA (3%) and either rabbit anti-BSA IgG (0.04 mg/ml) or sheep anti-BSA IgG (0.04 mg/ml).
Collect RAW264.7 cells from confluent plates and determine the number of cells per ml with a hemocytometer. It should be near 2 × 107 per 10-cm cell culture dish. Suspend 2 × 107 cells in cold serum-free medium and incubate on the pre-coated surfaces at 4°C for 1 h.
Warm to 37°C for 30 min to initiate cell spreading. As controls, plate macrophages on surfaces pre-coated with immune complexes and maintain at 4°C or plate on surfaces pre-coated with BSA alone. Under these conditions, the macrophages remain round and do not spread over the plate (see Kassas et al., 2017).
Subcellular fractionation
Scrape RAW264.7 macrophages from ten different 10-cm dishes in 40 ml PBS for each condition (4°C or 37°C) (Figure 1).
Pellet cells at 200 × g for 7 min and resuspend in 40 ml isolation medium (see Recipes).
Repeat the operation once to remove salts.
Suspend the pellet in 40 ml isolation medium containing 100 mM sucrose (slightly hypotonic medium) for effective homogenization, and pellet cells at 200 × g (Figure 1). Under hypotonic conditions, water flows into cells and causes them to swell, favoring plasma membrane rupture.
Transfer the pellet to a glass Dounce homogenizer and add 10 ml last supernatant to suspend the pellet.
Homogenize by 40 strokes with a tight-fitting pestle.
Combine the resulting mixture with the remaining supernatant.
Add 3.5 ml isolation medium containing 1.78 M sucrose (a hypertonic medium) to bring the homogenate back to an isotonic state, limiting further rupture of organelles.
Supplement with 2 mM MgCl2 to preserve nuclei in subsequent steps.
Centrifuge at 200 × g for 10 min to pellet nuclei/unbroken cells (save pellet as crude nuclear pellet).
Centrifuge the supernatant at 5,000 × g for 10 min to pellet mitochondria (save pellet as mitochondria).
Centrifuge the supernatant at 100,000 × g for 1 h to pellet ER, microsomes, and plasmalemma fragments (microsomal pellet).
Centrifuge the post-nuclear supernatant at 300 × g for 10 min to remove residual nuclei (Figure 1).
Centrifuge the post-mitochondrial supernatant at 5,000 × g for 10 min to remove residual mitochondria (Figure 1).
Wash and re-pellet the crude nuclear pellet in MgCl2-containing media. Wash and re-pellet mitochondria in MgCl2-free media.
Retain the supernatant from the 100,000 × g spin as the cytosolic fraction.
Prepare gradient media for each fraction as described in Recipes.
Centrifuge nuclei gradient media in an SW-41 rotor at 10,000 × g for 20 min, and mitochondrial and microsomal gradient media at 50,000 × g for 2 h (Figure 1).
Recover nuclei at the 30/35% OptiPrep interface; mitochondria at the 17.5/25% interface; plasmalemma and endoplasmic reticulum (ER) at the 10/17.5% and 17.5/25% interfaces, respectively; and microsomes at the dense 25/35% interface.
Freeze each sample in liquid nitrogen and store at -80°C.
Measure protein concentration in each sample by the Bradford assay (Table 1).
Note: We noted no significant differences in protein concentration among the different fractions obtained from RAW264.7 cells kept at 4°C or 37°C (Table 1).
Table 1. Amounts of protein in the different fractions isolated from 16 × 107 RAW264.7 macrophages under control conditions (4°C) or while undergoing frustrated phagocytosis (37°C). Values represent the mean obtained from three independent preparations ± S.D.
Protein amount (µg) Fraction 4°C 37°C Total lysate 46590 ± 235 45408.4 ± 203.8 Nuclei 636.5 ± 17.2 610.5 ± 21.5 Endoplasmic reticulum 462.5 ± 24.3 473.3 ± 19.3 Plasma membrane 781.5 ± 32.3 756.5 ± 41.8 Microsome 690.9 ± 45.3 668.6 ± 51.2 Mitochondria 2167.5 ± 121 1995.5 ± 104.2 Cytosol 28755 ± 135.2 28857 ± 256.5 Check the fraction purity by immunoblots using specific markers for the different fractions as described previously (Kassas et al., 2017; Tanguy et al., 2020). Suitable markers are the nuclei: Histone H3, the endoplasmic reticulum: calnexin, the plasma membrane: Na+/K+ ATPase, the microsome: cytochrome p450, mitochondria: HSP-60, and the cytosol: GAPDH.
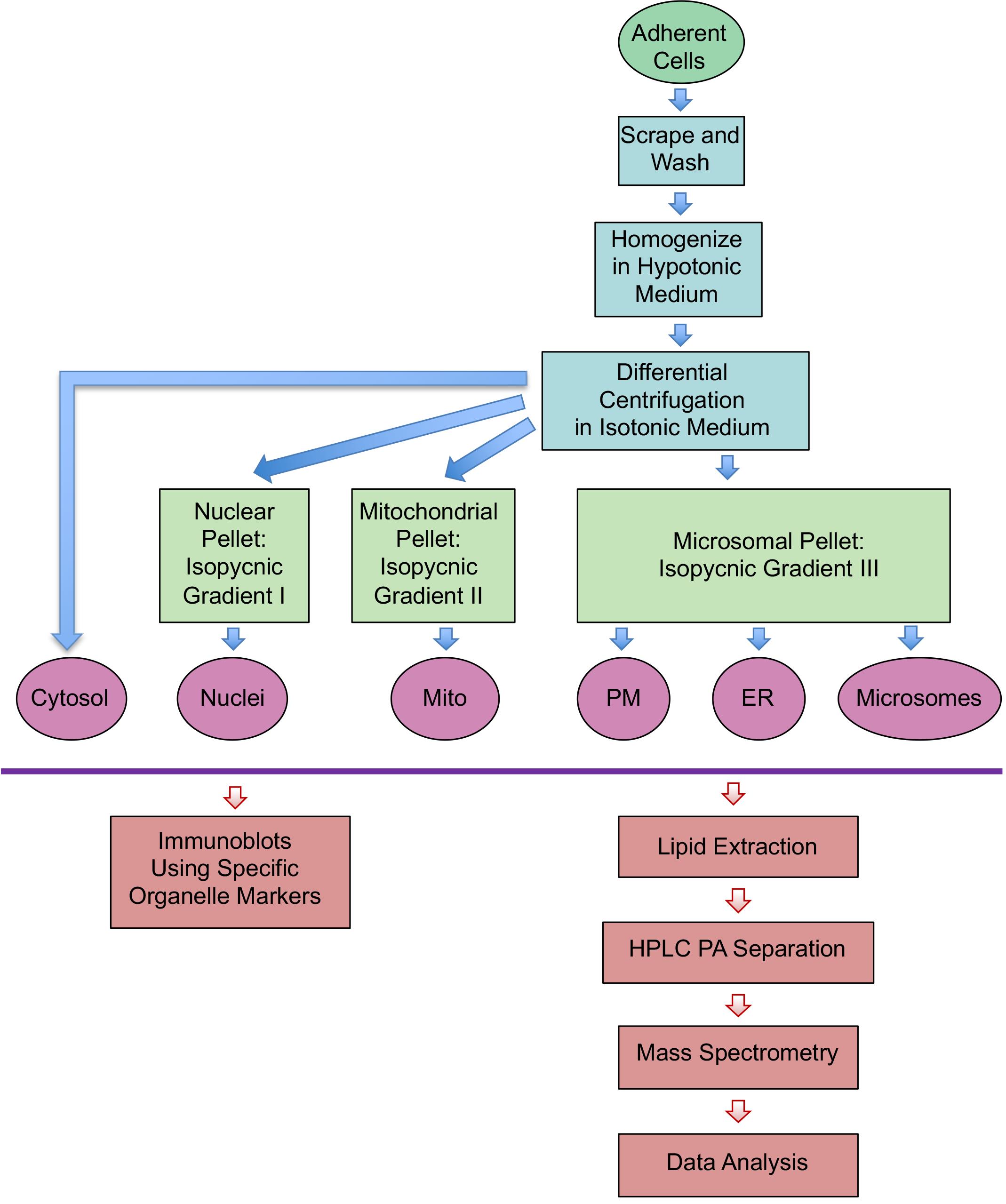
Figure 1. Schematic representation of the different steps from subcellular fractionation to lipidomics analysis. Endoplasmic reticulum (ER), Mitochondria (Mito), Plasma membrane (PM).
Phosphatidic acid extraction
Transfer the subcellular membrane fraction samples containing 200 µg protein in glass centrifuge tubes containing 1 ml water.
Add 2 ml chloroform/methanol 2/1 v/v and 100 µl 34:0_17:0 PA (17 µM) used as an internal standard, since it is not present in the samples due to the odd number of carbons.
Sonicate for 30 s at room temperature and mix vigorously.
Centrifuge for 5 min at 800 × g at room temperature to separate the organic and aqueous phases.
Collect the lower organic phase and transfer to clean glass tubes.
Add 2 ml chloroform to the upper aqueous phase.
Sonicate for 30 s and mix vigorously.
Centrifuge for 5 min at 800 × g at room temperature to separate the organic and aqueous phases.
Collect the lower organic phase and combine with the organic phase collected in Step D5.
Use the evaporator to dry the combined organic phases (Figure 1).
Mass spectrometry analysis dedicated to phosphatidic acid
Resuspend samples in 50 µl eluant A (LC solvent, see Recipes).
Transfer samples to glass vials suitable for injection.
Place the vials in the autosampler at 4°C.
Separate PA species on an LC 1,200 Infinity II system equipped with a Luna C8 150 × 1 mm column with the following gradient (Figure 1):
0 min start at 70% eluent A, 30% eluent B (see Recipes).
0 to 5 min ramp at 50% eluent A, 50% eluent B.
5 to 30 min ramp to 20% eluent A, 80% eluent B.
31 to 41 min hold at 5% eluent A, 95% eluent B.
42 to 52 min hold to 70% eluent A, 30% eluent B.
Note: Mass spectrometry analysis dedicated to phosphatidic acid is adapted from Buré et al. (2014).
Subject the HPLC eluates to coupled electrospray ionization in the negative ionization mode and to subsequent tandem MS analysis on a QTrap 6,500 mass spectrometer with the following settings optimized for this equipment to have a better intensity during fragmentation:
Dwell time 30 ms.
Declustering potential (DP) – 172 V.
Entrance potential (EP) – 10V.
Collision energy (CE) – 46V.
Collision cell exit potential (CXP) – 15V.
Quantitate the different PA species using the multiple reaction monitoring (MRM) transitions information found in Table 2 (see attached file). An example of an MRM chromatogram is shown in Figure 2.
Table 2. List of MRM transitions for PA. All MRM are made in negative mode. Q1 corresponds to the mass of the parent ions (PA is negatively charged) and Q3 to the mass of the fragment (ion daughter), ID corresponds to the specific transition: for example, PA28:0_14:0 corresponds to PA with 28 carbons and no unsaturation, losing the fatty acid containing 14 carbons and no unsaturation. IS indicates the internal standard.
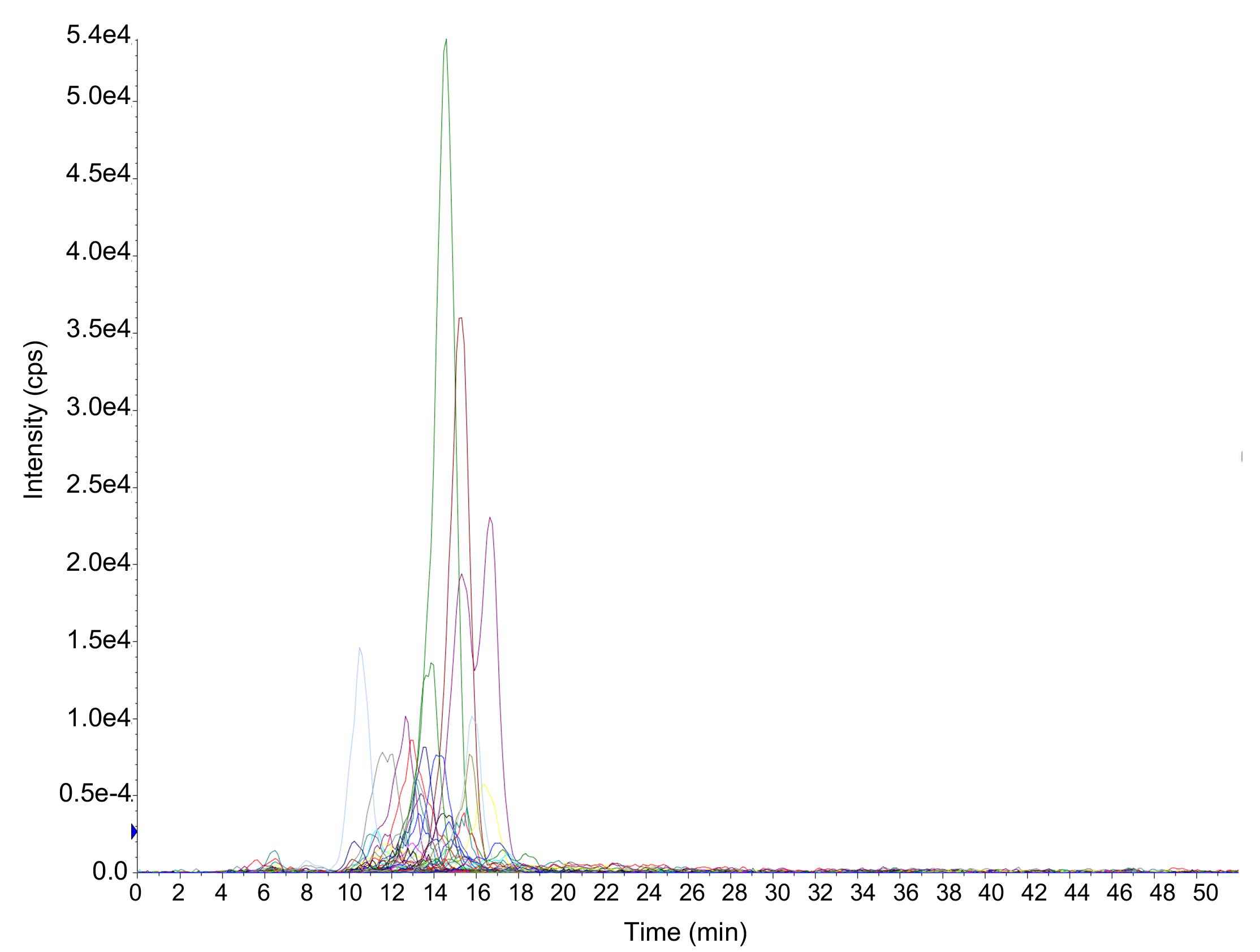
Figure 2. Example of MRM chromatograms of all search transitions for PA. Each color corresponds to a transition for a specific PA species, for example: green with a maximum at 14.5 min corresponds to PA36:1_18:1; maroon with a maximum at 15.2 min corresponds to PA38:2_18:1; purple with a maximum at 16.7 min corresponds to PA38:1_18:1; and blue with a maximum at 10.5 min corresponds to PA40:6_18:0. Intensities of the detected signals are expressed in counts per second (cps).
Data analysis
Define the integration parameters in Multiquant:
Integration Algorithm MQ4.
Gaussian Smooth Width 1.0 points.
Noise percentage: 40.0%.
Integrate all the samples using the Multiquant method and report the area under the peaks for each MRM (Figure 3).
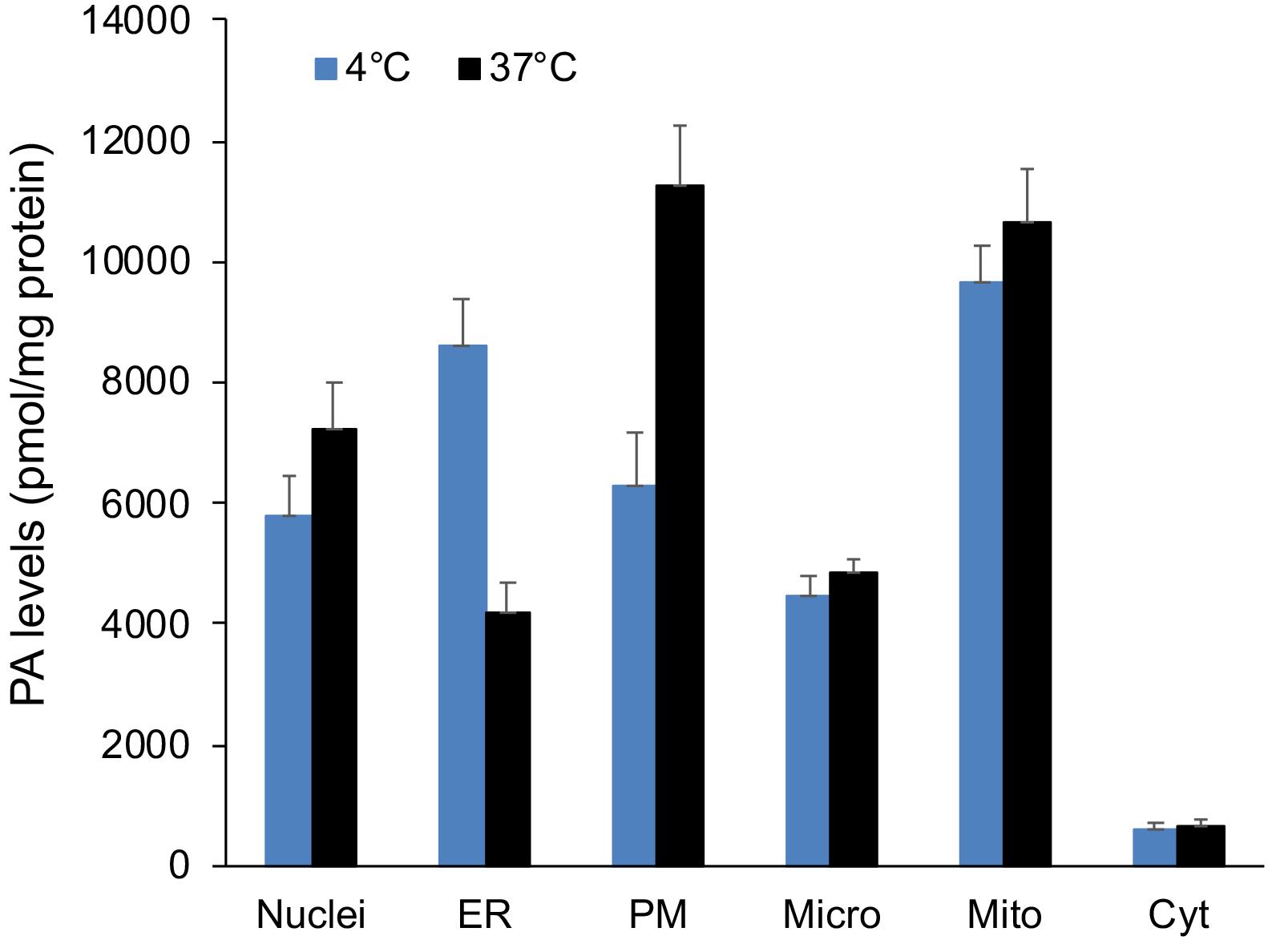
Figure 3. PA level in subcellular membrane fractions from RAW 264.7 macrophages undergoing frustrated phagocytosis. For each sample, the PA level was measured by duplicate analysis (each containing 200 µg protein) of each subcellular membrane fraction from cells either kept at 4°C or shifted to 37°C for 30 min and normalized to 1 mg protein. Results are presented as the mean of the total PA level per sample ± S.E.M. (n = 3) obtained from independent experiments.Sum the transitions corresponding to the same PA species, as for example, PA 32: 0 with PA32:0_14:0, PA32:0_16:0 and PA32:0_18:0.
Present the results as a ratio corresponding to the area of the PA species and the 34:0_17:0 PA used as an internal standard.
Notes
The analysis revealed that the PA level in the PM fraction increased, whereas that of the ER fraction dropped after RAW264.7 cells were switched from 4°C to 37°C (Figure 3). Among the 33 PA subspecies detected, 11 reached 2.5% of the total PA in at least one of the membrane fractions. The analysis of these most abundant PA showed a specific enrichment of each PA species in different membrane fractions, with a greater variability among fractions for the polyunsaturated omega-3 PA(40:6) (Figure 4).
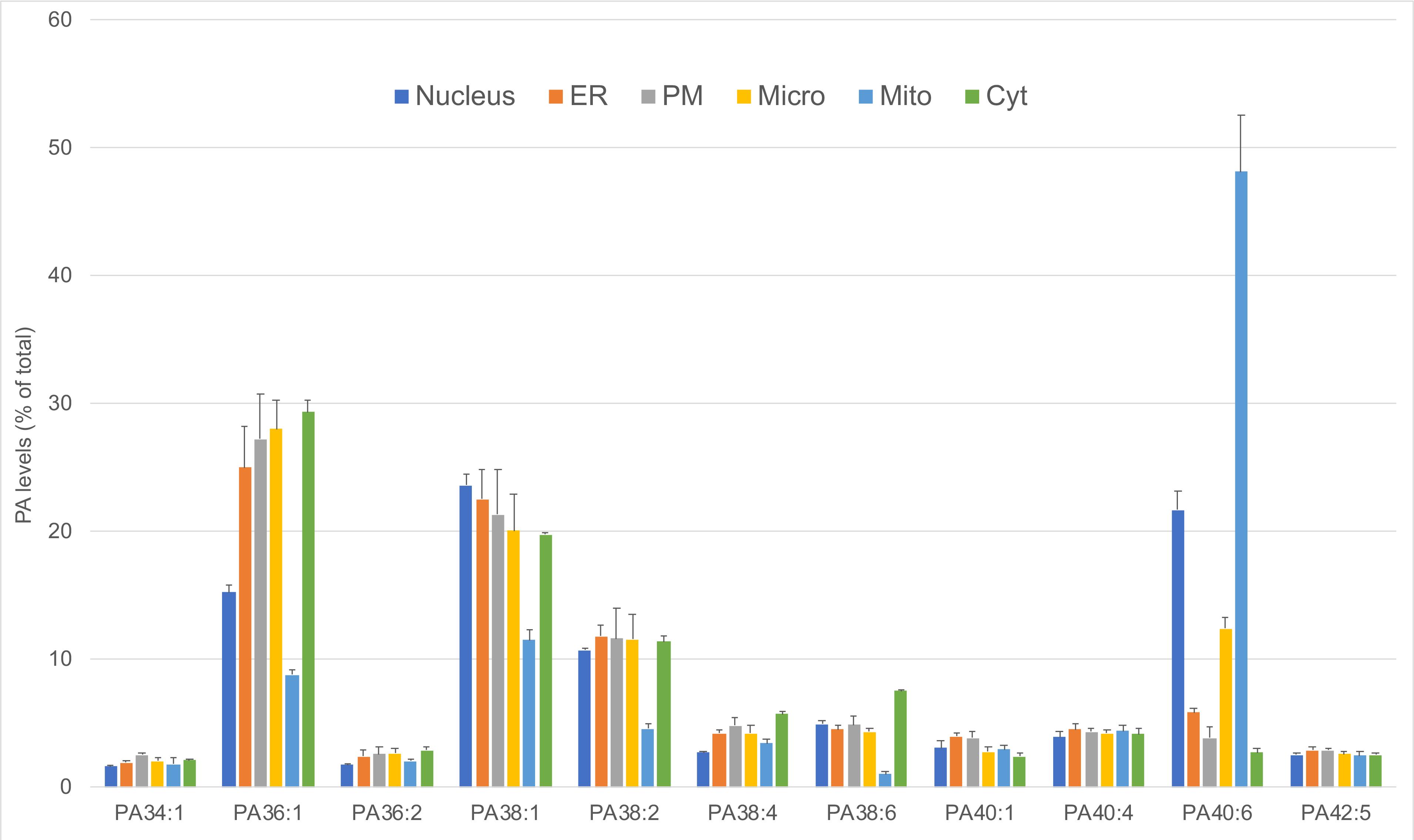
Figure 4. The 11 most abundant PA subspecies in subcellular membrane fractions from RAW 264.7 macrophages. For each sample PA subspecies, levels were measured by duplicate analysis (each containing 200 µg protein) of each subcellular membrane fraction from cells kept at 4°C, and the 11 that represented more than 2.5% of the total PA are shown. Results are expressed as the percentage of the total PA level in each sample ± S.D.
This protocol could be applied to other cell lines. The distribution among the fractions at the end may vary depending on the cell line. The general protocol could be applied after optimization to most cell lines since they all contain subcellular organelles. For instance, we have recently developed this protocol to measure PA species in plasma, Golgi, and secretory granule membrane fractions from neurosecretory PC12 cells (Carmon et al., 2020; Tanguy et al., 2020).
Recipes
Tissue culture freezing media
50% RPMI-Glutamax medium
40% FBS
10% DMSO
2× Laemmli buffer
125 mM tris-HCl (pH 6.8)
20% glycerol
2.5% SDS
2.5 mg/100 ml Coomassie Brilliant Blue G-250
Mix fresh before use: 950 µl of 2× Laemmli buffer with 50 µl β-mercaptoethanol
Isolation medium
250 mM sucrose
10 mM HEPES-Tris, pH 7.4
1 mM EGTA-Tris
Gradient media
For nuclei
Supplement isolation medium to 5 mM MgCl2, suspend crude nuclear pellet in 12 ml final volume 25% OptiPrep, and form three OptiPrep gradients by layering from the bottom up in three 14-ml thin-wall tubes: 4 ml 10% OptiPrep, 4 ml nuclei in 25% OptiPrep, 2.5 ml 30% OptiPrep, 1.5 ml 35% OptiPrep
For mitochondria
Suspend mitochondrial fraction in 6 ml final volume 35% OptiPrep and form three 14-ml tubes: 2 ml 10% OptiPrep, 4 ml 17.5% OptiPrep, 4 ml 25% OptiPrep, and 2 ml mitochondrial pellet in 35% OptiPrep
For remaining membranes
Suspend the microsomal pellet fraction in 6 ml final volume 35% OptiPrep and form three 14-ml tubes: 2 ml 10% OptiPrep, 4 ml 17.5% OptiPrep, 4 ml 25% OptiPrep, and 2 ml microsomal pellet in 35% OptiPrep
LC Solvent
Eluent A: Isopropanol/CH3OH/H2O (5/1/4) + 0.2% formic acid + 0.028% NH3
Eluent B: Isopropanol + 0.2% formic acid + 0.028% NH3
Acknowledgments
This work was supported by grants from the Fondation pour la Recherche Médicale (DEI20151234424) and the Agence Nationale pour la Recherche (ANR-19-CE44-0019) to N.V. The Metabolome Bordeaux facility was supported by the grant MetaboHUB-ANR-11-INBS-0010.
Competing interests
The authors declare no competing financial interests.
References
- Ammar, M. R., Kassas, N., Chasserot-Golaz, S., Bader, M. F. and Vitale, N. (2013). Lipids in Regulated Exocytosis: What are They Doing? Front Endocrinol (Lausanne) 4: 125.
- Bure, C., Ayciriex S., Testet E. and Schmitter, J. M. (2013). A single run LC-MS/MS method for phospholipidomics. Anal Bioanal Chem 405(1): 203-13.
- Carmon, O., Laguerre, F., Riachy, L., Delestre-Delacour, C., Wang, Q., Tanguy, E., Jeandel, L., Cartier, D., Thahouly, T., Haeberle, A. M., Fouillen, L., Rezazgui, O., Schapman, D., Haefele, A., Goumon, Y., Galas, L., Renard, P. Y., Alexandre, S., Vitale, N., Anouar, Y. and Montero-Hadjadje, M. (2020). Chromogranin A preferential interaction with Golgi phosphatidic acid induces membrane deformation and contributes to secretory granule biogenesis. FASEB J 34(5): 6769-6790.
- Corrotte, M., Chasserot-Golaz, S., Huang, P., Du, G., Ktistakis, N. T., Frohman, M. A., Vitale, N., Bader, M. F. and Grant, N. J. (2006). Dynamics and function of phospholipase D and phosphatidic acid during phagocytosis. Traffic 7(3): 365-377.
- Kassas, N., Tanguy, E., Thahouly, T., Fouillen, L., Heintz, D., Chasserot-Golaz, S., Bader, M. F., Grant, N. J. and Vitale, N. (2017). Comparative Characterization of Phosphatidic Acid Sensors and Their Localization during Frustrated Phagocytosis. J Biol Chem 292(10): 4266-4279.
- Labrousse, A. M., Meunier, E., Record, J., Labernadie, A., Beduer, A., Vieu, C., Ben Safta, T. and Maridonneau-Parini, I. (2011). Frustrated phagocytosis on micro-patterned immune complexes to characterize lysosome movements in live macrophages. Front Immunol 2: 51.
- Tanguy, E., Carmon, O., Wang, Q., Jeandel, L., Chasserot-Golaz, S., Montero-Hadjadje, M. and Vitale, N. (2016). Lipids implicated in the journey of a secretory granule: from biogenesis to fusion. J Neurochem 137(6): 904-912.
- Tanguy, E., Coste de Bagneaux, P., Kassas, N., Ammar, M. R., Wang, Q., Haeberle, A. M., Raherindratsara, J., Fouillen, L., Renard, P. Y., Montero-Hadjadje, M., Chasserot-Golaz, S., Ory, S., Gasman, S., Bader, M. F. and Vitale, N. (2020). Mono- and Poly-unsaturated Phosphatidic Acid Regulate Distinct Steps of Regulated Exocytosis in Neuroendocrine Cells. Cell Rep 32(7): 108026.
- Tanguy, E., Kassas, N. and Vitale, N. (2018). Protein(-)Phospholipid Interaction Motifs: A Focus on Phosphatidic Acid. Biomolecules 8(2). 20.
- Tanguy, E., Wang, Q., Moine, H. and Vitale, N. (2019a). Phosphatidic Acid: From Pleiotropic Functions to Neuronal Pathology. Front Cell Neurosci 13: 2.
- Tanguy, E., Tran Nguyen, A. P., Kassas, N., Bader, M. F., Grant, N. J. and Vitale, N. (2019b). Regulation of Phospholipase D by Arf6 during FcgammaR-Mediated Phagocytosis. J Immunol 202(10): 2971-2981.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Kassas, N., Fouillen, L., Gasman, S. and Vitale, N. (2021). A Lipidomics Approach to Measure Phosphatidic Acid Species in Subcellular Membrane Fractions Obtained from Cultured Cells. Bio-protocol 11(12): e4066. DOI: 10.21769/BioProtoc.4066.
Category
Immunology > Immune cell function > Macrophage
Neuroscience > Cellular mechanisms > Intracellular signalling
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.