- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In vivo CD40 Silencing by siRNA Infusion in Rodents and Evaluation by Kidney Immunostaining

Published: Vol 11, Iss 10, May 20, 2021 DOI: 10.21769/BioProtoc.4032 Views: 10365
Reviewed by: Fereshteh AzediAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2013
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The co-stimulatory molecule CD40 and its ligand CD40L play a key role in the regulation of immunological processes and are involved in the pathophysiology of autoimmune and inflammatory diseases. Inhibition of the CD40-CD40L axis is a promising therapy, and a number of strategies and techniques have been designed to hinder its functionality. Our group has broad experience in silencing CD40 using RNAi technology, and here we summarize protocols for the systemic administration of a specific anti-CD40 siRNA in different rodents models, in addition to the subsequent quantification of CD40 expression in murine kidneys by immunostaining. The use of RNAi technology with specific siRNAs to silence genes is becoming an essential method to investigate gene functions and is rapidly emerging as a therapeutic tool.
Graphic abstract:
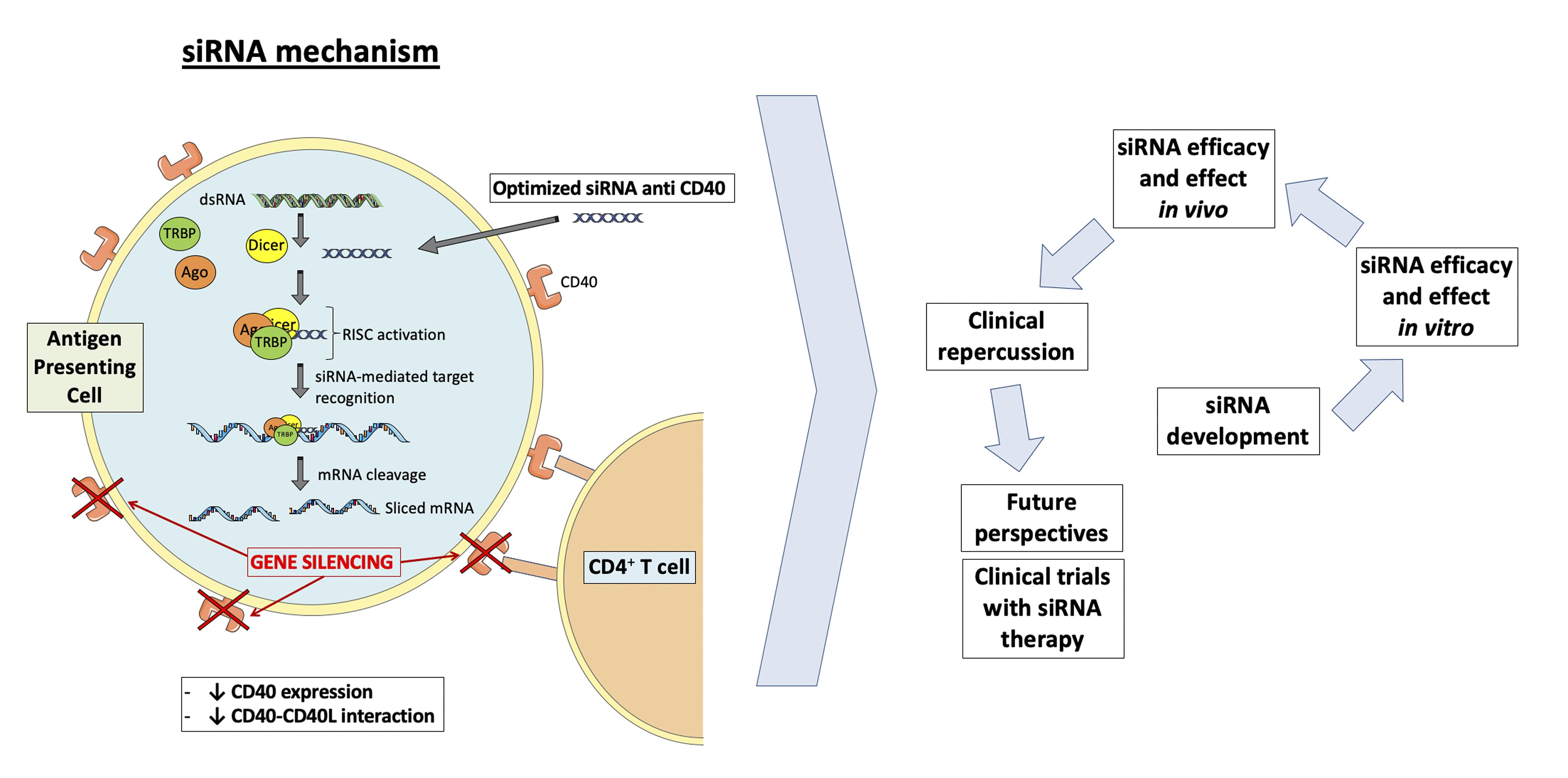
CD40 siRNA mechanism
Background
The co-stimulatory molecule CD40 and its ligand CD40L are one of the best characterized immune checkpoints involved in the pathophysiology of autoimmune and inflammatory diseases, including cancer, Graft-versus-Host-Disease, inflammatory bowel diseases, systemic lupus erythematosus (SLE), rheumatoid arthritis, type 1 diabetes mellitus, allograft rejection, and atherosclerosis (Lutgens et al; 2010; Ripoll et al., 2013; deRamón et al., 2015; Hueso et al., 2016; Hueso et al., 2019; Karnell et al., 2019). CD40 is a 43-50 kDa transmembrane protein belonging to the tumor necrosis factor (TNF) receptor superfamily and is expressed on the surface of many immune cells. The interaction of CD40 with its ligand CD40L (CD154) induces the trimerization of CD40 and stimulates downstream signaling, including the NF-κB pathway that upregulates proinflammatory genes (Elgueta et al., 2009). Thus, inhibition of the CD40-CD40L dyad is a promising therapy, and a number of strategies and techniques have been designed to hinder its functionality, such as the administration of small molecule inhibitors of the interaction of CD40 with TRAF6 (Bosmans et al., 2020), liposome-loaded anti-CD40 antisense oligonucleotides (ASO) (Arranz et al., 2013), anti-CD40 siRNAs (Pluvinet et al., 2004; deRamón et al., 2015; Hueso et al., 2016), or monoclonal antibodies (Remer et al., 2017). Currently, there are three clinical trials testing the safety and efficacy of the anti-CD40 monoclonal antibody, CFZ533, to prevent acute rejection in renal (NCT03663335) or liver (NCT03781414) transplant patients and to evaluate its effects on the kidney in patients with lupus nephritis (NCT03610516). Other clinical trials under development use an anti-CD40L antibody (NCT03605927) to prevent acute Graft-Versus-Host-Disease (NCT03605927), or an oncolytic adenoviral vector that expresses an anti-CD40 antibody (NCT03852511) to treat advanced or metastatic tumors. Our group has developed a chemically stabilized, cholesterol-conjugated small inhibitory RNA molecule against murine CD40 and reported the evaluation of its potency, distribution, and durability of effects following systemic administration (Ripoll et al., 2013; deRamón et al., 2015, Hueso et al., 2016, Hueso et al., 2019). Here, we summarize a protocol describing the systemic administration of this specific anti-CD40 siRNA in different mouse models.
Materials and Reagents
Consumables
Disposable gloves
RNAase-free 1.5 ml polypropylene tubes
96-well plates
0.5 ml polypropylene tubes
50 ml Falcon conical tubes (Corning, catalog number: 45352054)
70 μm nylon cell strainers (BD Biosciences , catalog number: 45352350)
Cell culture plates
Syringes
Towels
Sample collection tubes
23-25 G needles
2 ml RNAase-free Eppendorf tubes (Merck KGaA, catalog number: T2795)
Poly-L-lysine-coated slides (Merck KGaA, catalog number: P0425)
Whatman No. 1 filter paper
Reagents and Kits
Nuclease-free water (DEPC-treated water)
Absolute isopropanol (Merck KGaA, catalog number: I9516)
Tris base (Tris-hydroxymethyl-aminomethane; Bio-Rad Lab, catalog number: 161-0719)
3 M sodium acetate (Merck KGaA, catalog number: S7899)
Bromophenol Blue-Xylene Cyanole Dye Solution (Merck KGaA, catalog number: B3269-5mL)
N,N,N’,N’-tetramethylethylenediamine, TEMED (Bio-Rad, Hercules, catalog number: 161-0800)
Amonium persulfate, APS (Bio-Rad, Hercules, catalog number: 161-0700)
Methanol (Merck KGaA, catalog number: 82762)
Absolute ethanol (Obtained from general chemical providers)
Hydrogen peroxide (Obtained from general chemical providers)
D+ sucrose (AppliChem GmbH, catalog number: 200-334-9)
Ethylenediaminetetraacetic (EDTA, Merck KGaA, catalog number: E6758)
RNase-ZAP (Merck KGaA, catalog number: R2020)
BHT (Butylated hydroxytoluene; Merck KGaA, catalog number: W218405)
FACS lysing solution (Becton Dickinson, catalog number: 349202)
Xylene (mixture of isomers, VWR International, catalog number: 28975.360)
DPX mounting medium (VWR International, catalog number: 360294H)
Jelly from porcine skin (Sigma-Aldrich, catalog number: G1890)
Oil-Red O (Merck KGaA, catalog number: O0625)
RPMI 1640 medium (Biological Industries)
Note: Reagents 1-20 were stored at room temperature.
Opti-MEM (Themo Fisher, catalog number: 51985034)
Fetal bovine serum (Lonza Pharma&Biotech, catalog number: 14-802F)
Albumin ELISA kit (Active Motif)
DEPC (Diethyl Pyrocarbonate; Merck KGaA, catalog number: D5758).
40% acrylamide/Bis 29:1 (Bio-Rad, Hercules, catalog number: 161-0146)
Propidium iodine (StemCell Technologies, catalog number: 75002)
TRIzol (ThermoFisher Scientific, catalog number: 15596026)
DAB substrate (3,3’Diaminobenzidine tetrahydrochloride hydrate, Sigma-Aldrich, catalog number: D5637-5G)
Flow cytometry staining buffer (ThermoFisher Scientific, catalog number: 00-4222)
100 mM Penicilin/Streptomycin (ThermoFisher Scientific, catalog number: 15070-063)
Note: Reagents 21-30 were stored at 4°C.
200 mM L-glutamine (ThermoFisher Scientific, catalog number: 25030-024)
High-Capacity cDNA Reverse Transcription kit (ThermoFisher Scientific, catalog number: 4368814)
PureLinkTMRNA Mini kit (ThermoFisher Scientific, catalog number: 12183020)
5’ RACE system for rapid amplification of cDNA ends kit (ThermoFisher Scientific)
Oligofectamine 2000 (ThermoFisher Scientific, catalog number: 12252011)
GM-CSF (R&D systems, catalog number: 215-GM)
Lipopolysaccharides (LPS) from E. coli Serotype O111:B4 (Sigma-Aldrich, catalog number: L4391)
GeneEraser Luciferase Suppression-Test System (Stratagene/Agilent, catalog number: 240192)
Luciferase Assay System (Promega, catalog number: E1531)
Polyfect transfection reagent (Qiagen, catalog number: 301105)
Serum normal goat (Merck KGaA, catalog number: G9023) or horse 20% (Merck KGaA, catalog number: H0146)
Note: Reagents 31-41 are stored at -20°C.
Taqman gene expression assays (ABI/ThermoFisher Scientific) (Table 1)
Table 1. Commercial TaqMan probes used in this study
Name
of the gene tested
Probe code1
Gene unique identifier2
Position3
CD40
Mm00441891_m1
NM_011611
121
CD40L
Mm00441911_m1
NM_011616
348
IL1b
Mm01336189_m1
NM_008361
55
NLRP3
Mm00840904_m1
NM_145827
499
Apelin (Apln)
Mm00443562_m1
NM_013912
414
C3
Mm01232779_m1
BC029976
517
CD55
Mm00438377_m1
NM_010016
1154
FOXP3
Mm00475165_m1
NM_001199347
1307
IL6
Mm99999064_m1
NM_031168
233
IL10
Mm00439614_m1
NM_010548
232
MCP1 (Ccl2)
Mm00441242_m1
NM_011333
165
RANTES (Ccl5)
Mm01302428_m1
NM_013653
245
TLR3
Mm00628112_m1
NM_126166
280
TLR4
Mm00445273_m1
NM_021297
112
TLR9
Mm00446193_m1
NM_031178
106
AIM2
Mm01295719_m1
NM_001013779
869
18S rRNA
Hs99999901_s1
X03205
604(1) Commercial code of the Taqman probes used (ThermoFisher Scientific).
(2) Refseq/Genbank unique identifiers of the genes tested.
(3) Base position contained within the probe.
0.1 M citrate buffer, pH 6 (see Recipes), stored at room temperature
0.01 M citrate buffer (see Recipes), stored at room temperature
Oil-Red working solution (see Recipes), stored at room temperature
Phosphate-buffered saline (PBS), pH 7.5 (see Recipes), stored at room temperature
PBS-Triton (PBST) (see Recipes), stored at room temperature
PS buffer (see Recipes), stored at room temperature
Complete culture medium (see Recipes), stored at 4°C
Secondary antibodies (for a 1:200 dilution) (see Recipes), keep at -20°C
Annealing buffer 10× (see Recipes)
DEPC-treated water (see Recipes)
1 M Tris-HCl, pH 7.5 (see Recipes)
12% acrylamide gel (see Recipes)
Non-denaturing gel loading buffer (see Recipes)
4% PFA solution in PBS (see Recipes)
0.3% Oil-Red O stock solution (see Recipes)
Serum normal goat or horse 20% (see Recipes)
Primary antibodies
In this work we used the following specific primary antibodies:
Rabbit polyclonal anti-CD40 antibody (C-20, Santa Cruz Biotech, catalog number: sc-975)
Rabbit polyclonal anti-CD154 antibody (H215, Santa Cruz Biotech, catalog number: sc-9097)
Mouse monoclonal anti-DC-SIGN antibody (1B10, Santa Cruz Biotech, catalog number: sc-23926)
Rabbit polyclonal anti-NF-κB p65 antibody (anti-phospho-S536, Abcam, catalog number: Ab86299)
Anti-F4/80 antibody (Hycult Biotech, catalog number: HM1066)
Rabbit anti-mouse collagen-IV antibody (Chemicon international, catalog number: AB756p)
Goat polyclonal anti-human C3c antibody conjugated to FITC (Nordic-MuBio, catalog number: GAHu/C3c/FITC)
Furthermore, the following antibodies from BD Biosciences (San Jose, CA, USA) were used for flow cytometry:Anti-CD11c antibody (clone HL3)
Anti-CD11b antibody (clone M1/70)
Anti-CD40 antibody (clone HM40-3)
Anti-CD80 antibody (clone 16-10A1)
Anti-CD86 antibody (clone GL1)
Note: All primary antibodies were stored at -20°C, unless another temperature was specifically stated by the manufacturer.
Secondary antibodies and histological reagents
The following secondary antibodies were used in this work:
Alexa 488-labeled chicken anti-goat (ThermoFisher Scientific, catalog number: AB_2535870)
Goat anti-rabbit (ThermoFisher Scientific, catalog number: AB_2633280)
Alexa 555-labeled goat anti-mouse (ThermoFisher Scientific, catalog number: AB_2535844)
Alexa 546-labeled goat anti-rabbit (ThermoFisher Scientific, catalog number: AB_2633280)
Unconjugated goat anti-rat (Novus Biologicals)
Biotinylated horse anti-goat (Vector Laboratories, catalog number: BA-9500)
FITC-conjugated goat anti-mouse (Merck KGaA, catalog number: F0257)
VECTASTAIN Elite ABC HRP kit (Vector Laboratories, catalog number: PK-6100)
Avidin/Biotin blocking kit (Vector Laboratories, catalog number: PK-4001-NB)
Note: All secondary antibodies were stored at 4°C, unless another temperature was specifically stated by the manufacturer.
The following histological reagents were used:Harris hematoxylin solution (Sigma-Aldrich, catalog number: HHS32-1L)
Eosin Y solution, alcoholic (Sigma-Aldrich, catalog number: HT110132-1L)
Periodic acid (Sigma-Aldrich, catalog number: P0430-25G)
Schiff reagent (Sigma-Aldrich, catalog number: 3952016-500ML)
PAS staining kit (Sigma-Aldrich, catalog number: 101646)
Oil Red-O reagent (Sigma-Aldrich, catalog number: O0625-100G)
Tissue Tec OCT inclusion compound (Sakura FinetekEurope)
UltraCruz Tm mounting medium (Santa Cruz Biotech, catalog number: sc-24941)
DRAQ5 (ThermoFisher Scientific, catalog number: 65-0880-92)
Notes:
Used for nuclear counterstaining.
All histological reagents were stored at room temperature.
Animals
Six-month-old NZB/NZW (F1) female mice (The Jackson Lab, Charles River, Wilmington, MA, USA)
Six to eight-week-old male ICR mice (The Jackson Lab, Charles River, Wilmington, MA, USA)
Eight-week-old ApoE-/- female mice (The Jackson Lab, Charles River, Wilmington, MA, USA)
Note: Animals were housed in a room maintained at a constant temperature and given free access to water and a standard laboratory diet. To accelerate atherosclerosis, ApoE-/- mice were fed a Western diet that contained 0.2% cholesterol and provided 42% of the energy as fat (TD.88137; Harlan-Tekland, Madison, WI, USA). Animals were euthanized by inhalation of isoflurane. Protocols were approved by the Ethics Committee for Animal Research of UB-Bellvitge, and experiments were performed in accordance with the European legislation on Laboratory Animal Experiments.
siRNA oligonucleotides
In this work, we used a CD40-specific ds-siRNA homologous to both the mouse and rat CD40 mRNA sequence (herein antiCD40-siRNAChol) and a scrambled sequence (s/s) ds-siRNA as the control (herein s/s-control siRNAChol). Both were modified by conjugating a cholesterol (chol) molecule to the 3’ end of the sense strand by means of a pyrrolidine linker.
The siRNA sequences were as follows:
Anti-CD40 sense strand: 5’-GUGUGUUACGUGCAGUGACUU-3’
Anti-CD40 antisense strand: 5’-GUCACUGCACGUAACACACTG-3’
(s/s), control siRNA sense strand: 5’-ACUACAAGACUCGUGACCAUU-3’
(s/s), control siRNA antisense strand: 5’-UGGUCACGAGUCUUGUAGUUU-3’
To determine the transfection efficiencies and organ distributions of the siRNAs, a cholesterol-conjugated and an unmodified anti-CD40 siRNA were labeled with Cy5.5.
Note: All oligonucleotides were obtained from Microsynth AG (Balgach, Switzerland) and stored at -20°C.
Cell lines
The highly transfectable human embryonic kidney 293FT cell line (ThermoFisher Scientific, catalog number: R70007)
Primary dendritic cells (obtained from the bone marrow of ICR mice)
Equipment
Cell scrapers (Sarstedt AG & CO, Nümbrecht, GE, catalog number: 83.3950)
Scalpels
Forceps: Straight, serrated-tip forceps; straight or curved, serrated-tip fine forceps; and straight fine-tip forceps
Scissors: Straight, blunt scissors; straight, sharp, fine scissors; and micro-dissecting spring scissors
Automatic pipettes (1-10 μl, 20-200 μl)
Hemocytometer
Water bath
Jasco V-650 spectrophotometer (Easton, MD, USA, used to determine the melting point of siRNA duplexes)
Olympum autoanalyzer AU400 (Hamburg, Germany, used to determine urinary protein and creatinine concentrations)
TaqMan real-time PCR ABI Prism® 7700 (ThermoFisher Scientific, used for the qPCR experiments)
BD FACS Canto II cytometer (BD Biosciences, used in the flow cytometry experiments)
Zeiss SteREOLumar V12 microscope (Carl Zeiss AG, used for microscopy experiments)
Leica TCS-SL spectral confocal microscope (Leica Camera AG, used for microscopy experiments)
TD-20/20 luminometer (Turner Designs, used for luciferase assays)
T-25 ULTRA-TURRAXTM homogenizer (IKA®-Werke GmbH & Co)
NanoDrop 2000c spectrophotometer (ThermoFisher Scientific)
Tabletop centrifuge (Eppendorf Cooled Centrifuge, mode: 5424R)
Cell culture CO2 incubator (NuAire Autoflow NU 8700)
Software
Leica confocal software (Leica Camera AG, Wetzlar, Germany)
Image analysis software ProgResCapturePro 2.7.7 (JenoptiK AG, Jena, GE)
ImageJ v1.48 (NIH, Bethesda, MD, USA)
ExpressionSuite software v1.0 or later (ABI, ThermoFisher Scientific, Waltham, MA, USA)
SDS software v2.4 (ABI, ThermoFisher Scientific, Waltham, MA, USA)
FACS DIVA software (BD Biosciences, San Jose, CA, USA)
Procedure
Synthesis and preparation of siRNAs
Requirements: Disposable gloves, heated water bath, microcentrifuge, automatic pipettes (1-10 μl, 20-200 μl), Jasco V-650 spectrophotometer, RNAase-free 1.5-ml polypropylene tubes, ice, nuclease-free water, annealing buffer (10 mM Tris pH 7.5, 20 mM NaCl), 3 M sodium acetate (pH 5.2), isopropanol, 70% ethanol, oligonucleotides.
Design of anti-CD40 oligonucleotides.
Analyze the target mRNA sequence (herein murine CD40, NCBI accession X60592.1) to select a number of sequences that comply with the following structure, 5’-GN17C-3’. Nine different sequences were generated to select an optimal target site (Pluvinet et al., 2004). General rules to improve the effectivity of siRNA silencing are: 1) presence of G/C at the 5’ end of the sense strand; 2) presence of A/U at the 5’ end of the antisense strand; 3) presence of at least 5 A/U residues in the first 7 bases of the 5’ end of the antisense strand; 4) no runs of more than 9 G/C residues should be allowed in the sequence (Ui-Tei el al., 2004); 5) secondary structure at the target site should be kept to a minimum (Bohula et al., 2003; Kretschmer-Kazemi Far and Sczakiel., 2003).
Annealing of oligonucleotides to generate ds-siRNAs.
For oligonucleotide annealing, mix equimolar amounts of complementary sense and antisense strands of the different anti-CD40 and control siRNAs by mixing:
Oligonucleotide 1/sense strand (100 pmol/µl)………………………………………10 µl
Oligonucleotide 2/antisense strand (100 pmol/µl)……..…...……………………10 µl
2x oligo annealing buffer (10 mM Tris, pH 7.5, 20 mM NaCl)…………………50 µl
Nuclease-free water (up to) ………………………………………..………………………100 µl
Heat for 3 min at 90°C in a water bath and switch it off.
Let the tubes cool down slowly in the water bath to room temperature (<60 min). The final concentration should be 100 pmol/µl ds-siRNA.
Store the annealed ds-siRNA oligonucleotides at 4°C for immediate use or at -20°C for longer storage.
Determination of the melting temperature (Tm) of the ds-siRNAs.
Heat samples in the Jasco V-650 spectrophotometer using a linear temperature ramp of 0.5°C/min. Melting temperature is obtained as the maximum of the first derivative. The melting point of the unmodified CD40 ds-siRNA is 79°C and that of the cholesterol-derived CD40 ds-siRNA (antiCD40-siRNAChol) is 71°C.
Alcohol precipitation of ds-siRNAs.
Add a 0.1 volume of 3 M sodium acetate (pH 5.2) and 1 volume of isopropanol, mix.
Keep on ice for 5 min and spin down at the top speed in a microcentrifuge for 10 min.
Carefully aspirate the supernatant, wash the pellet with 0.5 ml cold 70% ethanol, and carefully remove all ethanol.
Air-dry the pellet for no longer than 15 min at room temperature and resuspend the ds-siRNA in nuclease-free water in 2-5-times the original volume.
Quantitate the siRNA concentration using a NanoDrop or equivalent. The final concentration should be 20-50 pmol/µl.
Analyze the siRNA on a non-denaturing 12% polyacrylamide gel (see Recipes) for size and integrity, and store at -20°C or -70°C.
Mix up to 5 μl oligonucleotides with 2 μl loading buffer (see Recipes).
Load the sample on a non-denaturing 12% polyacrylamide gel and subject to electrophoresis at 200-250 V.
Stop electrophoresis when the Bromophenol Blue dye front has migrated two-thirds of the way down the gel.
Stain the gel for 2-5 min in a 1 μg/ml solution of ethidium bromide.
Soak the gel for 2-5 min in water.
Visualize the siRNA using a UV transilluminator.
The siRNA should migrate as a 21-22 bp band that runs slightly behind the Bromophenol Blue dye front. A second, less intense band running behind the primary siRNA band may be apparent and represents one partially digested strand of siRNA. The underdigested RNA strand is 27 nt in length and does not create any non-specific effects when used to transfect cells.
Determination of the silencing ability of anti-CD40-siRNAChol using the GeneEraser Luciferase Suppression-test system
Requirements: Disposable gloves, 96-well plates, automatic pipettes (1-10 μl, 20-200 μl), cell scrapers (Sarstedt AG & CO, Nümbrecht, GE, catalog number: 83.3950), microcentrifuge, ice, 0.5-ml polypropylene tubes, pTarget-luc-rCD40, Oligofectamine, HEK-293 cells, Polyfect transfection reagent, 70% ethanol, 1× phosphate-buffered saline (PBS), 1× lysis reagent, luciferase assay reagent, TD-20/20 luminometer.
Generate the apTarget-luc-CD40 plasmid.
A blunt-end 506 bp fragment corresponding to the partial rat CD40 cDNA sequence (coding sequence of nucleotides 41-547; GenBank Acc. No AF241231) was blunt-end cloned into the plasmid pTarget-luc at the 3'UTR of the luciferase gene.
Transfect HEK-293 cells with siRNAs (100 nM).
Prepare a 100 nM solution of the ds-siRNAs, transfect confluent HEK-293 cells using Oligofectamine according to the manufacturer’s instructions.
After 6 h, transfect the above cells with 400 ng pTarget-luc-CD40 using Polyfect transfection reagent according to the manufacturer’s instructions. Normalize the transfection efficiency by co-transfecting 500 ng pCMV-bGal.
Incubate the cells for 48 h at 37°C in a humidified atmosphere with 5% CO2.
Prepare cell lysates for analysis.
Remove the growth medium from the cultured cells.
Rinse the cells in 1× PBS and remove as much as possible.
In a 96-well plate, dispense 20 µl/well 1× lysis reagent (1× lysis reagent is prepared by adding 4 vol. water to 1 vol. 5× lysis reagent from the kit).
Scrape the cells from the dish and transfer the solution to a microcentrifuge tube.
Pellet the cell debris by brief centrifugation and transfer the supernatant to a new tube.
Mix 20 µl cell lysate with 100 µl luciferase assay reagent and measure the light in the TD-20/20 luminometer.
Bone marrow-derived dendritic cell (DC) extraction, culture, activation, and transfection with siRNAs in vitro
Requirements: Disposable gloves, 50-ml Falcon conical tubes (Corning, catalog number: 45352054), 70-μm nylon cell strainers (BD Biosciences, catalog number: 45352350), autoclaved materials (forceps, scalpels, scissors), cell culture hood, ice, mice femurs and tibiae, hemocytometer, cell culture plates, sample collection tubes, Oligofectamine 2000, BD FACS lysing solution, complete RPMI 1640 medium (RPMI 1640 medium supplemented with 100 U/ml penicillin, 100 µg/ml streptomycin, 2 µM L-glutamine, 10% heat-inactivated FBS, and 20 ng/ml GM-CSF), Opti-MEM medium, propidium iodine (stock solution: 1.5 mM), siRNAs, LPS, cytometer.
Euthanize ICR mice according to the institutional guidelines and extract the femurs and tibiae.
Place each mouse onto a sterile surgical pad in a sterile hood.
Spray the mouse with 70% ethanol to avoid contamination of the samples.
Soak the femurs and tibiae in RPMI 1640 medium.
Harvest the bone marrow (BMC).
Note: All work should be performed under sterile conditions in a cell culture hood.
Cut the femurs at both ends with sterile scissors.
Transfer the bones to RPMI 1640 in a sterile Petri dish.
Vigorously flush the inside of the bones with 1 ml ice-cold RPMI 1640 medium using a 1-ml pipette. Flush 2-3 times until the bones are completely white.
Flush the bone marrow out onto a 70-μm nylon cell strainer placed in a 50-ml Falcon conical tube.
Smash the bone marrow through the cell strainer using a 5-ml plunger, and wash the strainer with 5 ml RPMI.
Centrifuge the cells at 250 × g for 8 min at 4°C and discard the supernatant.
Resuspend the cell pellet in 1 ml RBC lysis buffer and incubate for 5 min at room temperature.
Neutralize the lysis buffer by adding 5 ml FBS.
Centrifuge the cells at 250 × g for 8 min at 4°C, discard the supernatant, and resuspend in 5 ml RPMI 1640 medium. Take out an aliquot of cell suspension to count.
Determine the total number of cells obtained.
a. Dilute 10 μl bone marrow suspension in 90 μl 1× BD FACS lysing solution (see above).
b. Count 10 μl cell suspension using a hemocytometer.
Grow 5 × 106 cells.
Grow cells in 5 ml complete RPMI 1640 medium (see above) supplemented with 10 ng/ml IL-4 and 20 ng/ml GM-CSF in T25 flasks at 37°C in a humidified atmosphere with 5% CO2.
After 24 h, some of the cells adhere to the plate but many others are suspended in the culture medium. Colonies appear after 72 h. The number of adherent cells gradually decreases over time and on day 5, the suspended cells with dendritic protrusions gradually increase. On day 7, the suspended cells begin to aggregate and the protrusions elongate.
Change the culture medium on days 3 and 5.
Remove the medium carefully so as not to disturb the growing cells.
Briefly centrifuge (250 × g for 8 min) the removed volume of media since DCs are only loosely adherent. Resuspend the cell pellet in the same flask with 10 ml fresh RPMI 1640 supplemented with 20 ng/ml GM-CSF.
Harvest BMDCs (day 8)
Collect non-adherent cells by gently pipetting the culture medium, and transfer into sterile 50-ml centrifuge tubes. Discard the adherent cells that include macrophages.
Loosely adherent BMDCs become easily dislodged into suspension by this process, while macrophages remain adhered to the Petri dish.
Count the total number of cells obtained.
Transfect with siRNAs on day 8.
Incubate 1 × 106 immature BMDCs/ml with 2 µM unmodified siRNA-Cy5.5 or Chol-siRNA-Cy5.5 (see Section E of the Materials and Reagents section, above) using the cationic lipid Oligofectamine 2000 in Opti-MEM medium on 6-cm dishes.
Treat immature BMDCs with 100 ng/ml LPS on day 9 for 12 h.
Harvest BMDCs
Collect all the cells (detach cells with a scraper).
Centrifuge the cells at 250 × g for 8 min.
Remove the supernatant and resuspend the cell pellet in 5 ml PBS (wash 1)
Centrifuge the cells at 250 × g for 8 min.
Remove the supernatant and resuspend the cell pellet in 5 ml PBS (wash 2)
Centrifuge the cells at 250 × g for 8 min.
Resuspend the cell pellet in 1 ml RPMI 1640 supplemented with 10% FBS and 20 mM penicillin/streptomycin.
Count the cells.
Stain the DCs with a final concentration of ≤1 μg/ml propidium iodide (PI).
Prepare a 1 mg/ml (1.5 M) stock solution by dissolving solid PI in PBS. Protect from prolonged exposure to light.
Whenever possible, prepare and use the stock solution on the same day. If the stock solution must be made in advance, aliquot and store in tightly sealed vials at -20°C (generally stable for up to 1 month).
Resuspend 50 μl cell suspension in 50 μl flow cytometry staining buffer.
Add 5 μl PI staining solution per 100 μl cell suspension. Do not wash the cells after the addition of PI.
Incubate for 5–15 min on ice in the dark.
Analyze the samples immediately by flow cytometry.
Characterize the cell phenotype by flow cytometry.
Block non-specific Fc-mediated interactions with 0.5 μg anti-mouse CD16/CD32 per 100 μl for 15 min at 4°C before staining.
Antibody-binding kinetics are temperature dependent. Staining on ice may require longer incubation times.
Aliquot 50 μl cell suspension to each tube (one per antibody).
Combine the cell suspension with primary antibodies (the isotype control and the following antibodies: anti-CD11c (clone HL3), anti-CD11b (clone M1/70), anti-CD40 (clone HM40-3), anti-CD80 (clone 16-10A1), and anti-CD86 (clone GL1)) and the appropriate volume of flow cytometry staining buffer to reach a final staining volume of 100 μl. Vortex.
Incubate for at least 30 min on ice. Protect from light.
Wash the cells by adding 2 ml flow cytometry staining buffer to each tube.
Repeat the wash step twice by centrifuging the cells at 250 × g for 8 min at room temperature.
Remove the supernatant and resuspend the pellet in 100 μl flow cytometry staining buffer.
Transfer the cell suspension to polypropylene tubes for flow cytometry.
For storage of the samples before analysis, add 100 μl IC fixation buffer.
Analyze the cells according to fluorescence intensity of the above markers and acquire representative images for subsequent analysis/quantitation.
Evaluation of the silencing activity of antiCD40-siRNAChol in in vivo mouse models
Requirements: Gloves, scalpels, scissors, syringes, towels, cotton, sample collection tubes, 23–25 G needles, mice, siRNAs, PS buffer (see Recipes), isoflurane, isoflurane chamber, portable liquid nitrogen container, liquid nitrogen, and blood collection tubes.
Separate the mice into treatment and control groups.
Study the in vivo effect of the systemic administration of chol-siRNAs: Use 6-8-week old male ICR mice (4 mice per group). Administer 50 µg antiCD40-siRNAChol or s/s-control siRNAChol via i.p. injection (10 ml/kg in 0.2-µm-filtered PBS). At predefined time points (days 0, 1, 3, 5, 7, and 9), administer single doses of 5 µg LPS from E. coli via i.p. injection. Euthanize animals 4 h post-administration of LPS.
For the lupus nephritis study with 5-month-old NZB/NZW F1, set up the following groups:
CYP: Administer 50 mg/kg i.p. every 10 days (n = 9).
CTLA4: Administer 50 μg abatacept (Orencia, Bristol Myers Squibb) three times weekly (n = 9).
siCD40-1w: Administer 50 μg anti-CD40-siRNAChol i.p. once weekly (n = 9).
siCD40-2w: Administer 50 μg anti-CD40-siRNAChol i.p. twice weekly (n = 9).
Control: Administer 50 μg s/s-control siRNAChol i.p. twice weekly (n = 15).
For the atherosclerosis study with 8-week-old female ApoE-/- mice. Treat the mice twice weekly i.p. with 50 μg anti-CD40-siRNAChol, s/s-control siRNAChol, or vehicle (5 mice per group). Euthanize the animals at 8 weeks (basal group), 10 weeks, 14 weeks, or 24 weeks.
Tissue collection: urine and venous blood.
Place the animals in individual metabolic cages with water and the usual diet, and collect 24 h urine specimens before the onset of treatment and thereafter at monthly intervals.
Note: If mice do not produce urine in the given time, repeat the procedure on the following day in a warmer room.
Centrifuge the urine samples at 500 × g for 10 min. Collect the urine and retain the sediment for further studies; store the urine and sediment at -20°C.
Determine the weight of the mice twice monthly.
On a monthly basis, perform a non-terminal venous blood collection from the tail vein (without replacement of fluids) using short-term anesthesia by isoflurane inhalation.
Important points to remember for animal comfort:
Make the animal comfortable by maintaining the temperature at 24-27°C.
DO NOT rub the tails from the base of the tip as this will result in leukocytosis. If the vein is not visible, dip the tail into warm water (40°C).
Insert a 23-25 G needle into the blood vessel and collect blood using a capillary tube or syringe with a needle. In case of difficulties, cut a minimal surface of the skin, prick the vein with a bleeding lancet or needle and collect blood with a capillary tube or a syringe with a needle.
DO NOT try to collect blood more than three times. The maximum collection volume should be 0.2 ml blood per mouse. The estimated blood volume in an adult animal is 55-70 ml/kg body weight (Parasuraman et al., 2010).
After completing blood collection, stop the bleeding using pressure.
Wash the restraint frequently to avoid pheromonal-induced stress or cross infection.
At the end of the study, extract arterial blood by cardiac puncture.
Perform terminal anesthesia of animals in an isoflurane chamber.
Open the chest (thoracotomy) and obtain a blood sample directly from the ventricle, taking care not to collapse the heart.
Tissue collection: liver and kidneys
Euthanize all mice by inhalation of isoflurane.
Dissect out the kidneys and liver through the abdomen and wash them by perfusing ice-cold 1× PBS via the left ventricle.
Resect the kidneys. One kidney will be used for histological evaluation and the other for total RNA extraction.
For histological analysis, remove the upper third of the kidney to ensure that both cortical and juxtamedullary glomeruli are present and fix the piece in 5 ml PS buffer at 4°C for 24-48 h.
For immunofluorescence analysis, place a third of the kidney piece (to ensure that both cortical and medullary glomeruli are present) into the tissue mold and coat in OCT. Place it onto dry ice to freeze and store at -80°C.
For protein and RNA extraction, place 3 × 2 mm3 pieces of kidney cortex and liver into 0.5 ml plastic tubes and snap freeze them in liquid N2. Store at -80°C. For long-term tissue storage for RNA extraction, place the tissue in five volumes of RNA stabilization reagent and store at -80°C.
RNA extraction and purification from kidney tissue.
Requirements: Disposable gloves, laboratory fume hood, RNase-free pipettes and pipette tips, sterile plasticware, cold TRIZolTM, rotor-stator homogenizer (ULTRA-TURRAXTM), ice, round-bottomed RNase-free tube for tissue homogenization, RNase-free water (water treated with DEPC, see Receips), tabletop centrifuge, 2 ml RNAase-free Eppendorf tubes, NanoDrop, PureLink RNA mini kit, chloroform, 70% ethanol (100% ethanol mixed with RNAse free water), vortex.
Add 1 ml cold TRIzol to 50-100 mg renal tissue in a round-bottomed RNase-free tube, homogenize on ice. Avoid foaming by keeping the tip of the probe submerged in the lysis solution while holding the tip against the tube wall.
Incubate for 5 min at room temperature to allow complete dissociation of the nucleoprotein complex.
Add 0.2 ml chloroform and vortex.
Incubate for 2-3 min at room temperature.
Centrifugate the sample for 15 min at 12,000 × g, 4°C.
The mixture separates into a lower red phenol-chloroform phase, an interphase, and a colorless upper aqueous phase. Carefully transfer the aqueous phase to a 2-ml Eppendorf tube (important: avoid transferring any of the interphase or organic layer).
Add one volume 70% ethanol and vortex.
Transfer ≤700 μl sample to the spin cartridge and purify total RNA according to the kit instructions (in this case the PureLink RNA mini kit).
Quantify the total RNA using the NanoDrop.
First strand DNA synthesis (reverse transcription).
Requirements: Ice, RNase-free water, tabletop centrifuge, Eppendorf tubes, High-Capacity cDNA Reverse Transcription kit, thermal cycler, 70% ethanol, vortex.
The reverse transcription reaction is performed according to the instructions of the kit used (in this case the High-Capacity cDNA Reverse Transcription) using 500 ng RNA.
Determine CD40 mRNA expression in the kidney and liver by qPCR.
Set up reactions with CD40 primers (Mn_00441891_m1; Taqman gene expression assays from ABI/ThermoFisher Scientific, Waltham, MA, USA).
Analysis of anti-CD40-siRNAChol-directed cleavage products by 5’RACE.
Requirements: Gloves, total RNA (from section C1), thermocycler, cDNA synthesis kit, 5’RACE kit.
Synthesize first strand cDNA from 5 μg total RNA (kidney) using the High-Capacity cDNA RT Kit with the gene-specific primer #1 (GSP1): 5’-GCCGACTGGGCAGGGATGACAGACG.
Ligate the adaptor oligonucleotide from the 5’RACE kit to the 5’ end of the cDNA.
Specifically amplify cleavage products using a primer complementary to the adaptor (5’RACE kit) and the nested gene-specific primer #2 (GSP2): GSP2: 5’-AGCCAGGGATACAGGGCGTGTGC. Use the following PCR program: 1 m, 94°C × 60 s and 35 cycles of 94°C × 30 s, 55°C × 30 s and 72°C × 60 s, followed by a final extension of 72°C × 5 min
Check for the presence of a 310 bp amplification product, corresponding to CD40 mRNA specifically cleaved by the anti-CD40 siRNA.
Use 20 μl PCR product for analysis on a 1% ethidium bromide-stained agarose gel by electrophoresis with a corresponding DNA molecular weight marker.
Histological evaluation of renal lesions in siRNA-treated animals.
In this section, we will prepare kidney samples for standard histological analysis (embedded in paraffin), immunofluorescence analysis (in OCT), and protein and RNA analysis (stored in RNA stabilization solution).
Requirements: One of the kidneys obtained in section D2, gloves, ice-cold 1× PBS, tissue molds and cassettes, forceps, paraffin, paraffin dispenser, cold plate, PS buffer (see Recipes), 15% sucrose, 4% paraformaldehyde (PFA), OCT, dry ice, RNA stabilization reagent, high-density polyethylene (HDPE), 50% ethanol, 70% ethanol, 96% ethanol, absolute ethanol, deionized water (MilliQ or similar), microtome.
For standard histological analysis.
Remove the upper third of a kidney to ensure that both cortical and juxtamedullary glomeruli are present and fix the piece in 5 ml PS buffer at 4°C for 24-48 h. Place the tissue in a labeled cassette (use a pencil, as solvents will dissolve the ink) and prepare for paraffin embedding.
Dip cassettes in a wide-mouth high-density polyethylene (HDPE) 1 L jar filled with 15% sucrose for 24 h before embedding in paraffin blocks.
Dip in 50% ethanol for 2-3 days.
Dip in 70% ethanol for a minimum of 3 h (but can be maintained for up to 7 days).
Dip in 96% ethanol overnight.
Dip in absolute ethanol for a minimum of 3 h.
Wash with clearing agent (xylene) for 1 h at room temperature.
Perform a first paraffin wax at 60°C overnight.
Perform a second paraffin wax at 60°C for a minimum of 3 h.
Place a small amount of molten paraffin in the mold (dispense from a paraffin reservoir). Use warm forceps, transfer the tissue into the mold cut side down, as it was placed in the cassette.
Transfer the mold to a cold plate and gently press the tissue flat. Paraffin will solidify in a thin layer that holds the tissue position.
When the tissue is in the desired orientation, add the labeled tissue cassette to the top of the mold as a backing. Press firmly.
Hot paraffin is added to the mold from the paraffin dispenser. Be sure that there is enough paraffin to cover the face of the plastic cassette. If necessary, fill the cassette with paraffin while cooling, keeping the mold full until solid.
Paraffin solidifies in 30 min. The paraffin block can then be removed and sectioned. If the wax cracks or the tissues are not well aligned, melt them again and start over. Tissue blocks can be stored at room temperature for years.
Tissues are sectioned using a microtome (Leica RM 2155).
Turn on the water bath and check that the temperature is 45°C. Use 1 L fresh deionized water and add 1 g jelly from porcine skin (G1890, Sigma-Aldrich, Sant Louis, MO, USA). Place a fresh blade on the microtome (blades may be used to section up to 10 blocks, but replace if sectioning becomes problematic). The blade should be angled at 5°. Blocks to be sectioned are placed face down for about 15 min on an ice block (cold wax allows thinner sections). After cutting, use forceps to pick up the ribbons of sections and float them on the surface of the water in the water bath to allow for expansion of the sample.
Insert the block into the microtome with the wax block facing the blade, and align the vertical plane. Set the dial to cut 10-μm sections in order to plane the block; once cutting smoothly, set to 2–3 μm-thick sections. Face the block by cutting it down to the desired tissue plane and discard the paraffin ribbon.
If the block is ribboning well, cut another four sections, pick them up with forceps or a fine paint brush, and float them on the surface of the water in the 45°C water bath. Float the sections onto the surface of clean glass slides.
If the block is not ribboning well, place it back on the ice block to cool for a longer period to harden the wax.
If the specimens are fragmented when placed in the water bath, then it may be too hot.
Place the slides on the warming block in a 60°C oven for 10 min (so the wax starts to melt) to bond the tissue to the glass. Slides can be stored overnight at room temperature.
For immunofluorescence analysis.
Place the other pole of the kidney into a tissue mold, coat in OCT, and place it onto dry ice to freeze. Store at -80°C.
Place a small amount of molten OCT (tissue freezing medium, Leica Ref = 14020108926, Leica Biosystems, Richmond, IL) in the mold.
Using forceps, transfer the tissue to the mold, cut side down.
Pour liquid N2 into a polystyrene box containing a 50-ml tube rack (do not submerge the tube rack in the liquid N2). Place the OCT mold on the tube rack and freeze using the vapor.
Wrap with foil and snap freeze in liquid N2 until storage at -80°C.
For protein and RNA extraction.
Place 3 × 2 mm3 pieces of kidney cortex into 0.5-ml plastic tubes and snap freeze in liquid N2. Store at -80°C. For long-term tissue storage for RNA extraction, place the tissue in five volumes of RNA stabilization reagent and store at -80°C.
Direct immunofluorescence analysis of IgG and C3 deposits in the kidney.
Requirements: OCT-embedded kidney samples, gloves, ice-cold 1× PBS, cryostat, poly-L-lysine-coated slides (Merck KGaA, catalog number: P0425) or FLEX IHC microscope slides (Agilent, Ref. K8020, Santa Clara, CA, USA), 4% PFA (see Recipes), acetone, blocking solution, conjugated primary antibodies (FITC-conjugated goat anti-mouse IgG, FITC-conjugated goat anti-mouse C3), UltraCruz Tm mounting medium, and DRAQ5.
Prepare the kidney slides for immunofluorescence analysis.
Place the OCT compound mold containing frozen kidney (section D3) at -20°C for 2 h prior to sectioning. Ensure that the [cut] surface of the kidney is carefully placed against the bottom of the OCT mold to enable well-oriented tissue sections.
Use a cryostat to cut 5 µm-thick sections and place them on the surface of poly-L-lysine-coated slides or FLEX IHC microscope slides. Ensure that there are no folds or holes in the tissue section, which can distort tissue morphology. As positive IF controls, use samples of human parotid glands or ganglions; and as negative controls, omit the primary antibodies from the staining process. For the isotypic control, use an irrelevant immunoglobulin of the same isotype, species, and concentration as the primary antibody.
Upon removal from the cryostat, keep at room temperature for 20 min and then fix the slides by submerging into ice-cold pure acetone for 20 min.
Wash the slides 3 times with 100 µl PBS for 10 min each.
Wash the slides twice with distilled water for 5 min each.
Incubate the slides in blocking solution (20% normal goat serum in 1× PBST + 0.2% jelly from porcine skin) overnight at 4°C.
Wash the slides 3 times with 100 µl PBS for 10 min each.
Incubate the sections with primary antibodies (FITC-conjugated goat anti-mouse IgG at 1:300, and FITC-conjugated goat anti-mouse C3 at 1:50) in PBST-jelly buffer containing 1% normal serum. Incubate the slides for 1 h at room temperature in a humidified chamber to avoid the tissue drying out, which would lead to non-specific binding and high background staining.
Wash the slides 3 times in PBS for 5 min each. Primary antibodies can be saved for subsequent experiments.
Mount the samples with a drop of mounting medium containing 1 µg/ml DRAQ5.
Analysis of CD40 expression in the kidney by immunostaining with horseradish peroxidase (HRP).
Requirements: Gloves, paraffin blocks, pressure cooker, timer, 1× PBS, 0.01 M citrate buffer pH 6, 1% Triton X-100 in PBS, 0.1% fish jelly, bovine serum albumin (BSA), xylene, absolute ethanol, primary antibodies (optimal dilutions and incubation times should be determined for each primary antibody prior to use), biotinylated secondary antibody (135 μl normal serum + 45 μl biotinylated secondary antibody from the ABC staining kit in 10 ml PBS), humidified chamber, ABC (avidin-biotin complex) peroxidase standard staining kit (prepare reagent 30 min before use), DPX mounting medium, DAB substrate.
Controls: Prepare a negative control with diluent alone (without antibodies) and a positive control with a tissue known to contain the antigen of interest.
Prepare kidney slides for HRP analysis.
Deparaffinize the samples from section D5 by performing standard xylol-ethanol washes.
Wash the slides 3 times in 1% PBS for 5 min each.
Block endogenous peroxidase activity with a methanol wash (30% methanol in PBS + 1% peroxide hydrogen) for 10 min.
Note: Place the slides on a flat surface. Do not allow the slides to touch each other. Do not allow the sections to dry out.
Wash three times in 1% PBS for 5 min each.
Facilitate antigen retrieval by heating the samples for 5 min in a pressure cooker (on a plastic rack for slides) containing 10 mM citrate buffer, pH 6. This will break protein cross-links after formalin fixation.
Allow to cool slowly at room temperature for 30 min.
Wash twice in 1% Triton X-100 in PBS (PBST) for 5 min each.
To minimize cross-reactivity and reduce non-specific binding caused by hydrophobic interactions, pre-incubate with 20% normal goat (NGS) or horse serum in PBS-Triton + 0.2% jelly at 4°C for 2 h. Remove excess fluid from the slides using a brisk motion and carefully wipe each slide around the sections.
Incubate with primary antibody (anti-CD40 at 1/100, anti-DC-SIGN at 1/50; anti-NF-κB at 1/1,000) in 1% normal goat serum. Apply 100 µl to each slide, covering the tissue sections; tilt each slide from side to side. Incubate in a humidity chamber overnight at 4°C.
Keep at room temperature for 30 min.
Wash 3 times in 1× PBST for 5 min each.
Add 100 µl biotinylated secondary antibodies (at 1/200 + 1% NGS from the Vectastain ABC kit in PBST-jelly). Incubate in the humidified chamber for at least 45 min at room temperature. During this step, it is recommended to prepare the substrate mixture (reagent A = avidin + Reagent B = biotinylated HRP).
Wash 3 times in PBST for 5 min each. Wash well to remove traces of sodium azide as this will inhibit peroxidase activity when developing.
Add the substrate mixture at 1/100 and incubate for 45 min.
Wash 3 times in PBST for 5 min each.
Wash 3 times in PBS for 5 min each.
Incubate the tissue sections with 50 µl Vector DAB substrate in the dark for 3 min or until the desired color reaction is observed when monitored under the microscope. Terminate the reaction before background staining appears in the negative controls by rinsing gently with distilled water from a wash bottle. Discharge DAB in the corresponding recipient. All plasticware that come into contact with DAB must be treated with diluted bleach.
Wash in running tap water for 5 min.
Wash in distilled water.
Counterstain with hematoxylin:
Add hematoxylin (1 vol. hematoxylin solution + 2 vol. distilled water).
Wash with tap water for 5 min.
Wash in distilled water.
Wash with 70% ethanol for 5 min.
Perform three washes with 96% ethanol for 5 min each.
Perform three washes with absolute ethanol for 5 min each.
Perform three washes with xylene for 5 min each.
Mount with DPX.
Microscope examination:
Sections should be independently examined by two blinded pathologists, and at least 10 fields from each kidney section should be studied at high magnification.
Determine the extent of renal damage. Assess typical glomerular active lesions of lupus nephritis: mesangial expansion, endocapillary proliferation, glomerular deposits, extracapillary proliferation, and interstitial infiltrates; as well as tubule-interstitial chronic lesions: tubular atrophy and interstitial fibrosis using a Zeiss SteREOLumar V12 microscope from Carl Zeiss AG (Oberkochen, Germany). Grade the lesions semi-quantitatively using a scoring system from 0 to 3 (0 = no changes, 1 = mild, 2 = moderate, and 3 = severe changes).
Quantitate the number of positive cells for each of the markers studied using semi-quantitative evaluation of expression from 0 to 4 (0 = no staining, 1 = staining in <25% of the sample, 2 = staining in 25-50%, 3 = staining in 50–75%, and 4 = staining in 75-100%) in the different compartments of the kidney (glomeruli, vessels, and interstitium). NFκB p65 immunostaining was considered positive when located inside the nuclei.
Evaluation of atherosclerotic lesions in mouse aortas.
Requirements: ApoE-/- mice, isoflurane chamber, dissection tools (scissors: straight blunt-ended scissors, straight sharp fine scissors, and micro-dissecting spring scissors; tissue forceps: straight serrated-tip forceps, straight or curved serrated-tip fine forceps, and straight fine-tip forceps), syringes, 23 G and 27 G needles, tubes (one EDTA-coated tube, a serum tube, and two lithium heparin-coated tubes), ice-cold PBS, deionized water, RNAzap, 70% ethanol, ice, liquid N2, microscope, cork bed, pinning bed, needles, scalpel, Oil Red O, minutien pins 0.1-mm, Petri dishes, 6× magnification microscope.
Check the extension of ATH lesions in the entire aorta length.
Euthanize mice with isoflurane.
Spray each mouse with 70% ethanol to avoid contamination of the samples.
Excise whole aortas (as modified from Centa et al., 2019) and prepare samples for microscope analysis:
Make a midline incision with scissors from the jugular notch to the pubic bone.
Exsanguinate the mouse by cardiac puncture through the thorax wall (use a 23 G needle). This procedure usually yields 500 μl blood from a 20-week-old mouse. Collect 4 tubes (one EDTA-coated tube, a serum tube, and two lithium heparin-coated tubes). Keep them at room temperature.
Open the abdominal cavity. Cut the parietal peritoneum with scissors in the midline and laterally on both sides.
Open the diaphragm and the chest cavity (cut the rib cage as laterally as possible).
Make an incision in the right auricle for perfusion fluid drainage. Insert a 27 G needle through the apex of the heart in the cranial direction. Keep the needle fixed in the left ventricle while slowly perfusing with 10 ml ice-cold PBS over a minimum of 2 min.
Dissect the liver, spleen, and kidneys.
Cut the trachea and oesophagus on the right side of the heart without damaging the aortic arch. Cut the diaphragm and structures attaching the viscera to the retroperitoneum. Leave the heart, aorta, and kidneys in situ. Fold away the lungs and viscera caudally and cover with a napkin to begin retroperitoneal microdissection of the abdominal aorta (perform microdissection under a microscope at 6× magnification).
Dissect the aortic bifurcation (lift the surrounding tissue with forceps and cut under tension with scissors). Dissect the abdominal aorta cranially. Cut the abdominal branches from the aorta and free the aorta proximally through the aortic hiatus in the diaphragm.
Remove the adipose tissue covering the thoracic aorta. Dissect dorsally of the thymus (carefully) to free the aortic arch with branches. Continue dissecting the carotid arteries as distally as possible in the thoracic cavity. Neck dissection should include carotid bifurcation.
Clean the instruments by sequential rinses in deionized water, RNase decontamination solution, 70% ethanol, and PBS before cutting the aorta.
Lift the heart by the apex with the forceps. Cut the aorta close to the heart and place the whole heart in a tube with PBS. The heart may be stored on ice for a couple of hours before continuing with processing and cryo-mounting of the aortic root.
Cut the aortic arch and place half in a tube containing 1 ml 4% formaldehyde overnight at 4°C.
Dissect the remaining descending aorta, place in a tube, and snap freeze in liquid N2 for RNA analysis.
Place the heart on a cork bed with the ventral side facing up. Fix the heart to the cork with a needle through the apex. Hold the base of the heart with anatomical forceps. Cut away the apical 2/3 of the heart between the two auricles with a scalpel (angled 20° caudally in the sagittal plane and 20° cranially in the transversal plane).
Embed the aortic root in OCT compound (see section F). Store the specimens in zip lock bags at 80°C until cryo-sectioning.
“En face” analysis of the aortic arch and braquiocephalic artery.
Clean the aorta from the remaining periadventitial adipose tissue under a microscope (since Oil Red O stains most hydrophobic and neutral lipids such as triglycerides, diacylglycerols, and cholesterol esters, it is crucial to remove all such tissue at this point). Use scissors and forceps without manipulating or damaging the aorta. Always keep the aorta moist by applying additional PBS when needed.
Place the cleaned and fixed aorta in a 1.5-ml tube (one aorta per tube).
Add 1 ml 78% methanol to each tube and place on a tilted roller with gentle movement for 5 min. Replace the methanol solution and repeat this step twice.
Discard the methanol and add 1 ml fresh Oil Red O.
Incubate the tube on the tilted roller for 50-60 min.
Transfer the aorta to a clean tube and wash twice with 1 ml 78% methanol for 5 min each on the tilter roller.
Discard the methanol and refill the tube with 1 ml PBS.
Note: If necessary, at this step, aortas can be stored at 4°C.
Prepare a pinning bed: Place a sheet of 25 × 25 mm paraffin-wax film, wrapped with black electrical insulation tape, on a cork bed to make a dark background for the aorta. Place a label on the backside of the pinning bed and use a lead pencil to write the mouse identification number (normal pen ink will disappear in the staining process).
Transfer the fixed aortic arch to the pinning bed and place a drop of PBS on top.
Remove any small stained remnants of adventitial fat (carefully by microscope examination).
Cut open the aorta longitudinally to expose the intimal surface. Introduce the tips of a pair of micro-dissection spring scissors into the artery lumen and cut the outer curvature of the aortic arch from the ascending arch to the left subclavian artery. Begin to cut the outer curvature of the ascending arc in the distal direction and continue to cut open the branches, including the brachiocephalic artery. Continue to cut along the length of the thoracic aorta.
Cut open the lesser curvature and fold open the aorta to display the intimal surface.
Pin the open arch to the pinning bed using the blunt end of minutien insect pins. Gently bend the pins away from the specimen when in place. Pin the aorta flat on the bed without stretching the specimen. Store the pinned arch facing downwards in a Petri dish filled with PBS at 4°C.
Acquire images with a Zeiss steREOLumar V12 microscope connected to a RGB camera (ProgRes C F scan). Place a ruler next to the aorta for calibration of the image.
Quantitate the extension of atherosclerotic plaques.
Use image analysis software (ImageJ) to determine the lesion area and total intima surface. Lesion quantitation should be performed in a blinded fashion, and it is advisable that a second investigator confirms the results.
Calculate the total arch area: In ImageJ, select the “polygon selection” tool and encircle the total arch area by repetitive clicks. Select “measure” in the analyze menu to display the total arch area in the result window.
Calculate the lesion-free area: In ImageJ, select the “freehand selection” tool and encircle all plaques (stained in an orange-red color) in the arch area while pressing the Alt key. Click “measure” in the analyze menu to display the lesion-free area in the result window.
Calculate the relative lesion area by subtracting the lesion-free from the total arch area and dividing the result by the total arch area.
Cryo-sectioning of the aortic root.
Set the cryostat temperature at -20°C and section thickness to 10 μm. Mount the OCT block containing the aortic root on the specimen holder with the ventricular tissue facing outward. While starting to cut, fine-tune the alignment of the section surface to be parallel to the specimen holder.
Remove the surrounding excess OCT to make it easier to collect the sections without folds. The aortic root should be positioned perpendicularly to the knife blade.
Collect initial control sections on ordinary microscope slides and discard. The first sections should only contain heart muscle tissue. Progress the sectioning by 200 μm. Collect a section and check the progress under a light microscope.
When getting closer to the left ventricle outflow tract, check every 100 μm under the microscope. When initial indications of a vessel wall are observed, slow down the pace to 50 μm. When the first aortic valve appears, this will be point zero for collecting sections. It can be difficult to see exactly when the valves appear, but an exact localization is crucial to perform comparations of lesions in the same region.
Tilt the specimen toward the point zero valve to align the section plane with the two other valves. This is crucial for obtaining true cross-sections of the aorta. Make a drawing of the aortic root, indicating the valves as they appear, and count every 10-μm section that is cut from the point zero onward. When a second aortic valve appears, slightly tilt the specimen again from the valve to align the specimen with the third valve. The distance from where the first aortic valve leaflet appeared to where all 3 of the aortic valve leaflets appeared together did not exceed 80 μm. The first 5-μm thick section with the 3 aortic valve leaflets was mounted onto a glass slide. The adjoining 8 sections, including the first, were collected one by one onto 8 glass slides and marked in order from 1 to 8. The 9th section was collected onto the first glass slide, the 10th onto the second glass slide, and so forth, until each of the 8 glass slides held 8 sections. The 8 frozen sections collected on each glass slide covered a distance of 400 μm.
Fix the sections collected for Oil Red O staining in 4% formaldehyde for 10 min and for immunohistochemistry or immunofluorescence with ice-cold pure acetone for 10 min. Dry at room temperature for 30 min. Store the sections at -20°C.
Capture the images directly using an RGB camera (ProgRes C F scan) attached to a light microscope (NIKON E800). Save high resolution images, preferably in tagged image file (TIFF) format.
Morphometric image analysis using ImageJ.
The lesion quantitation should be performed in a blinded fashion, and it is advisable that a second investigator confirms the obtained results.
Use the area quantitation feature in the image analysis software to define the total vessel area by encircling the external elastic lamina of the aortic vessel. In ImageJ, select the “polygon selection” tool and encircle the area by repetitive clicks. Then select “measure” in the analyze menu. The total vessel area is displayed in the result window.
Continue to quantitate the ATH lesions in the intimal layer of the vessel, defined by the internal elastic lamina and the luminal boundary. Usually, lesions on the aortic valves are excluded from measurement. In ImageJ, select the “freehand selection” tool and encircle all plaques while pressing the Alt key. Select “measure” in the analyze menu to display the lesion-free vessel area in the result window.
Calculate the relative lesion area by subtracting the lesion-free area from the total vessel area and dividing the result by the total vessel area. Calibrate the results in the image analysis software according to the magnification used in order to obtain the absolute lesion area in µm2.
Data analysis
The cholesterol-conjugated, specific anti-CD40 siRNA demonstrated efficient transfection efficiency in dendritic cells (DCs) without decreasing cell viability (evaluated by propidium iodide). Here, we demonstrate the inhibition of CD40 expression and the subcellular localization of the anti-CD40-siRNAChol in the cytoplasm of DCs at 45 min post-transfection (Ripoll et al., 2013). The specificity of siRNA cleavage of CD40 mRNA was confirmed by 5’RACE.
In the silencing experiments (Ripoll et al., 2013), we firstly matured DCs with LPS (confirmed by an increase in CD40, CD80, and CD86 cell surface expression as measured by flow cytometry) and transfected them with anti-CD40-siRNAChol, which caused a 35% decrease in CD40 expression as compared with the scrambled controls, as well as a significant reduction in the release of TNFα, MCP1, and IL6. In addition, LPS increased CD40 mRNA expression by almost 20-fold (in the kidney) and 35-fold (in the liver) as compared with control levels in a mouse model. CD40 mRNA expression returned to baseline after 24 h in the kidney and after 48 h in the liver. A single anti-CD40-siRNAChol administration reduced renal and hepatic CD40 mRNA expression by 65% and 60%, respectively, for 3 days as compared with control values, with the effects persisting for up to 5 days.
In the lupus nephritis model (Ripoll et al., 2013), Cy5.5-labeled anti-CD40-siRNAChol was localized in tubular cells from the kidney (Figure 1). In this disease model, we studied the inhibitory effects of anti-CD40-siRNAChol on animal survival, renal function, inflammatory mediators, anti-DNA antibody levels, and renal lesions. For these experiments, mice were distributed into one of five groups subjected to different treatments:
CYP group (n = 9): 50 mg/kg i.p. CYP every 10 days
CTLA4 group (n = 9): 50 µg i.p CTLA4 (ORENCIA, Abatacept, Bristol Myers Squibb, Uxbridge, UK) three times a week
Anti-CD40-siRNAChol-1w group (n = 9): 50 µg i.p. anti-CD40-siRNAChol once a week
Anti-CD40-siRNAChol-2w group (n = 8): 50 µg i.p. anti-CD40-siRNAChol twice a week
Control group (n = 15): 50 µg i.p. ss-control siRNAChol twice a week
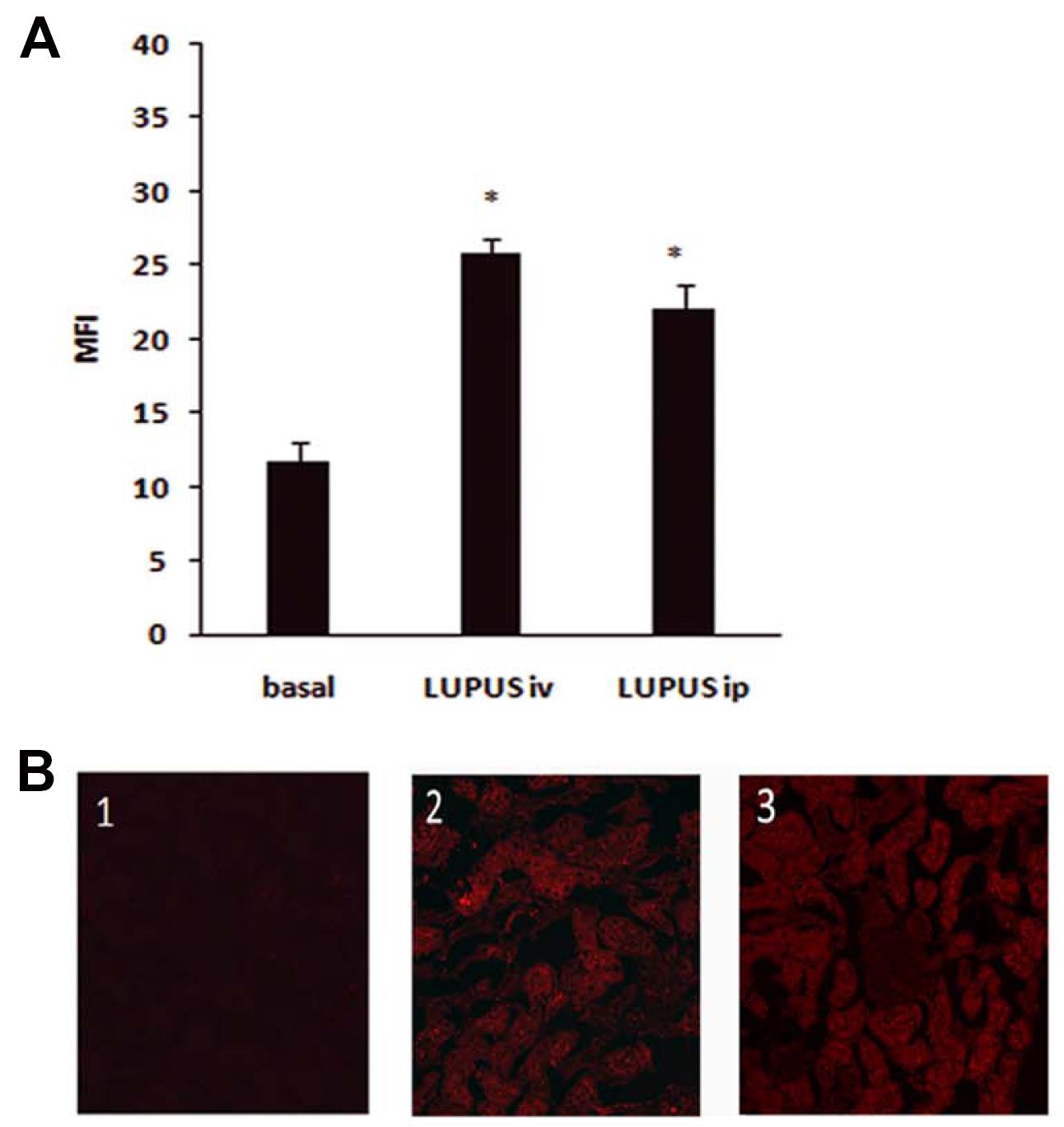
Figure 1. In the lupus nephritis model, Cy5.5-labeled anti-CD40-siRNAChol was localized in tubular cells from the kidney. A. Quantitation of renal internalization of Chol-siRNA administered i.v./i.p. in lupus mice. B. Representative kidney photomicrographs (×400) of: 1) basal autofluorescence; 2) i.v. administration; and 3) i.p. administration. Data are expressed as the mean ± SEM of four separate experiments. *P <0.05 vs. basal. doi:10.1371/journal.pone.0065068.g005.
Animal survival (by the Kaplan-Meier method) was 100% for the CYP, CTLA4, and CD40-2w groups; 88% for the CD40-siRNA-1w group, and 73% for the untreated group at the end of follow-up. Mice treated with siCD40-2w showed no increase in proteinuria or albuminuria and displayed a dose-dependent reduction in total IgG, anti-dsDNA antibodies, and all IgG fractions. The levels of IgM, IgA, and IgE, as well as pro-inflammatory cytokines such IL2, TNF, IFNγ, MCP1, and IL6, were also reduced in the anti-CD40-siRNAChol-2w group. Histological lesions were graded semi-quantitatively using a scoring system from 0 to 3 (Control = 8 ± 1.5; CYP = 4.3 ± 1.1; CTLA4 = 1.6 ± 0.7; anti-CD40-siRNAChol-1w = 3.8 ± 1.5; anti-CD40-siRNAChol-2w = 1.6 ± 0.6), and IgG and C3 glomerular deposits were reduced in all treated groups (Figures 2 and 3). The anti-CD40-siRNAChol-2w group showed an absence of extra-capillary proliferation, interstitial infiltrates, tubular atrophy, and interstitial fibrosis. Infiltrating CD3+ cells in the tubule-interstitial space were significantly reduced in all treatment groups except siCD40-1w. Furthermore, kidney expression levels of CD40, C3 (a manifestation of local complement synthesis), and pro-inflammatory cytokines were reduced in anti-CD40-siRNAChol-2w mice (Figure 4). In addition, a significant reduction in CD40 protein expression was observed in the interstitial, glomerular, and vascular compartments of the kidneys and in the circulation in the antiCD40-siRNAChol-2w group.
Finally, we used the CD40 silencing strategy to reduce the progression of atherosclerotic (ATH) lesions in ApoE-/- mice (Hueso et al., 2016). We distributed the mice into 8 treatment groups:
Basal B/8w (n = 5)
ss-control siRNAChol/10 w (n = 5)
ss-control siRNAChol/24w (n = 5)
Scrambled oligonucleotide control SC/14w (n = 5)
Anti-CD40-siRNAChol/10w (n = 5)
Anti-CD40-siRNAChol/14w (n = 5)
Anti-CD40-siRNAChol/24w (n = 10)
Vehicle (n = 5)
The “en face” analysis of whole aorta stained with Oil Red O confirmed that the number and extension of ATH plaque areas decreased in anti-CD40-siRNAChol/24w as compared with control values. Less F4/80 infiltrating macrophages were detected in the vessel walls from anti-CD40-siRNAChol/24w animals, suggesting a role for CD40 in the recruitment of macrophages to the plaque. Finally, less NF-κB+ cells were detected in the intima of anti-CD40-siRNAChol/24w mice, indicating that the protective effect of CD40 silencing may be mediated by NF-κB signaling. Since the strategy aimed to systemically silence CD40, a reduction in the splenic populations of CD3+CD40+ (T-lymphocytic) and CD11b+CD40+ (monocytic) cells was observed in the anti-CD40-siRNAChol/24w animals, suggesting that the reduction in atherosclerotic lesions may be associated with anti-inflammatory mechanisms in the vessel wall via systemic effects.
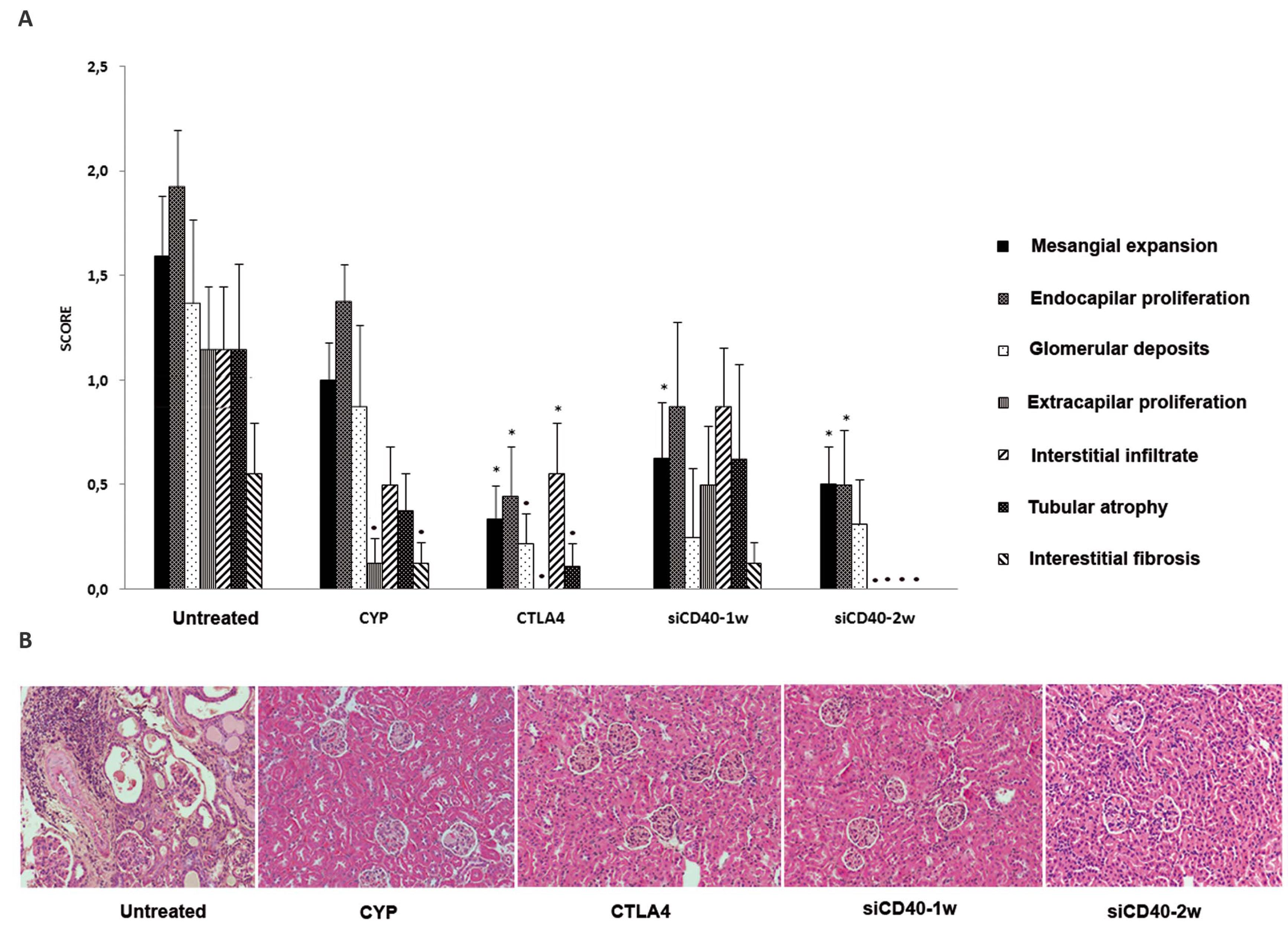
Figure 2. Histopathological lesions in the lupus nephritis model silenced by CD40 siRNA. A. Costimulatory blockade reduced the lesions in lupus nephritis. B. Representative photomicrograph (×200) of the renal histology from each group. Data are expressed as the mean ± SEM. *P < 0.05 vs. untreated, P < 0.01 vs. untreated. doi: 10.1371/journal.pone
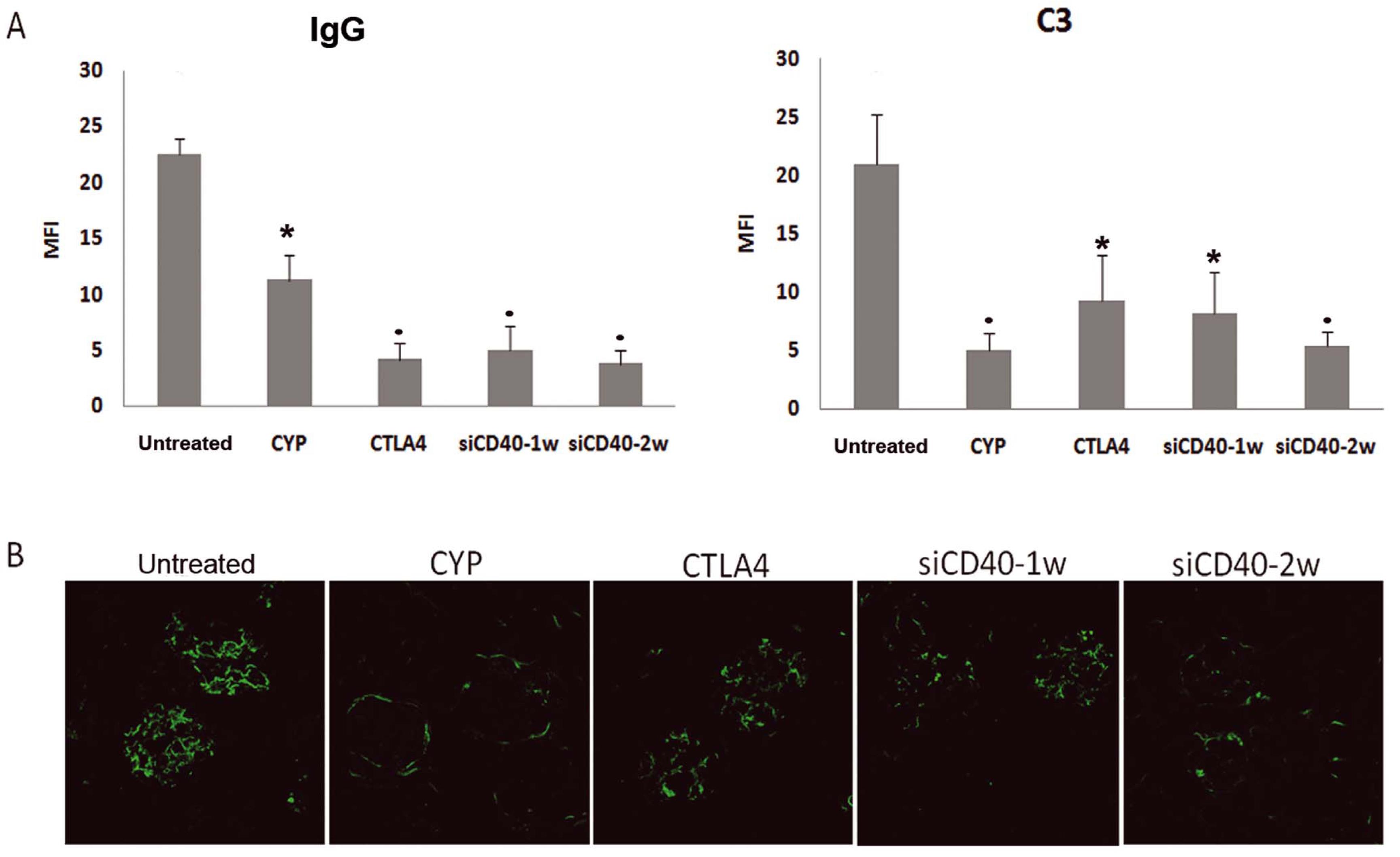
Figure 3. Immunohistochemical analysis of renal IgG and C3 in the lupus nephritis model silenced by CD40 siRNA. A. Deposits of renal IgG and C3 were quantitated by confocal microscopy (MFI). All treatments reduced glomerular deposits. B. Representative photomicrographs of C3 deposits (×630) for each group. Data are expressed as the mean ± SEM. *P < 0.05 vs. untreated, P < 0.01 vs. untreated. doi:10.1371/journal.pone.0065068.g006.
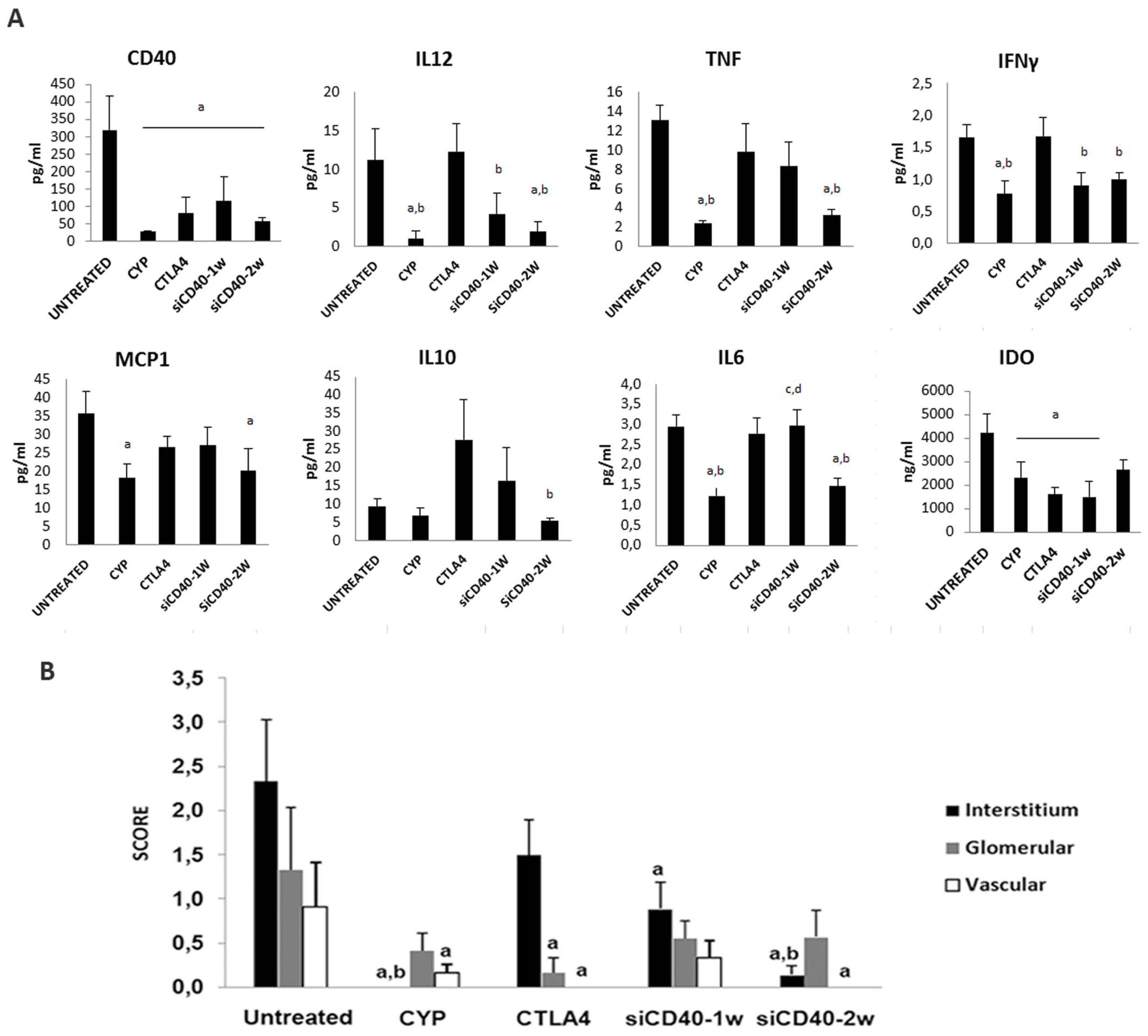
Figure 4. Systemic circulating inflammatory cytokines and local CD40 immunostaining. A. Lupus nephritis promoted the overexpression of CD40 in the serum; this immune modulatory protein was reduced in all therapies. Lupus nephritis also induced an increase in other inflammatory cytokines; treatment with anti-CD40 siRNA reduced some of them. B. Immune localization and quantitation of CD40 protein in different kidney compartments; Chol-siRNA reduced CD40 expression, especially in interstitial cells and vessels. Data are expressed as the mean ± SEM. a P < 0.05 vs. untreated; b vs. CTLA4; c vs. CYP; and d vs. siCD40-2w. doi:10.1371/journal.pone.0065068.g007.
Recipes
Annealing buffer 10×
1 M Tris-HCl (pH 7.5) 5 ml
5 M NaCl 2 ml
DEPC-treated water 43 ml
DEPC-treated water
Diethyl pirocarbonate (DEPC) 0.1% 1 ml
Distilled water 1,000 ml
Mix well and leave at room temperature for 1 h
Autoclave
Let cool to room temperature prior to use
1 M Tris-HCl, pH 7.5
Tris 121.14 g
Distilled water 800 ml
Adjust the pH to 7.5 with the appropriate volume of concentrated HCl
Bring the final volume to 1 L with deionized water
12% acrylamide gel
For 15 ml, enough for a 13 cm × 15 cm × 0.75 mm thick gel:
10× TBE 1.5 ml
40% acrylamide (acrylamide: bis acrylamide = 29:1) 4.5 ml
Distilled and deionized water 9 ml
Stir to mix, then add:
10% ammonium persulfate 150 μl
TEMED 15 μl
Mix briefly after adding the last 2 ingredients and pour the gel immediately
Non-denaturing gel loading buffer
Sucrose 50%
Bromophenol Blue 0.25%
Xylene cyanole 0.25%
4% PFA solution in PBS
Caution: Formaldehyde is toxic
Requirements: Gloves, safety glasses, fume hood
Place PBS 1× 800 ml in a glass beaker on a stir plate in a ventilated hood
Heat to 60°C while stirring (take care that the solution does not boil)
Add 40 g paraformaldehyde power
Slowly raise the pH by adding 1 N NaOH dropwise from a pipette until the solution clears
Cool the solution and filter
Adjust the volume of the solution to 1 L with 1× PBS
Recheck the pH and adjust with small amounts of diluted HCl to 6.9
Solution can be aliquoted and frozen or stored at 2.8°C for up to one month
0.1 M citrate stock buffer, pH 6
Distilled water 800 ml
Citric acid (MW = 192.1 g/mol) 11.341 g
Sodium citrate (MW = 294.1 g/mol) 12.044 g
Adjust the solution to the final desired pH using HCl or NaOH
Add distilled water to 1,000 ml
Store at room temperature (shelf life up to 3 months)
0.01 M citrate buffer
0.1 M citrate buffer 100 ml
Distilled water to 900 ml
Complete culture medium
RPMI 1640 medium 500 ml
100 U/ml penicillin
100 μg/ml streptomycin
2 M L-glutamine
10% heat-inactivated and filtered FBS
20 ng/ml GM-CSF
0.3% Oil Red O stock solution
Oil Red O 0.3 g
Isopropanol 99% 100 ml
Dissolve the solution at 56°C for 1 h
Filter the solution through Whatman No. 1 filter paper and keep at 4°C
This solution is stable for 1 year
Oil Red O working solution
Prepare in a fume hood
Stock solution 60 ml
Distilled water 40 ml
Filter the solution through Whatman No. 1 filter paper
The solution is stable for no longer than 2 h and must be prepared 15 min before use
Phosphate-buffered saline (PBS), pH 7.5
0.1 M phosphate
0.15 M NaCl
PBS-Triton (PBST)
Triton-X 2 ml
PBS 1,000 ml
PBST/Jelly
Jelly from porcine skin 0.1 g
PBST 50 ml
PS Buffer
Formalin 10% 200 ml
Water, quality MilliQ or similar 200 ml
Heat 60-70°C
Add 1 pellet NaOH
Cool in water to 24°C
Add 37.5 g D+ sucrose
Add 2 ml 0.5 M EDTA and 2.5 ml BHT (butylated hidroxytoluene)
Adjust to pH 7.4 with 1 M NaOH
Add MilliQ water to 500 ml and filter using filter paper with a medium filtration rate particle retention of 10-20 µm
Secondary antibodies (for a 1:200 dilution)
Antibody 0.5 μl
Serum normal goat (or horse) 20% 1 μl
PBST/Jelly 98.5 μl
Serum normal goat or horse 20%
Serum from normal goat or horse 20 μl
PBST/Jelly 80 μl
Acknowledgments
This study was partially funded by Instituto de Salud Carlos III (Co-funded by the European Regional Development Fund. ERDF, a way to build Europe) through the projects PI11/00556, PI14/00762, and PI18/01108 and by REDinREN (12/0021). We thank REDinREN and the CERCA program/Generalitat de Catalunya for institutional support.
Competing interests
There are no conflicts of interest.
Ethics
The experiments were carried out in accordance with EU legislation on animal experimentation and were approved by CEEA: Animal Experimentation Ethics Committee, the Institutional Ethics UB Committee for Animal Research.
References
- Arranz, A., Reinsch, C., Papadakis, K. A., Dieckmann, A., Rauchhaus, U., Androulidaki, A., Zacharioudaki, V., Margioris, A. N., Tsatsanis, C. and Panzner, S. (2013). Treatment of experimental murine colitis with CD40 antisense oligonucleotides delivered in amphoteric liposomes. J Control Release 165(3): 163-172.
- Bohula, E. A., Salisbury, A. J., Sohail, M., Playford, M. P., Riedemann, J., Southern, E. M. and Macaulay, V. M. (2003). The efficacy of small interfering RNAs targeted to the type 1 insulin-like growth factor receptor (IGF1R) is influenced by secondary structure in the IGF1R transcript. J Biol Chem 278(18): 15991-15997.
- Bosmans, L. A., Bosch, L., Kusters, P. J. H., Lutgens, E. and Seijkens, T. T. P. (2020). The CD40-CD40L Dyad as Immunotherapeutic Target in Cardiovascular Disease. J Cardiovasc Transl Res 3..
- Centa, M., Ketelhuth, D. F. J., Malin, S. and Gistera, A. (2019). Quantification of Atherosclerosis in Mice. J Vis Exp(148).
- de Ramon, L., Ripoll, E., Merino, A., Lucia, M., Aran, J. M., Perez-Rentero, S., Lloberas, N., Cruzado, J. M., Grinyo, J. M. and Torras, J. (2015). CD154-CD40 T-cell co-stimulation pathway is a key mechanism in kidney ischemia-reperfusion injury. Kidney Int 88(3): 538-549.
- Elgueta, R., Benson, M. J., de Vries, V. C., Wasiuk, A., Guo, Y. and Noelle, R. J. (2009). Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev 229(1): 152-172.
- Hueso, M., de Ramon, L., Navarro, E., Ripoll, E., Cruzado, J. M., Grinyo, J. M. and Torras, J. (2016) Silencing of CD40 in vivo reduces progression of experimental atherogenesis through an NF-κB/miR-125b axis and reveals new potential mediators in the pathogenesis of atherosclerosis. Atherosclerosis 255:80-89.
- Karnell, J. L., Rieder, S. A., Ettinger, R. and Kolbeck, R. (2019). Targeting the CD40-CD40L pathway in autoimmune diseases: Humoral immunity and beyond. Adv Drug Deliv Rev 141: 92-103.
- Kretschmer-Kazemi Far, R. and Sczakiel, G. (2003). The activity of siRNA in mammalian cells is related to structural target accessibility: a comparison with antisense oligonucleotides. Nucleic Acids Res 31(15): 4417-4424.
- Lutgens, E., Lievens, D., Beckers, L., Wijnands, E., Soehnlein, O., Zernecke, A., Seijkens, T., Engel, D., Cleutjens, J., Keller, A. M., Naik, S. H., Boon, L., Oufella, H. A., Mallat, Z., Ahonen, C. L., Noelle, R. J., de Winther, M. P., Daemen, M. J., Biessen, E. A. and Weber, C. (2010). Deficient CD40-TRAF6 signaling in leukocytes prevents atherosclerosis by skewing the immune response toward an antiinflammatory profile. J Exp Med 207(2): 391-404.
- Parasuraman, S., Kumar, E., Kumar, A. and Emerson, S. (2010). Free radical scavenging property and diuretic effect of triglize, a polyherbal formulation in experimental models. J Pharmacol Pharmacother 1(1): 38-41.
- Pluvinet, R., Petriz, J., Torras, J., Herrero-Fresneda, I., Cruzado, J. M., Grinyo, J. M. and Aran, J. M. (2005) RNAi-mediated silencing of CD40 prevents leukocyte adhesion on CD154-activated endothelial cells. Blood 104(12): 3642-3646.
- Remer, M., White, A., Glennie, M., Al-Shamkhani, A. and Johnson, P. (2017). The Use of Anti-CD40 mAb in Cancer. Curr Top MicrobiolImmunol 405: 165-207.
- Ripoll, E., Merino, A., Herrero-Fresneda, I., Aran, J. M., Goma, M., Bolanos, N., de Ramon, L., Bestard, O., Cruzado, J. M., Grinyo, J. M. and Torras, J. (2013). CD40 gene silencing reduces the progression of experimental lupus nephritis modulating local milieu and systemic mechanisms. PLoS One 8(6): e65068.
- Ui-Tei, K., Naito, Y., Takahashi, F., Haraguchi, T., Ohki-Hamazaki, H., Juni, A., Ueda, R. and Saigo, K. (2004). Guidelines for the selection of highly effective siRNA sequences for mammalian and chick RNA interference. Nucleic Acids Res 32(3): 936-948.
- Hueso, M., Casas, A., Mallén, A, de Ramón, L., Bolaños, N., Varela, C,. Cruzado, JM., Torras, J., Navarro, E. (2019). The double edge of anti-CD40 siRNA therapy: It increases renal microcapillar density but favours the generation of an inflammatory milieu in the kidneys of ApoE−/− mice. J Inflamm (Lond) 16: 25-34.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Hueso, M., Mallen, A., Ripoll, E., de Ramon, L., Bolaños, N., Valera, C., Guiteras, J., Checa, J., Navarro, E., Grinyo, J. M., Cruzado, J. M., Aran, J. M. and Torras, J. (2021). In vivo CD40 Silencing by siRNA Infusion in Rodents and Evaluation by Kidney Immunostaining. Bio-protocol 11(10): e4032. DOI: 10.21769/BioProtoc.4032.
Category
Molecular Biology > RNA > RNA interference
Immunology > Animal model > Mouse
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.