- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

CRISPR/Cas9-mediated Precise SNP Editing in Human iPSC Lines
Published: Vol 11, Iss 12, Jun 20, 2021 DOI: 10.21769/BioProtoc.4051 Views: 6606
Reviewed by: Jaira Ferreira de VasconcellosGiovanna PiovaniAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2020


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Human induced pluripotent stem cells (hiPSCs) have been extensively used in the fields of developmental biology and disease modeling. CRISPR/Cas9 gene editing in iPSC lines often has a low frequency, which hampers its application in precise allele editing of disease-associated single nucleotide polymorphisms (SNPs), especially those in the noncoding parts of the genome. Here, we present a unique workflow to engineer isogenic iPSC lines by SNP editing from heterozygous to homozygous for disease risk alleles or non-risk alleles using a transient and straightforward transfection-based protocol. This protocol enables us to simultaneously obtain pure and clonal isogenic lines of all three possible genotypes of a SNP site within about 4 to 5 weeks.
Keywords: Induced pluripotent stem cellsBackground
The application of CRISPR/Cas9 gene editing to iPSC cells provides unrivalled potential in the fields of developmental biology, disease modeling, and regenerative medicine. However, since iPSCs are not especially acquiescent to the traditional strategies used in CRISPR/Cas9-gene editing, the editing efficiency is often low, especially for homology-directed repair (HDR)-mediated editing of single nucleotide polymorphisms (SNPs). Special experimental methods must be developed to circumvent this limitation; for instance, using simultaneous CRISPR/Cas9 mutagenesis and reprogramming of iPSCs (Howden et al., 2015; Tidball et al., 2018), advanced cell sorting techniques such as FACS (Forsyth et al., 2006; Miyaoka et al., 2014), or recombination of mutation-carrying cassettes though the presence of long homology arms in targeting plasmids (Hendel et al., 2014). Nevertheless, these highly specialized protocols for gene editing in iPSC lines are time and/or resource-consuming, which persists as a critical limiting factor in generating isogenic iPSC lines via CRISPR/Cas9-mediated precise SNP editing.
Here, we present a demonstrated workflow and detailed procedures that allow direct editing of iPSCs using the CRISPR/Cas9 system with an HDR technique by simple liposome-based transfection, followed by antibiotic selection to enrich the edited iPSCs and single clonal selection to obtain pure iPSC isogenic lines of all three possible genotypes. This method was adapted from Ran et al. (2013) with demonstrated results in our recent publications (Forrest et al., 2017; Zhang et al., 2018 and 2020). Notably, this protocol worked out very well for our unique CRISPR/cas9 SNP editing design from a heterozygous iPSC line to isogenic pairs homozygous for disease risk alleles or non-risk alleles, which makes the functional interpretation of an edited SNP more reliable by directly comparing isogenic lines of all three different genotypes (Forrest et al., 2017; Zhang et al., 2018 and 2020).
Materials and Reagents
Materials
1.5 ml Eppendorf tubes (VWR, catalog number: 89000-028)
4-w NunclonTM Delta MultiDishes (Thermo Scientific, catalog number: 62407-068)
6-well culture plates (Thermo Fisher, catalog number: 140675)
96-well culture plates (Corning, Falcon®, catalog number: 353072)
Standard 60 × 15 mm dishes w/vent (Fisher Scientific, catalog number: 12-565-95)
15 ml centrifuge tubes (VWR, catalog number: 21008-216)
2 ml cryogenic vials with closures, polypropylene (Corning®, catalog number: 89089-764)
10 μl racked tips, low-retention sterile (VWR, catalog number: 10017-062)
200 μl racked tips, low-retention sterile (VWR, catalog number: 76322-150)
1,000 μl low-retention tips (VWR, catalog number: 10017-090)
BioDot 96-well non-skirted PCR plates (Dot Scientific, catalog number: 650-PCR)
Reagents
mTeSR1 (STEMCELL, catalog number: 85850)
mFreSR Cryopreservation Medium (STEMCELL, catalog number: 05855)
ReLeSR (STEMCELL, catalog number: 05872)
Matrigel® hESC-Qualified Matrix (Corning®, catalog number: 354277)
FuGENE® HD Transfection Reagent (Promega, catalog number: E2311)
Accutase (STEMCELL, catalog number: 07920)
Primocin (Invitrogen, ant-pm-1)
Y-27632 dihydrochloride (R&D Systems, catalog number: 1254/1)
QIAprep Spin Miniprep Kit (250) (Qiagen, catalog number: 27106)
Qiaprep EndoFree Plasmid Maxi Kit (10) (Qiagen, catalog number: 12362)
BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific, catalog number: 4337455)
DyeEx 2.0 kit (Qiagen, catalog number: 63204)
HotStarTaq DNA Polymerase (250 U) (Qiagen, catalog number: 203203)
PlasmidSafe Exonuclease (Lucigen, catalog number: E3101K)
QuickExtractTM DNA Extraction Solution (VWR, catalog number: 76081-768)
FastDigest BbsI (Thermo Fisher Scientific, catalog number: ER1101)
T7 ligase (NEB, catalog number: M0318S)
ATP solution, 100 mM (NEB, catalog number: N0451)
dNTP solution mix, 10 mM (NEB, catalog number: N0447S)
Opti-MEM I, 100 ml (Thermo Fisher Scientific, catalog number: 31985062)
Shrimp alkaline phosphatase (Thermo Fisher Scientific, catalog number: 783901000UN)
PCRX Enhancer System (Thermo Fisher, catalog number: 11495017)
Isopropanol (Sigma-Aldrich, catalog number: 190764)
Nuclease-free water, PCR grade (Thermo Fisher Scientific, catalog number: AM9937)
Teknova DNA/RNA Resuspension Buffer (TE buffer) (VWR, catalog number: 100216-886)
Invitrogen One Shot® Stbl3TM Chemically Competent E. coli cells (Thermo Fisher Scientific, catalog number: C737303)
1 kb DNA ladder (Promega, catalog number: G5711)
Equipment
C1000 Touch Thermo Cycler (Bio-Rad, model: 1851197)
Sorvall Legend XTR Centrifuge (Thermo Scientific, catalog number: 75217420)
Benchtop refrigerated centrifuge 5430R (Eppendorf, catalog number: 022620601)
Heracell 150i Tissue Culture Incubator (Thermo Fisher Scientific, catalog number: 51026283)
ABI 3730 DNA Analyzer (Thermo Fisher Scientific, model: 3730S)
Nalgene Mr. Frosty Freezing container (Sigma-Aldrich, catalog number: C1562)
Software
ApE – A plasmid Editor (M Wayne Davis, https://jorgensen.biology.utah.edu/wayned/ape/)
Procedure
Experimental planning
Cell line selection
Generally, the experiment will be the most convenient if the starting cell line is heterozygous at the desired SNP site to be edited. In this way, we can synthesise two single-stranded DNA oligonucleotides (ssODNs), each carrying a different genotype, and perform two separate SNP editing experiments simultaneously, thereby obtaining edited homozygous cell lines in both allele directions at the end of the experiment (Figure 1A). It is also possible to obtain homozygous cell lines of the opposing genotype by starting from a homozygous cell line when the circumstances do not allow a high degree of freedom, such as using cell lines derived from clinically diagnosed patients (Figure 1B). In such cases, it would be beneficial to increase the number of clones picked in Sections E and F to maximize the chance of detecting a positive one. Whilst there would be a chance that one of the genotypes (heterozygous or homozygous of the opposite genotype) does not appear in the first round of CRISPR/Cas9 editing, obtaining the homozygous cell line of the opposite genotype is usually sufficiently satisfactory for the purpose of the experiment. Alternatively, a heterozygous line obtained from the first round of editing can be used for another round of CRISPR/Cas9 editing with the same ssODN to obtain a homozygous line of the opposite genotype. In all cases, the genotype of the starting cell line at the desired SNP site needs confirmation before proceeding to Section B. To confirm the genotype at the specific SNP site, use 30 ng genomic DNA extracted from the starting cell line and perform Sanger sequencing by proceeding with Steps E4-E5. Alternatively, online databases, next-sequencing results, or cell line-associated patient metadata may also provide genotype information at the SNP site to be edited.
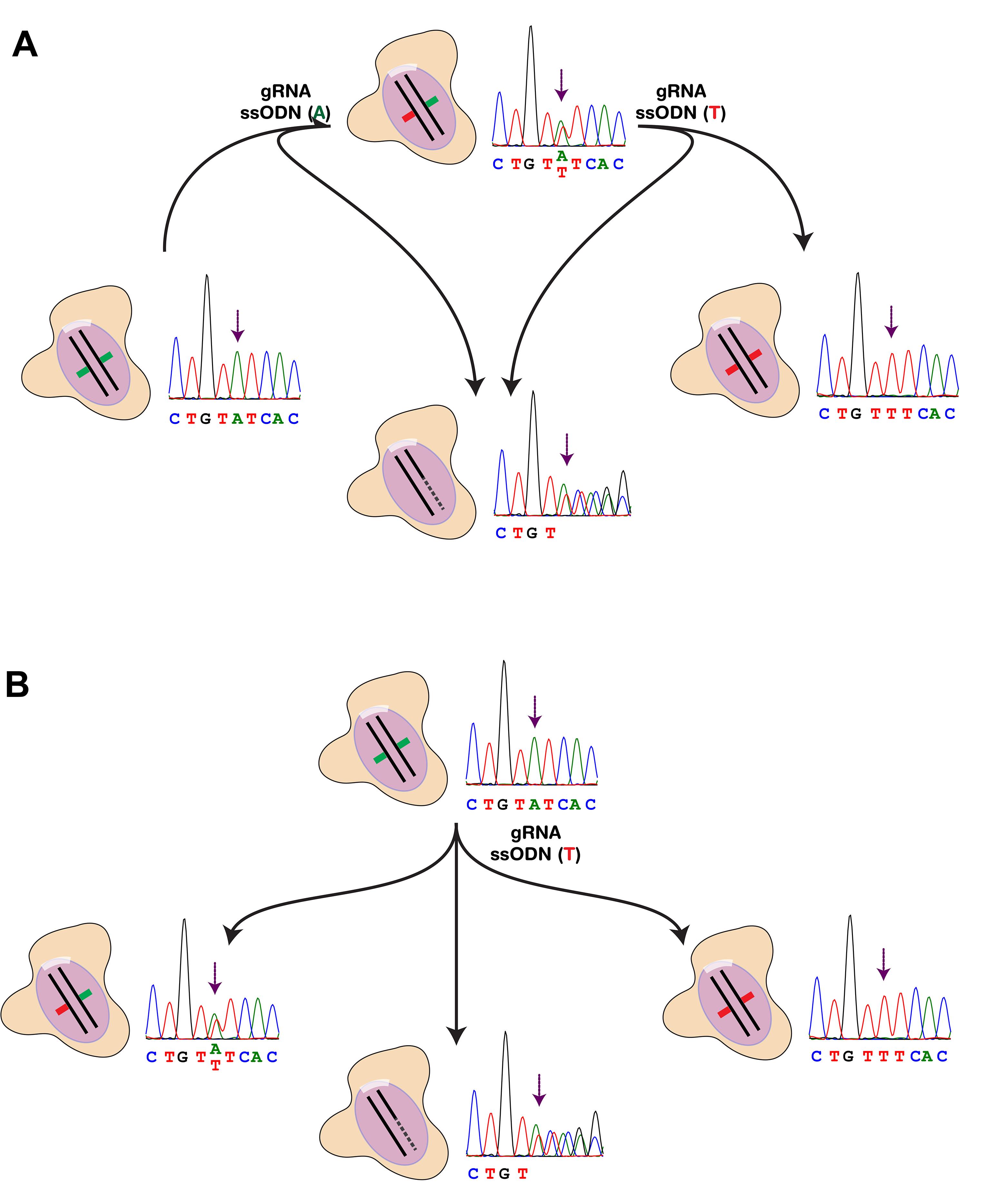
Figure 1. SNP editing strategies using SNP site rs2027349 as an example. A. SNP editing starting from a heterozygous cell line. Two ssODNs carrying both alleles at the SNP site (A/T) are used to edit the heterozygous line (top) to the opposite direction and generate two homozygous cell lines of different genotypes (middle). Cells that have CRISPR/Cas9-mediated DNA cleavage events but not ssODN integration show the presence of indels in the form of two overlapping peak sets proximal to the target site (bottom). B. SNP editing starting from a homozygous cell line. Only one ssODN carrying the alternative allele (T) is required to generate both the heterozygous (middle-left) and homozygous (middle-right) cell line of the opposite genotype. Note that the chance of obtaining the edited homozygous cell clones is significantly lower than that of the heterozygous one.Technical requirements. The overall experiment incorporates three main parts: plasmid cloning and preparation; iPSC maintenance, transfection, and subcloning; and Sanger sequencing. Whilst it is desirable that one operator be acquainted with all these skills, this protocol has been designed to work as a collaboration project with several members, each with their own specialization.
gRNA design and vector preparation
Use dbSNP (https://www.ncbi.nlm.nih.gov/snp/) or any genome browser to fetch the DNA sequence proximal to the target SNP for gRNA design. Generally, the sequence will extend 65 bp both upstream and downstream of the target SNP, for a total of 131 bp. Save the sequence as it will be used to synthesise the ssODN with homology arms flanking the target SNP site. This sequence will also be used in a gRNA design tool such as Benchling (https://www.benchling.com) to generate the gRNA sequence (20 bp) for the introduction of a double-strand break (DSB). Pick the gRNA with the highest efficiency score while maintaining a reasonable specificity score. It is also found that gRNA sequences with a cleavage site proximal to the target SNP location may have better recombination efficiency (see Figure 2 for the schematic figure as well as an output example from Benchling for the gRNA and ssODN used to edit SNP rs2027349). In the meantime, design a pair of PCR primers covering the target SNP using Primer3 (http://primer3.ut.ee/) or similar online tools. The PCR product should be 400-600 bp in size, and the SNP is at least 150 bp from either end of the product.
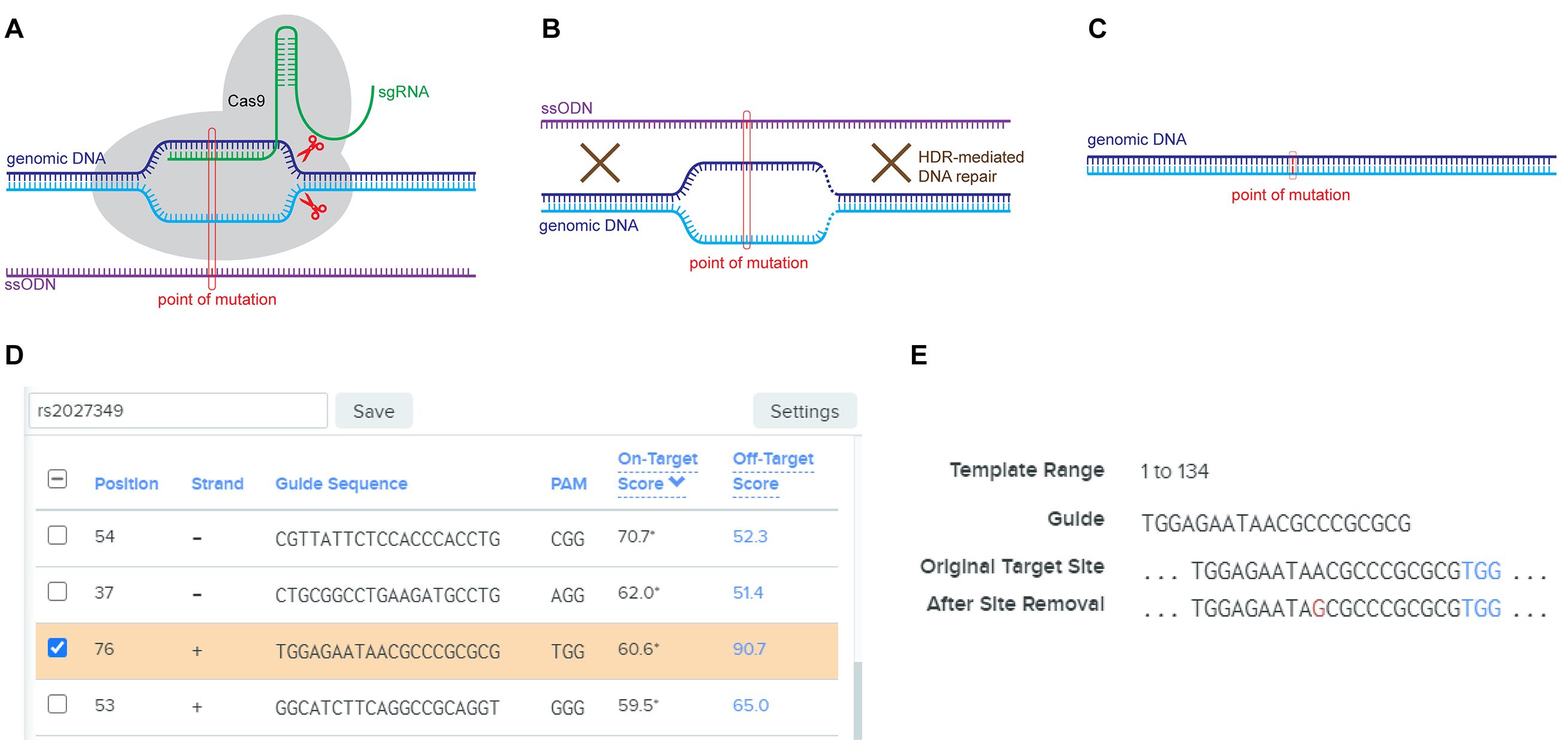
Figure 2. Schematic of HDR-mediated SNP editing. A. gRNA-introduced double-strand breaking event. The single guide RNA (sgRNA) binds to a specific genomic DNA sequence and introduces a DSB in the genomic DNA double-strand. The ssODN is shown in parallel to highlight the mutation site; B. HDR-mediated DNA repair event using ssODN as the template. After creating the DSB (dashed line), the HDR-mediated DNA repair starts and, during this process in some cells, ssODN carrying the mutant allele is used as the template for the final repaired product; C. Final product after the repair using ssODN as the template, with the point of mutation highlighted; D. Sample gRNA candidate output list from Benchling using human GRCh38 genomic sequence chr1:150,067,554-150,067,687. Here, the highlighted item contains SNP rs2027349 as …AATA[A]CGCC…; E. Sample ssODN output window from Benchling using the same genomic sequence above, showing conversion of the SNP itself from A to G (in red).Order the following oligonucleotides from a vendor (such as IDT or Sigma-Aldrich):
ssODN, 131 nt in length, single-stranded with the target SNP of the edited allele located at base position 66. Do not order the complementary strand. Two ssODNs carrying both alleles are required if starting from a heterozygous cell line, while one ssODN carrying the opposite allele is required if starting from a homozygous cell line. Order in 4 nmol Ultramer Oligo dry format with standard desalting if from IDT.
sgRNA-top: 5’-[Phos]-CACCgNNNNNNNNNNNNNNNNNNNN;
sgRNA-bottom: 5’-[Phos]-AAACNNNNNNNNNNNNNNNNNNNNc;
in which N stands for the gRNA sequence. Note the tailing c at the end of sgRNA-bottom oligo. DO NOT include the PAM sequence in the sgRNA oligos.
Both oligos can be ordered in 5′-phosphorylated formats (recommended) to avoid the in-house PNK phosphorylation step (the efficiency of which varies). Order in 25 nmol dry DNA Oligo format with standard desalting if from IDT and resuspend to 100 μM with TE buffer upon arrival for long-term storage at -20°C.
Sequencing primers, as mentioned above. The PCR primers can be used as sequencing primers if the SNP is not too near/far from either end. Order in 25 nmol dry DNA Oligo format with standard desalting and resuspend to 100 μM with TE buffer upon arrival for long-term storage at -20°C.
U6-Forward primer (5’-gagggcctatttcccatgatt) for sequencing the gRNA insert of the final plasmid construct. Order in 25 nmol dry DNA Oligo format with standard desalting and resuspend to 100 μM with TE buffer upon arrival for long-term storage at -20°C.
Assemble the pSpCas9(BB)-2A-Puro (PX459) V2.0 (Addgene, 62988) construct with customized gRNA insert.
Mix well the following components in a PCR tube with a cap at room temperature. Do not use diethyl pyrocarbonate (DEPC)-treated water throughout the experiment as it may inhibit the reaction.
Component Amount (μl) sgRNA top (100 μM) 1 sgRNA bottom (100 μM) 1 T4 ligation buffer, 10× (NEB) 1 Water, PCR grade 7 Total 10 Use a thermocycler with a heated lid and set the following parameters to anneal the oligos. Ramp down at the minimum ramp rate (e.g., 0.1°C/s). Throughout this protocol, a thermocycler with a heated lid should be used (with lid heating function turned on) unless otherwise specified.
Step # Temperature Time Note 1 95°C 5 min Minimum ramp rate 2 25°C 2 min Dilute the annealed oligos by adding 1 μl to 199 μl PCR-grade water, mix well.
Perform the cut-ligation reaction by mixing well the following components in a PCR tube with cap. We recommend setting up a negative control experiment by substituting the diluted oligos with water, whose function will be introduced later.
Component Amount (μl) pSpCas9(BB)-2A-Puro (PX459) V2.0, 100 ng/μl 1 Diluted annealed oligos from step A3c 2 Tango buffer, 10× 2 DTT, 10 mM 1 ATP, 10 mM 1 FastDigest BbsI 1 T7 ligase 0.5 Water, PCR grade 11.5 Total 20
Then set up the thermocycler parameters as follows:Step # Temperature Time Note 1 37°C 5 min 2 21°C 5 min 3 / / Return to step #1, 6 cycles Treat the ligation product and negative control with PlasmidSafe exonuclease to digest any residual linearized DNA by assembling the following components in a PCR tube with cap and mixing well. This heat-inactivated reaction can be stored at -20°C for at least 1 week or -80°C indefinitely if it cannot be used immediately.
Component Amount (μl) Ligation product from Step A3d 11 PlasmidSafe buffer, 10× 1.5 ATP, 10 mM 1.5 PlasmidSafe exonuclease 1 Total 15
Then set up the thermocycler as follows:Step # Temperature Time Note 1 37°C 30 min 2 70°C 30 min 3 4°C hold Transformation of competent cells. We recommend the use of RecA- E. coli strains such as Invitrogen Stbl3 (recA13) or NEB Stable (recA1), since the inserted gRNA and its associated gRNA scaffold may include unstable sequences. As a guideline, use 2 μl product from Step A3e per transformation (include a negative control) following the vendor’s instructions. After heat shock, it is necessary to add recovery media (such as SOC or NEB 10-beta/Stable Outgrowth Medium) at 32°C for 1 h with shaking before spreading on LB/Amp+ agar plates, since the amount of initial DNA input is low.
Incubate at 32°C for 16 h and check for colonies the next day. A large number of colonies on the negative control plate usually suggests that the BbsI digestion did not work. Pick 4-6 colonies from the plate with annealed oligos added and inoculate 4 ml LB/Amp+ media with each colony. Use a new LB/Amp+ agar plate to record all the inoculated colonies and their corresponding tube numbers. Incubate at 37°C overnight.
Isolate plasmid DNA using spin columns such as those from a Qiagen QIAprep Spin Miniprep Kit and send for sequencing using the U6-Forward primer to identify positive clones. In the meantime, keep the record plate from Step A3g at 4°C until all the positive clones have been identified by sequencing.
Inoculate 250 ml LB/Amp+ with the colonies of positive clones from the record plate to prepare transfection-grade plasmid. Culture and extract the plasmid using a Qiagen EndoFree Plasmid Maxi Kit to ensure complete removal of endotoxins according to the manufacturer’s instructions. Dilute the purified plasmid to 1 μg/μl or appropriate in endo-free TE buffer (provided in the maxi-prep kit) as 50-μl aliquots and store at -20°C until use.
Maintenance of human iPSCs for editing
Grow healthy undifferentiated human iPSCs in 6-well plates coated with Matrigel. Generally, for each well of the 6-well plate, use 2 ml mTeSR1 culture media and replace every 24 h. Observe the colony morphology every day and watch for signs of differentiation.
Pass the cultured iPS cells when confluency reaches 60-70% (approximately 4-7 d). Dissociate colonies from the old plates by treating each well with 1 ml ReLeSR and incubating for 3-5 min with the plate lid on according to the technical manual. Dilute the dissociated cell aggregates and transfer 1/10-1/20 of the cells to a new well pre-coated with Matrigel in 2 ml mTeSR1. For details, please refer to Section 3 of the Technical Manual: Maintenance of Human Pluripotent Stem Cells in mTeSRTM1, StemCell Technologies TM (https://www.stemcell.com/media/files/manual/10000005505-Maintenance_of_Human_Pluripotent_Stem_Cells_mTeSR1.pdf).
Transfect human iPSCs with the gRNA-integrated Cas9 vector and ssODN for precise gene editing.
Seed iPSCs in 60-mm Petri dishes for transfection.
As a guideline, each well of the 6-well plate at 60-70% confluence has approximately 1 × 106 iPSCs. On the other hand, each 60-mm Petri dish requires 4 × 105-5 × 105 iPSCs for seeding. Multiply the total number of iPSC wells for seeding by 1.2 to compensate for loss during collection and counting.
Prepare enough 60-mm cell culture grade Petri dishes coated with Matrigel for iPSC seeding. Generally, prepare one Petri dish for each clone that will be generated, plus one dish for transfection efficiency evaluation.
When iPSCs in 6-well plates reach a confluence of 60-70%, calculate the number of iPSCs required for transfection. Check cell morphology and manually scrape off any differentiated colonies under a stereomicroscope before dissociation. Do not use a culture with more than 20% of the colonies with signs of differentiation. Remove culture media from the plates and add 1 ml Accutase per well. Place the plates back into the incubator and wait for 10 min to dissociate cells.
After 10 min, add 1 ml room-temp mTeSR to each well of the 6-well plate. Smack the plate against the palm to dislodge any remaining cells from the bottom gently without spilling media. Collect media from all wells into a 15-ml sterile Falcon tube and gently pipet up and down 5-7 times using a regular 1-ml tip or glass pipette to mix well and break up the clumps.
Determine the cell density using either a hemocytometer or automatic cell counter such as a ThermoFisher Countess 3. The cell density at this stage should be approximately 3 × 105-6 × 105 cells/ml. In the meantime, keep the Falcon tube containing the cells at room temperature; do not place cells on ice. Calculate the total number of cells collected.
Centrifuge the Falcon tube at 200 × g for 5 min at room temperature to precipitate the cells. Carefully remove all media and add an equal volume of fresh mTeSR1. Resuspend the cell pellet using a wide-bore 1-ml tip or glass pipet. Centrifuge the cell suspension again at 200 × g for 5 min at room temperature.
Calculate the final volume of mTeSR1 required for seeding iPSCs into 60-mm Petri dishes, plus one dish for a transfection control. Each dish will need 4-5 × 105 cells suspended in 4 ml mTeSR1. Add the ROCK inhibitor Y27632 to a final concentration of 5 μM in the media to increase viability after replating. If multiple dishes (say, transfections) are required, it may be more convenient to transfer partially resuspended cells into a larger 50-ml Falcon tube and add more mTeSR1 after transfer. Remove all media from Step D1f and replace with mTeSR1 containing Y27632. Dilute the cells to the desired concentration and fill each 60-mm dish with 4 ml fully resuspended iPSCs as single cells. Incubate at 37°C, 5% CO2 for a minimum of 12 h.
Transfect iPSCs with the gRNA-integrated Cas9 vector and ssODN.
1 h before transfection, check cell growth and replace media with 4 ml fresh antibiotic-free mTeSR1.
For each 60-mm dish, prepare the following mix in a 1.5-ml tube. Pipet up and down to mix well. For the control dish, replace the pSpCas9(BB)-2A-Puro construct with the pEGFP-N2 plasmid.
Component Amount (μl) pSpCas9(BB)-2A-Puro (PX459) V2.0 with gRNA insert,
1 μg/μl
3 ssODN, 1 μg/μl 3 Opti-MEM I Reduced Serum Medium 473 Total 479 Add 21 μl FuGene HD transfection reagent (1:3.5 DNA: reagent ratio, for a total of 500 μl) to the mix from Step D2b and briefly vortex (2-3 s) the tube to mix. Incubate the final mix at room temperature for 6 min.
Add the DNA:FuGENE:Opti-MEM I mix to the 60-mm dish and gently swirl to mix. Do not pipet up and down since this interferes with the transfection process. Incubate at 37°C, 5% CO2 for 24 h.
Select positive clones using puromycin and evaluate the transfection efficiency.
Select positive clones using puromycin
Note: Also read Step D2.
For all dishes transfected with the pSpCas9(BB)-2A-Puro construct, add 4 ml mTeSR1 supplemented with 0.5 μg/μl puromycin per dish to selectively enrich the cells transfected with the pSpCas9(BB)-2A-Puro construct, which carries a puromycin resistance cassette. The concentration of puromycin used may need optimization on a cell line-specific basis since cell lines may have very different puromycin tolerance levels (ranging from 0.3 to 0.7 μg/μl). Incubate the dishes at 37°C, 5% CO2 for 24 h.
Carefully remove media and any dead cells or cell debris from the dishes. Add 4 ml mTeSR1 supplemented with 0.3 μg/μl (or as determined by the previous assay) puromycin per dish. Incubate the dishes at 37°C, 5% CO2 for another 24 h.
Carefully remove media and any dead cells or cell debris from the dishes. Withdraw puromycin selection from this time (72 h post-transfection). Add 4 ml mTeSR and replace the culture media daily. Incubate the dishes at 37°C, 5% CO2 for 5-10 d until the iPSC colonies reach the proper size for clone selection (1.5-2 mm in diameter, Figure 3).

Figure 3. Example of an iPSC colony for clone selection
Evaluate the transfection efficiency by EGFP expression. This step should be performed in parallel with Step D1.
For the dish transfected with the pEGFP-N2 plasmid, carefully replace the culture media with 4 ml fresh mTeSR without puromycin. Incubate the dish at 37°C, 5% CO2 for 24 h.
Replace culture media with another 4 ml fresh mTeSR without puromycin. Incubate the dish at 37°C, 5% CO2 for another 24 h.
Count several fields of view to assess the percentage of EGFP+ cells. The transfection efficiency, as calculated by the percentage of EGFP+ cells, should be at least 40-50%.
Isolate individual iPSC colonies for genotype identification of positive clones.
On the day of colony picking, prepare one flat-bottomed 96-well plate coated with Matrigel (referred to as Plate A). Add 80 μl mTeSR1 containing 10 μM ROCK inhibitor Y27632 to each well of Plate A. Prepare another uncoated 96-well plate (referred to as Plate B, flat-bottomed wells).
Replace the media in the 60-mm dish with fresh ROCK inhibitor-free mTeSR1.
Set a P100 or P200 pipette at 50 μl. Attach a P200 tip and use the tip to scrape off a single colony. Aspirate the scraped-off colony or its fragments (if the colony breaks during the process) using the pipet and transfer all its contents (50 μl) to one well of Plate A. Gently pipet up and down 3 times to break the cell clump. Without replacing the pipet tip, transfer 50 μl from Plate A into the corresponding well of Plate B (Say, A1 well to A1 well, etc.) Repeat this step until all the colonies of interest on the 60-mm Petri dish have been picked up.
Spin down both plates at 200 × g for 5 min at room temperature with the lids on.
Keep Plate A at 37°C, 5% CO2 for 24 h to allow iPSCs to reattach to the well. Continue to incubate the plate and replace the media daily (100 μl mTeSR1 per well). Plate A will be used for further clone expansion after genotyping.
Use Plate B for DNA extraction.
Remove Plate B carefully from the centrifuge without shaking and place it on ice. Carefully remove media from the wells without disturbing the cell pellets using either a pipet or a multichannel pipet (set at 200 μl). DO NOT use vacuum aspiration as the cell pellets are not firmly attached to the bottom. Repeat this step to remove media from all wells on the plate.
Add 16 μl quick DNA extraction buffer (Lucigen/VWR) to each well containing the cell pellet. Set a P10 or P50 multichannel pipet at 10 μl. Carefully pipet up and down 5 times to mix; minimize bubble formation. This step resuspends cells and performs cell lysis for subsequent use.
Transfer all (16 μl) the mixed cell suspension to a 96-well PCR plate and seal using either s striped cap or film. Mark the plate as “Extracted DNA” with name and date. The product from this step should be stored at -20°C if not immediately used (1-2 weeks) or -80°C for extended storage. Set up the thermocycler as follows and discard Plate B.
Step # Temperature Time Note 1 65°C 15 min 2 68°C 15 min 3 98°C 10 min 4 4°C hold
Sample preparation for DNA sequencing on a Sanger sequencer.
Prepare the PCR reaction mix using the quick-isolated DNA from the previous step as the template. Assemble the reactions on ice according to the following chart, multiply by the number of samples for each component and further multiply by 1.1 to compensate for potential loss during reagent transfer.
Component Amount (μl) 10× PCR Buffer 1 10× PCR Enhancer 1 50 mM MgSO4 0.3 10 mM dNTP 0.2 Forward primer (10 μM) 0.2 Reverse primer (10 μM) 0.2 Qiagen HotStarTaq DNA polymerase 0.1 Water, PCR grade 5.5 Total 8.5 Mix all components WITHOUT the DNA template in a 1.5-ml tube on ice. Use a new 96-well PCR plate and dispense 8.5 μl reaction mix into each well until all the desired wells have been filled. Subsequently, add 1.5 μl extracted DNA to each well (a total of 10 μl). Pipet up and down using a P10 pipet set at 5 μl to mix well while avoiding bubble formation. This operation will be more convenient if performed with a multichannel pipet.
Seal the top of the PCR plate using a striped cap or film and mark the plate as “PCR product”. Set up the thermocycler as follows. The annealing temperature (3) and extension time (4) may require optimization, as the operator sees appropriate.
Step # Temperature Time Note 1 95°C 10 min Taq activation 2 95°C 30 s 3 53°C 90 s Or as appropriate 4 72°C 60 s Or as appropriate 5 / / Go to 2, 40 cycles 6 72°C 10 min Final extension 7 4°C hold While the PCR reaction is running, set up one or more 1.25% TAE/TBE agarose gels with a sufficient number of wells to hold at least 20 samples for PCR efficiency inspection.
Assemble the shrimp alkaline phosphatase (SAP) reaction mix on ice to remove single-stranded DNA and dNTPs from the PCR product according to the following chart. Multiply by the number of samples for each component and further multiply by 1.1 to compensate for potential loss during reagent transfer.
Component Amount (μl) 10× SAP buffer 0.5 Shrimp alkaline phosphatase (SAP) 0.5 Exonuclease I 0.1 Water, PCR grade 3.9 Total 5 After the PCR reaction has finished, use a new 96-well PCR plate and dispense 5 μl SAP mix into each well until all the desired wells have been filled. Subsequently, transfer 5 μl (1/2 of the total volume) product from the plate marked “PCR product” to the corresponding well in the 96-well plate holding the SAP mix (a total of 10 μl). Pipet up and down to mix well while avoiding bubble formation. Mark the plate as “SAP product” and set up the thermocycler as follows.
Step # Temperature Time Note 1 37°C 50 min 2 95°C 15 min 3 4°C hold While the SAP reaction is running, add 1.5 μl 6× loading dye into selected wells of the “PCR product” plate and load their contents onto the TAE/TBE gel prepared in Step E4g. Run the gel at 100 V with an appropriate molecular ladder for at least 25 min. Check the shape, number, and clarity of the bands in each lane. Take images and keep them for the records.
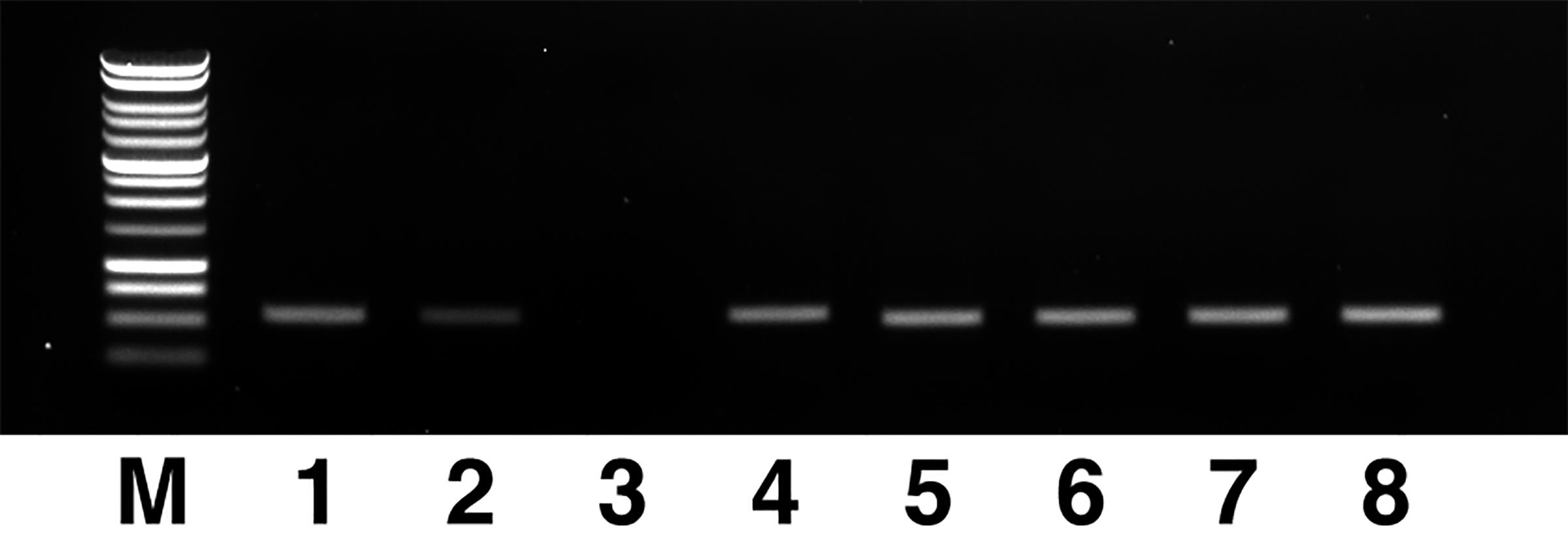
Figure 4. Sample agarose gel electrophoresis results of PCR products. Lane 3 has too little DNA to be acceptable. Lane 2, albeit of reduced brightness, is still acceptable for DNA content. M: molecular marker, 1 kb, Promega G5711.After taking gel images of the PCR reactions, count the number of “good” reactions (a single band with the correct molecular size and sufficient DNA amount as evaluated by the band’s brightness, see Figure 4) and prepare the mix for Sanger sequencing. Assemble the Sanger reaction mix on ice according to the following table. Multiply the volume of each component by the count of “good” reactions only and further multiply the results by 1.1 since the BigDye Terminator reagent is expensive.
Component Amount (μl) BigDye Terminator V3.1 1 Sequencing primer (10 μM) 1 Water, PCR grade 3 Total 5 After the SAP reaction has finished, use a new 96-well PCR plate and dispense 5 μl Sanger sequencing mix into each well until all the desired wells have been filled. Subsequently, transfer 5 μl (1/2 of the total volume) product from the plate marked “SAP product” to the corresponding well of the 96-well plate holding the Sanger sequencing mix (a total of 10 μl). Pipet up and down to mix well while avoiding bubble formation. Mark the plate as “Sequence PCR product” and set up the thermocycler as follows. The annealing temperature (3) may require optimization, as the operator sees appropriate.
Step # Temperature Time Note 1 96°C 10 s 2 55°C 5 s Or as appropriate 3 60°C 4 min 4 / / Go to 2, 25 cycles 5 4°C hold Purify the sequencing PCR product using a Qiagen DyeEx 2.0 Spin Kit (best for ≤ 16 samples). If the DyeEx 2.0 Kit is not available or there are more than 16 samples, skip Step E5j and resume at Step E5k.
Loosen the top cap and snap off the bottom closure of the spin column. Insert the spin column into a collection tube and centrifuge at 750 × g for 3 min.
Transfer the spin column to a new 1.5-ml tube labeled with the sample name. Aspirate the sequencing PCR product from the corresponding well and slowly apply the reaction product directly onto the center of the slanted gel bed surface. Centrifuge again at 750 × g for 3 min.
Discard the spin column and place the 1.5-ml tubes in a centrifugal vacuum device (such as a SpeedVac) for 30–60 min until all the liquid has evaporated. Do NOT turn on the heating during the vacuum process. After drying, add 15 μl sequencing grade formamide to the tube and pipet up and down 10 times to dissolve the DNA. Proceed to Step E5kvii.
Purify the sequencing PCR product by isopropanol precipitation.
Note: (!) Denotes that this step is critical and one should proceed with caution.
Add 40 μl 75% isopropanol to each well and gently pipet up and down 5 times to mix. This operation will be more convenient if performed with a multichannel pipet.
Incubate at room temperature for 20 min, then centrifuge at 3,000 × g for 30 min in a swing-bucket centrifuge at room temperature (20°C). Turn on the cooling function of the centrifuge if available since the air inside the centrifuge may get hot fast.
Carefully remove the plate from the centrifuge without shaking. Invert the plate onto 3-4 layers of paper towel very gently to decant the isopropanol. Repeat the process once with another fresh 3-4 layers of paper towel and keep the plate inverted for 10 s to remove as much isopropanol as possible. Do not use pipetting or a vacuum to remove any remaining isopropanol from the wells.
Flip the 96-well plate back slowly, and carefully add 150 μl 75% isopropanol to each well to wash the pellet.
Important: Do NOT mix! This operation will be more convenient if performed using a multichannel pipet. Centrifuge the plate again at 3,000 × g for 10 min.
Carefully invert the plate on the paper towel stack as in Step E5kiii to decant the isopropanol. Keep the plate inverted (!), carefully transfer it onto a new paper towel stack, and centrifuge again at 300 × g (!) for 1 min to eliminate the residual isopropanol.
Carefully remove the 96-well plate from the centrifuge and flip it back. Add 15 μl sequencing-grade formamide into each well and pipet up and down 5 times to mix. Avoid the formation of bubbles inside the well.
Seal the plate with an adhesive film and denature the DNA at 95°C for 2 min on a thermocycler. Immediately place the plate on ice and incubate for 2 min.
Centrifuge the plate at 2000 × g for 1 min. The plate is now ready for Sanger sequencing injection on an ABI 310/3730/3730xl/3500 sequencer.
Expand positive clones with the desired genotypes.
Examine the sequencing spectrum files using ApE or ABI GeneMapper to identify positive candidate clones for expansion (see Section A, Figure 1, and Figure 5). The expanded iPSCs will be used for further single-cell subcloning to obtain pure clones.
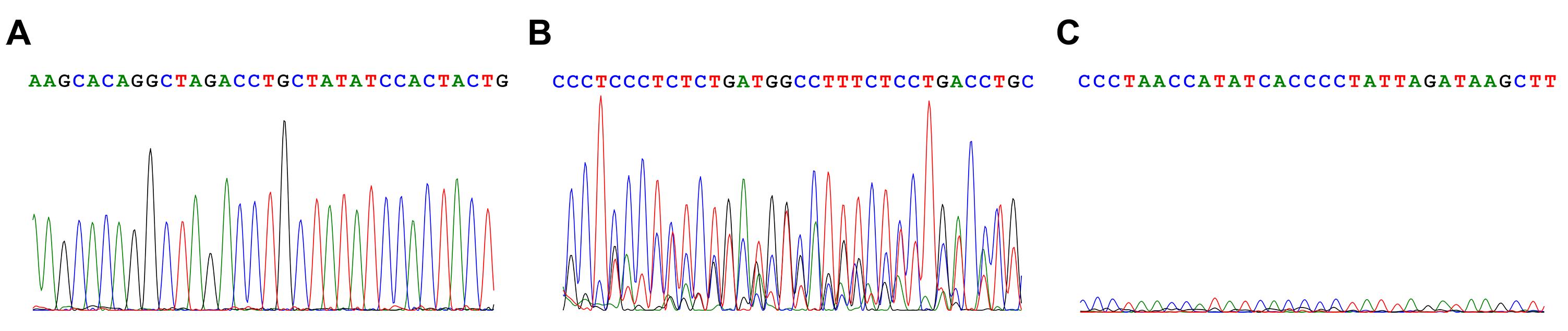
Figure 5. Sample sequencing spectrums of good and bad quality. A. A good result, showing clear discrete peaks. B. A bad result, showing multiple overlapping peak sets, implying the presence of multiple PCR products in the sequencing input. C. A bad result, showing a very weak peak signal.When the colonies reach the proper size (1.5-2 mm in diameter or 60-70% confluence in the wells) in Plate A (from Step E3a), dissociate the iPSC colonies by firstly removing all media from the wells. Gently add 50 μl ReLeSR to each well, aspirate out quickly, and stand for 5 min at room temperature.
Add 100 μl mTeSR1 to each well and carefully dislodge the cell clumps from the bottom of the well by tapping the plate and scraping using a regular P200 pipet tip. Transfer the contents of each well into one well of a Matrigel-coated 4-well or 24-well plate and add mTeSR1 containing 5 μM ROCK inhibitor to a final volume of 500 μl. Continue culture at 37°C, 5% CO2 in an incubator with daily refreshing of mTeSR1 until the cells reach 60-70% confluence in the wells.
Single-cell subcloning to obtain pure isogenic lines.
Carefully remove media from the wells and subsequently add 400 μl Accutase per well for 10 min to dissociate the cells from the bottom. Gently pipet up and down 5 times using a P1000 tip to make a single-cell suspension and then add another 400 μl mTeSR1 to neutralize the Accutase. Take 10 μl suspension to count the cell density using the trypan blue method and centrifuge the remaining cells at 250 × g for 5 min.
Remove the supernatant and resuspend the cells in 1 ml mTeSR1 containing 5 μM ROCK inhibitor Y-27632. Isolate the volume equivalent to 2000 live cells (as determined in the previous step) and replate into a 60-mm Matrigel-coated dish in 4 ml mTeSR1 containing 5 μM ROCK inhibitor Y27632. Continue to culture any remaining cells in 4/24-well plates as backups. Continue culture at 37°C, 5% CO2.
Replace half the media in the 60-mm dish with ROCK inhibitor-free mTeSR1 48 h post replating. Continue culture at 37°C, 5% CO2 in an incubator with daily refreshing of mTeSR1 until the colony size reaches 1.5-2 mm in diameter. Repeat the colony picking procedure as described under Step E3. Pick approximately 30 colonies from each 60-mm dish. Repeat the genotyping procedure for the selected colonies as described under Steps E4-E5.
Once pure clones have been identified by Sanger sequencing, repeat Step F1 to transfer each colony from the 96-well plates to 2 (!) wells of the Matrigel-coated 4/24-well plates until the confluence reaches 60-70%. Continue to monitor the cell growth as different clones may have different growth speed at this stage.
Remove media from the wells and add 400 μl ReLeSR to each well. Remove ReLeSR from the wells and incubate for 5 min at room temperature.
Coat enough 6-well plates with Matrigel and add 2 ml mTeSR1 per well. Add 300 μl mTeSR1 to each well of the 4/24-well plate treated with ReLeSR and gently pipet up and down 5 times using a wide-bore pipet tip to dissociate the cell clumps. Aliquot 100 μl cell clump suspension to each well of the 6-well plate (3 wells in total) and gently shake to mix. Prepare 4-6 wells for each clone. Continue to incubate until the confluence reaches 60-70%.
Freezing cell lines for cryopreservation.
Closely observe the cell growth in 6-well plates until the confluence reaches 60-70%. It is recommended that all wells on the same plate be processed in one run.
Add 1 ml ReLeSR per well of the 6-well plate. Remove ReLeSR from the wells and incubate for 3-5 min at 37°C, 5% CO2 in an incubator.
Add 1 ml room temperature mTeSR1 per well of the 6-well plate treated with ReLeSR and dislodge the cells by tapping the plate firmly. Transfer the dislodged cell suspension to a sterile 1.5-ml Eppendorf tube and centrifuge at 200 × g for 5 min at room temperature.
Carefully remove as much supernatant as possible without touching the cell pellet at the bottom. Add 1 ml room temperature mFreSR to the Eppendorf tube and carefully resuspend the cell pellet using a wide-bore pipet tip by pipetting up and down 5 times. Transfer the tube contents to a pre-labeled cryovial tube. Confirm the tube is firmly sealed and place it in a Mr Frosty Freezing Container. Generally, prepare as many tubes (usually 4-6 tubes as determined in Steps F2d-F2f) as possible for each clone. Repeat this step until all the cells have been transferred to cryovials.
Place the Mr Frosty Freezing Container containing cryovials in a -80°C freezer for at least 24 h (but no longer than 72 h) to freeze the contents. Move all the vials to liquid N2 for long-term storage. Keep detailed records.
Data analysis
Spectrum analysis of Sanger sequencing results
Successful identification and isolation of precise-edited, pure isogenic clones heavily rely on proper analysis of Sanger sequencing results. As a guideline, firstly remove samples with low signal intensities, aberrant patterns (such as “merged” large peaks rather than individual peaks when nucleotide bases of the same type appear consecutively in a sequence, see Figure 5). Subsequently, positive clones are identified by examining the genotype at the desired SNP site.
Evaluation of gRNA and ssODN efficiencies
Since the ssODN integration and subsequent precise SNP editing requires CRISPR/Cas9-mediated DNA cleavage to take precedence, the efficiencies of CRISPR/Cas9-mediated DNA cleavage and ssODN integration require independent evaluation. Generally, as most CRISPR/Cas9-mediated DNA breaks are not appropriately repaired and produce indels, the percentage of indel-containing clones (see Figure 1) generated can serve as an indicator of the efficiency of the gRNA sequence used in the experiment. The percentage of indel-containing clones is approximately 60-80% for a successful gRNA sequence design. The integration of ssODN is relatively stable at approximately 5% of CRISPR/Cas9-mediated DNA cleaving events. If the number of positive clones is low, calculate the likelihood of the two events separately from Sanger sequencing results to determine which oligo sequence may be the cause and require redesign.
Acknowledgments
Funding: R01MH106575, R01MH116281, R01AG063175. We acknowledge the concept of origin as developed by Ran et al. (2013) and further methodology demonstration in Zhang et al. (2020) (Doi: 10.1126/science.aay3983).
Competing interests
The authors declare no conflicts of interests.
Ethics
The NorthShore University HealthSystem Institutional Review Board (IRB) approved the study.
References
- Forrest, M. P., Zhang, H., Moy, W., McGowan, H., Leites, C., Dionisio, L. E., Xu, Z., Shi, J., Sanders, A. R., Greenleaf, W. J., Cowan, C. A., Pang, Z. P., Gejman, P. V., Penzes, P. and Duan, J. (2017). Open Chromatin Profiling in hiPSC-Derived Neurons Prioritizes Functional Noncoding Psychiatric Risk Variants and Highlights Neurodevelopmental Loci. Cell Stem Cell 21(3): 305-318 e308.
- Forsyth, N. R., Musio, A., Vezzoni, P., Simpson, A. H., Noble, B. S. and McWhir, J. (2006). Physiologic oxygen enhances human embryonic stem cell clonal recovery and reduces chromosomal abnormalities. Cloning Stem Cells 8(1): 16-23.
- Hendel, A., Kildebeck, E. J., Fine, E. J., Clark, J., Punjya, N., Sebastiano, V., Bao, G. and Porteus, M. H. (2014). Quantifying genome-editing outcomes at endogenous loci with SMRT sequencing. Cell Rep 7(1): 293-305.
- Howden, S. E., Maufort, J. P., Duffin, B. M., Elefanty, A. G., Stanley, E. G. and Thomson, J. A. (2015). Simultaneous Reprogramming and Gene Correction of Patient Fibroblasts. Stem Cell Reports 5(6): 1109-1118.
- Miyaoka, Y., Chan, A. H., Judge, L. M., Yoo, J., Huang, M., Nguyen, T. D., Lizarraga, P. P., So, P. L. and Conklin, B. R. (2014). Isolation of single-base genome-edited human iPS cells without antibiotic selection.Nat Methods 11(3): 291-293.
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A. and Zhang, F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8(11): 2281-2308.
- Tidball, A. M., Swaminathan, P., Dang, L. T. and Parent, J. M. (2018). Generating Loss-of-function iPSC Lines with Combined CRISPR Indel Formation and Reprogramming from Human Fibroblasts. Bio-protocol 8(7): 2794.
- Zhang, S., Moy, W., Zhang, H., Leites, C., McGowan, H., Shi, J., Sanders, A. R., Pang, Z. P., Gejman, P. V. and Duan, J. (2018). Open chromatin dynamics reveals stage-specific transcriptional networks in hiPSC-based neurodevelopmental model. Stem Cell Res 29: 88-98.
- Zhang, S., Zhang, H., Zhou, Y., Qiao, M., Zhao, S., Kozlova, A., Shi, J., Sanders, A. R., Wang, G., Luo, K., Sengupta, S., West, S., Qian, S., Streit, M., Avramopoulos, D., Cowan, C. A., Chen, M., Pang, Z. P., Gejman, P. V., He, X. and Duan, J. (2020). Allele-specific open chromatin in human iPSC neurons elucidates functional disease variants.Science 369(6503): 561-565.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Zhang, H. and Zhang, S. (2021). CRISPR/Cas9-mediated Precise SNP Editing in Human iPSC Lines. Bio-protocol 11(12): e4051. DOI: 10.21769/BioProtoc.4051.
- Zhang, S., Zhang, H., Zhou, Y., Qiao, M., Zhao, S., Kozlova, A., Shi, J., Sanders, A. R., Wang, G., Luo, K., Sengupta, S., West, S., Qian, S., Streit, M., Avramopoulos, D., Cowan, C. A., Chen, M., Pang, Z. P., Gejman, P. V., He, X. and Duan, J. (2020). Allele-specific open chromatin in human iPSC neurons elucidates functional disease variants.Science 369(6503): 561-565.
Category
Stem Cell > Pluripotent stem cell > Cell pluripotency
Stem Cell > Pluripotent stem cell > Cell-based analysis
Cell Biology > Cell engineering > CRISPR-cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.