- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Fluorescence Dequenching-based Liposome Leakage Assay to Measure Membrane Permeabilization by Pore-forming Proteins
Published: Vol 11, Iss 10, May 20, 2021 DOI: 10.21769/BioProtoc.4025 Views: 7158
Reviewed by: Guangzhi ZhangHimanshu SharmaShailesh Kumar
Original research article
The authors used this protocol in:

Apr 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Pore-forming toxins (PFTs) have been discovered in a wide range of organisms. Their functions are essential to the survival or virulence of many species. PFTs often interact with lipid membranes. Large unilamellar vesicles (LUV), also known as liposomes, have been commonly used as reliable membrane models for testing PFTs activity. Liposomes have great adaptability in size, lipid composition, and loading cargo. Incorporating the fluorescent dye/quencher pair, 8-Aminonaphthalene-1,3,6-Trisulfonic Acid (ANTS) and p-Xylene-Bis-Pyridinium Bromide (DPX), in liposomes is an effective approach for measuring membrane leakage. When ANTS and DPX are encapsulated in a liposome, the fluorescence of ANTS is quenched by DPX. However, disruption of liposome integrity and subsequent leakage result in measurable fluorescence emitted by ANTS. Here, we report our protocol for optimal liposome preparation for measuring liposome leakage by fluorescence dequenching.
Keywords: Fluorescence dequenchingBackground
Pore-forming toxins (PFTs) are a family of virulence factors produced by microbial pathogens for host invasion (Alouf et al., 2005). The major action of PFTs is forming pores on lipid membranes, resulting in membrane lysis and/or translocation of other effector proteins into host cells (Bischofberger et al., 2009). Liposomes have long been used as a biological membrane model for protein-membrane interactions due to their great adaptability in size, lipid composition, and loading cargo (Chatterjee and Agarwal, 1988). A common way of using liposomes to measure pore formation or membrane leakage is to encapsulate ions or fluorescent dyes inside liposomes. Upon liposome rupture, the released ions or fluorescent dyes emit measurable signals. We first used a K+ release assay to measure pore formation on liposomal membranes by the anthrax toxin (Sun et al., 2007 and 2008; Sun and Collier, 2010). Later, the fluorescence dye and quencher pair ANTS/DPX caught our attention (Nieva et al., 1989; Ruiz-Argüello et al., 1996). Compared with the K+ release assay, which requires a bulky K+ probe and frequent changes of costly probe membranes, the ANTS/DPX dequenching assay featured higher sensitivity, more accurate and consistent measurements, and lower cost. Therefore, over the past ten years, we have transitioned to the ANTS/DPX dequenching assay. With this assay, we have successfully measured the membrane permeabilizing activity (MPA) of Mycobacterium tuberculosis (Mtb) virulence factors. The two effector proteins, 6-kDa early secreted antigenic target (ESAT-6) or EsxA and 10-kDa culture filtrate protein (CFP-10) or EsxB have been tested with various experimental designs and settings (De Leon et al., 2012; Ma et al., 2015; Peng et al., 2016; Zhang et al., 2016; Aguilera et al., 2020; Ray et al., 2019). EsxAB is a heterodimer implicated in penetrating the phagosomal membrane of macrophages, allowing for translocation of Mtb into the cytosol (De Leon et al., 2012; Ma et al., 2015; Zhang et al., 2016; Aguilera et al., 2020). Recently, we have identified a new lipid composition for liposomes that greatly enhances the MPA of EsxA (Ray et al., 2019; Vazquez-Reyes et al., 2020). Here, we describe in detail the fluorescence dequenching based liposome leakage assay with our new lipid composition.
Materials and Reagents
Screw cap glass vials with PTFE screwcap (Spectrum Chemical, catalog number: 985-92215)
Cuvette spinbar (VWR, catalog number: 58949-030)
Quartz UV cuvette (Fireflysci, catalog number: 1FLUV10)
515 nm filter (Andover Corporation, catalog number: 515FG05-50S)
1 ml syringe (BD, catalog number: 309659)
9’’ Pasteur pipet (Fisher scientific, catalog number 13-678-6b)
Saint-Gobain Tygon tubing (Saint-Gobain, catalog number: ACF00017F)
Glass tube rack (Fisher Scientific, catalog number: K7492100100)
HiTrap® desalting columns (Millipore-Sigma, catalog number: GE17-1408-01)
Gel loading pipet tips (Fisherbrand, catalog number: 02-707-139)
10 mm filter supports (Avanti Polar Lipids, catalog number: 610014)
0.2 µm membrane (Whatman, catalog number: 800281)
Aluminum foil (Fisherbrand, catalog number: 01-213-100)
1 ml sample loop (Thermo Scientific, catalog number: 03-052-382)
Chloroform (Fisher Chemical, catalog number: c298)
1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) (Avanti Polar Lipids, catalog number: 850375)
1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl) iminodiacetic acid) succinyl] (nickel salt) (18:1 DGS-NTA(Ni) (Avanti Polar Lipids, catalog number: 790404)
1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1'-rac-glycerol) (sodium salt) (POPG) (Avanti Polar Lipids, catalog number: 840457)
1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC) (Avanti Polar Lipids, catalog number: 850457)
Ethanol (Fisher Chemical, catalog number: a4094)
HPLC grade water (Fisher Chemical, catalog number: W5-4)
Tris-base (Fisher Chemical, catalog number: BP152)
Sodium chloride (Fisher Chemical, catalog number: BP358)
Sodium hydroxide (Fisher Chemical, catalog number: S318)
Hydrochloric acid (Fluka, catalog number: 7647)
Dry ice
4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid, N-(2-Hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES) (Fisher Bioreagents, catalog number: BP310)
8-Aminonaphthalene-1,3,6-trisulfonic acid, disodium salt (Setareh Biotech, catalog number: 6951)
p-Xylene-bis-pyridinium bromide (DPX) (Setareh Biotech, catalog number: 6271)
Triton X-100 (Sigma-Aldrich, catalog number: X100)
Sodium acetate (Sigma-Aldrich, catalog number: S2889)
Methanol (Fisher, catalog number: A453-500)
Gel filtration buffer (see Recipes)
pH 4 buffer (see Recipes)
Equipment
Glass tubes (VWR, catalog number: 89000-502)
50 ml beaker (Pyrex, catalog number: 02-540G)
Lab mounting stand (Humboldt MFG Company, catalog number: H-21217)
Nitrogen gas tank
Gas tank regulator (VWR, catalog number: 55850-277)
Large top desiccator (Corning Life Sciences, catalog number: CLS3120250)
Hot plate/stirrer (Thermo Fisher Scientific, catalog number: SP131325)
Extruder set with heating block (Avanti Polar Lipids, catalog number: 610000)
pH meter (Mettler Toledo, catalog number: 30266626)
Vortex (VWR, catalog number: 58816-121)
Scale (Mettler Toledo, catalog number: ML54)
AKTA FPLC system (General Electric, catalog number: UPC-900 and P920)
Fluorometer (ISS, catalog number: K2)
Water bath (VWR, catalog number: 13271-086)
Procedure
Drying phospholipids
If working with lipid powders, create a lipid stock solution.
Measure 50-150 mg of lipids into a glass vial with a screw cap.
Add 5-15 ml of 3:2 chloroform:methanol as needed to maintain concentration at 10 mg/ml for ease of usage.
Note: Cover the vial using PTFE lined caps; otherwise, the chloroform may dissolve plastics and contaminate the sample.
Measure the appropriate amount of lipids (10 mg will be enough for approximately 30 reactions) into a glass tube.
Note: Liposomes can be made of single lipid forms [e.g., 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1'-rac-glycerol) (sodium salt) (POPG), and 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC)] or their combinations. For example, the Ni2+-chelating lipid, 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl) iminodiacetic acid) succinyl] (nickel salt) or (DGS-NTA(Ni)), can attach His-tagged proteins to the membranes (Sun et al., 2007 and 2008; Jacquez et al., 2014). The new liposome developed for measuring the MPA of EsxAB is composed of POPC to POPG at a 4:1 molar ratio.
Use a clamp stand to hold the vial in place.
Using a second clamp, position a Pasteur pipette approximately 2-5 mm away from the liquid in the tube.
Connect the tubing to the end of the Pasteur pipette and open the valve to allow gentle airflow.
Evaporate the chloroform from the vial until there is no liquid visible.
Remove the vial from the stand and place it inside a vacuum chamber at room temperature for at least 3 h or overnight.
Store the dry lipids at -20 °C. Lipids may be stored under these conditions for up to a month.
Preparing column and FPLC for desalting
A 5-ml desalting column is needed for removing the excess ANTS/DPX and for buffer exchange. If necessary, two desalting columns can be connected in tandem to improve desalting results.
Pass at least 5 column volumes (CV) or, in this case, 50 ml of water through the column to remove the ethanol from the column.
Pass at least 5 CV (50 ml) of gel filtration buffer through the column.
Prepare the machine for desalting by passing 50 ml of water through the machine at 5 ml/min.
Pass 50 ml of gel filtration buffer through the machine at 5 ml/min.
Connect the column to the machine and pass an additional 100 ml of gel filtration buffer.
Clean the injection valve by injecting 1 ml of gel filtration buffer.
Repeat Step B7 at least two more times.
Preparing the lipid-ANTS/DPX solution
Prepare a freezing solution in a beaker by adding dry ice to > 95% ethanol. The freezing solution is used to flash freeze the lipids containing ANTS/DPX. Alternatively, liquid nitrogen can be used to flash freeze. It is ideal to keep the freezing solution below -50 °C.
Fill another beaker with lukewarm water for thawing the lipids.
Re-suspend lipids in 5 mM HEPES (pH 7.4), 1 ml for every 10 mg of lipids.
Vortex until the solution is well suspended.
Measure the solution volume and add 50 mM of ANTS and DPX into the tube.
Mix by pipetting up and down and cover the tube with aluminum foil.
Place the tube into the freezing solution and allow it to freeze.
Move the tube into warm water to thaw the lipids.
Repeat Steps C7-C8 for five more freeze/thaw cycles.
Note: Six freeze/thaw cycles are usually sufficient to solubilize the lipids. If problems occur during the extrusion, such as difficulties in passing the liquid through the membranes, perform more freeze/thaw cycles.
Producing liposomes via extrusion
Assemble the extruder according to the manufacturer’s specifications.
Draw up 1 ml of gel filtration buffer using one of the syringes and pass the buffer through the extruder until the liquid has been transferred to the other syringe.
Note: You should feel a little bit of resistance. If there is no resistance, it is possible the membrane has moved during assembly or has torn.
Remove the buffer from the syringe.
Draw up to 1 ml of liposomes into one of the syringes.
Pass the liposomes through the membrane until the liquid has been transferred from one syringe to the other.
Repeat Step D5 for a total of 20 times.
Inspect the fluid after extrusion; it should have changed from an opaque to a transparent, yellow-tinged liquid (Figure 1).

Figure 1. Comparison of the lipid solution before and after extrusion. A. The lipid solution before extrusion has an opaque color. B. The lipid solution becomes more transparent after extrusion.Dispense the lipids into a clean glass tube.
Removal of excess fluorescent dyes via desalting chromatography
After pre-equilibration of the column and machine and cleaning the injection loop, inject the liposomes into the injection loop.
Run the desalting program at a 0.5 ml/min flow rate, using gel filtration buffer, and collect 0.5 ml per fraction.
Allow 50 ml of buffer to pass through the column.
The liposomes will be eluted out of the column after approximately 2 ml have passed through.
Collect fractions containing the liposomes, usually in six tubes for a total 3 ml of liposomes (Figure 2).
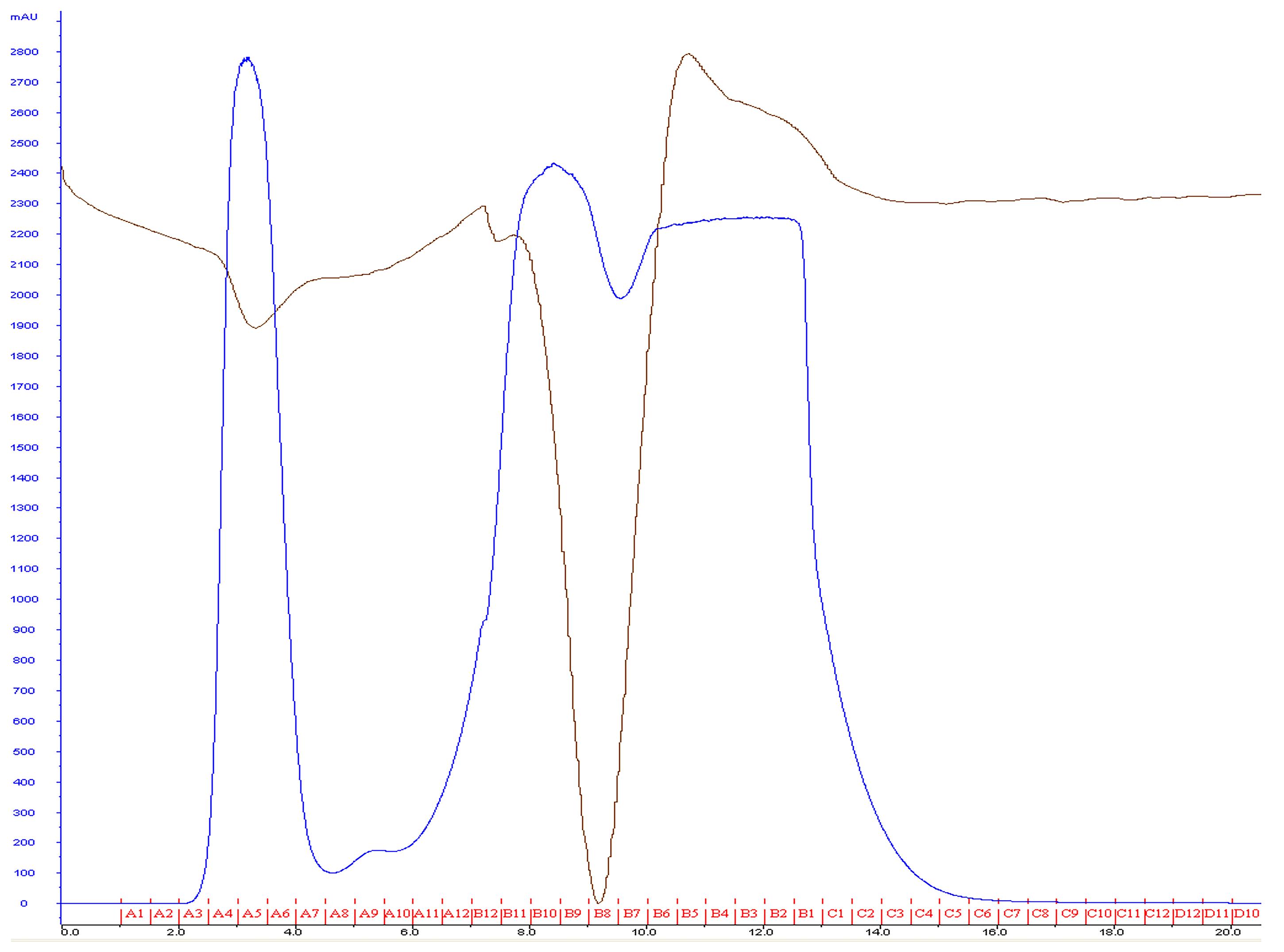
Figure 2. Most lipids will elute after flowing approximately 2 ml of buffer through the column. A typical yield of 3 ml is expected using this procedure. The blue line represents mAu, and the brown line represents conductance.Combine all liposome samples, cover with aluminum foil, and store at 4 °C.
Liposomes at this stage can be stored for approximately 1-3 weeks.
Liposomes prepared with this procedure are usually consistent in concentration. However, technical replicates with the same batch of lipids are needed for every biological replicate to obtain consistent results.
Clean the column by passing at least 5 CV + 30 ml (the volume of the machine) of water through the machine.
Pass at least five CV in addition to 30 ml of 20% ethanol through the machine for column storage.
Liposome leakage assay
The quality of the liposomes should be tested before analyzing the protein of interest by adding 2% (v/v) Triton X-100, which completely lyses the liposomes, quickly revealing a strong fluorescence signal.
Set fluorometer excitation to 350 nm and record emissions at 520 nm and continue to measure at 1-second intervals for 5-10 min.
Cross polarizers are applied at the excitation and emission paths to reduce background. If necessary, apply a 515 nm long-pass filter at the emission path to reduce background scatter.
If available, connect a water bath to the water inlet and outlet of the sample chamber to maintain a constant temperature. This is especially important with proteins that interact at the physiological temperature, as is the case with EsxAB.
To a UV cuvette, add 1.15 ml of gel filtration buffer, 150 µl of 1 M NaAc buffer (pH 4), and 100 µl of liposomes.
Place the sample cuvette inside the sample chamber and close the lid.
After approximately 30 s, use a gel loading pipette tip to inject 100 µl of 2% Triton X-100 into the cuvette through the hole on the cover lid. The ANTS fluorescence should increase at least 5-fold (Figure 3). This will serve as a positive control.
The fluorescence intensity will drop for a second or two due to the dilution of the mix inside the cuvette, but it will begin to rise shortly after.
A negative control consists of the sample prepared with liposomes and the gel filtration buffer.
Continue experimenting by testing cytolytic proteins; prepare the cuvette as in Step F5 (Figure 3).
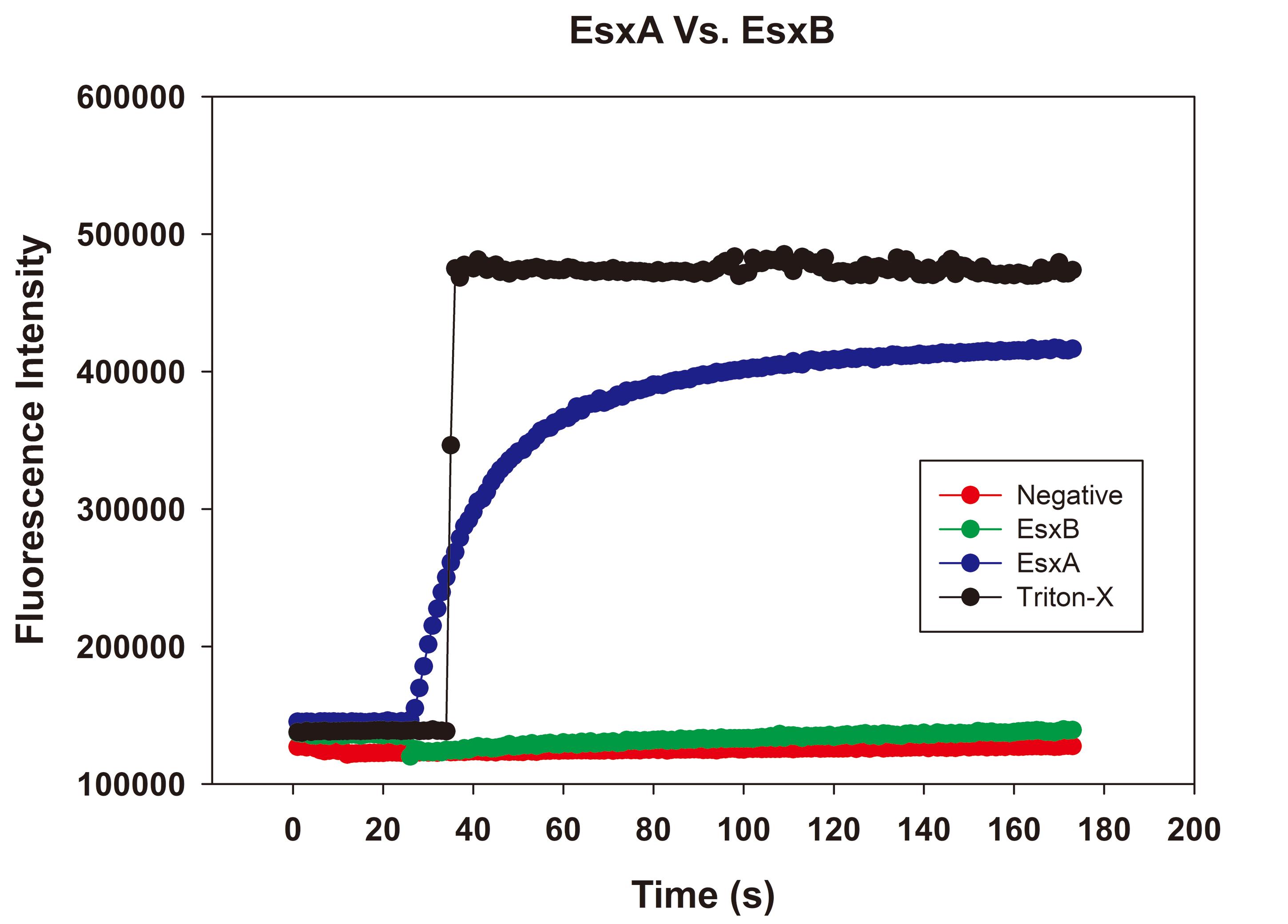
Figure 3. Raw plots of an ANTS/DPX assay. The background fluorescence intensity is approximately 100,000 units. The fluorescence intensity increased 5-fold after adding 2% Triton X-100. With EsxA, a protein known to have MPA, we observed a gradual increase in fluorescence. EsxB, a protein without MPA, did not increase fluorescence intensity. The negative control consisting of the only gel filtration buffer did not increase fluorescence intensity.Place the UV cuvette in the sample chamber and close the lid.
After about 30 s, use a gel loading pipette tip to add 100 µl of protein into the cuvette.
Protein concentrations can vary, but a total of 100 µg is usually used in our experiments.
Depending on the nature of the experiment, the protein can be loaded into the cuvette instead of the pH 4 buffer. Then, after 30 s, adding the pH 4 buffer will cause EsxAB to lyse the membrane.
Data analysis
The fluorescence intensity is normalized at the time point the protein was added by subtracting the lowest intensity value from all other values, removing the background fluorescence.
The lowest intensity value is now deemed the measurement at T = 0, and all values from T = 0 until the end of the experiment are averaged for all replicates.
The dilution of the lipids by adding the sample results in a small drop in fluorescence. Measuring from the moment the protein is added allows determination of the increase in fluorescence as the liposomes are lysed.
The average intensity is graphed against the time to obtain a curve (Figure 4A).
The plateau followed by a one-phase association can also be used to obtain an association constant.
The average of the highest fluorescence intensity values for each sample are graphed in a bar graph (Figure 4B).
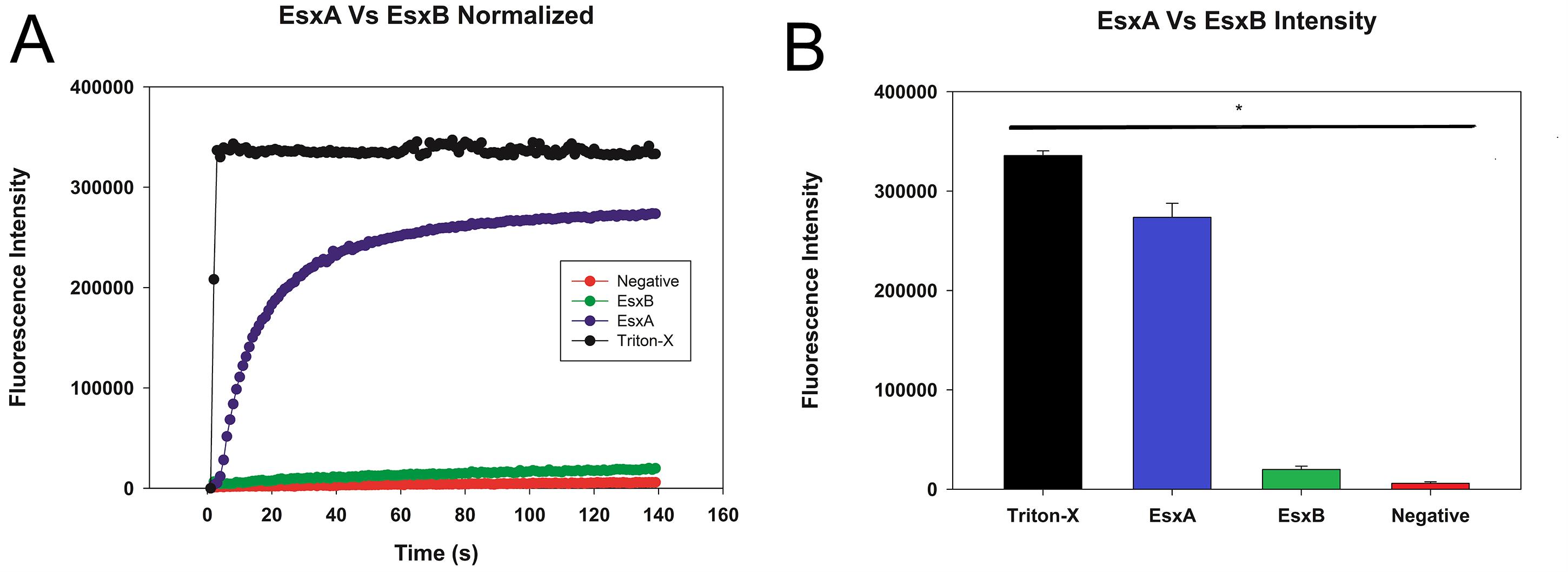
Figure 4. Plot of sample liposome experiment after normalization. A. The data were normalized to remove background fluorescence and to account for the drop in fluorescence caused by the dilution of the liposomes by the sample. B. The peak fluorescence was graphed with error bars representing the standard deviation. Statistical significance is represented here as well.The values are tested for normality using the Shapiro-Wilk test.
If the data fails to differ from normal, it is tested for significance with a One-way ANOVA; otherwise, a Tukey-Kramer test is performed.
Recipes
Gel filtration buffer
20 mM Tris, pH 7.4
10 mM NaCl
pH 4 Buffer
1 M NaAC, pH 4.01
Acknowledgments
This study was supported by grants from NIGMS (SC1GM095475 to J. Sun), National Center for Research Resources (5G12RR008124), and National Institute on Minority Health and Health Disparities (G12MD007592). Javier Aguilera was supported by a National Institutes of Health Grant (R25GM069621 to R. Aguilera) via the RISE program for graduate students. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This protocol was adapted from Nieva et al.,1989 (DOI: 10.1021/bi00444a032) and optimized for the EsxA/EsxB complex.
Competing interests
Authors have no competing interests to declare.
References
- Aguilera, J., Karki, C. B., Li, L., Vazquez Reyes, S., Estevao, I., Grajeda, B. I., Zhang, Q., Arico, C. D., Ouellet, H. and Sun, J. (2020). Nα-Acetylation of the virulence factor EsxA is required for mycobacterial cytosolic translocation and virulence. J Biol Chem 295(17): 5785-5794.
- Alouf, J. E., Ladant, D., and Popoff, M. R. (2005). The comprehensive sourcebook of bacterial protein toxins. 3rd Edition. Academic Press. ISBN: 9780080456980.
- Bischofberger, M., Gonzalez, M. R. and van der Goot, F. G. (2009). Membrane injury by pore-forming proteins. Curr Opin Cell Biol 21(4): 589-95.
- Chatterjee, S. N. and Agarwal, S. (1988). Liposomes as membrane model for study of lipid peroxidation. Free Radic Biol Med 4(1): 51-72.
- De Leon, J., Jiang, G., Ma, Y., Rubin, E., Fortune, S. and Sun, J. (2012). Mycobacterium tuberculosis ESAT-6 exhibits a unique membrane-interacting activity that is not found in its ortholog from non-pathogenic Mycobacterium smegmatis. J Biol Chem 287(53): 44184-91.
- Jacquez, P., Lei, N., Weigt, D., Xiao, C. and Sun, J. (2014). Expression and purification of the functional ectodomain of human anthrax toxin receptor 2 in Escherichia coli Origami B cells with assistance of bacterial Trigger Factor.Protein Expr Purif 95: 149-155.
- Ma, Y., Keil, V. and Sun, J. (2015). Characterization of Mycobacterium tuberculosis EsxA membrane insertion: roles of N- and C-terminal flexible arms and central helix-turn-helix motif. J Biol Chem 290(11): 7314-22.
- Nieva, J. L., Goni, F. M. and Alonso, A. (1989). Liposome fusion catalytically induced by phospholipase C. Biochemistry 28(18): 7364-7.
- Peng, X., Jiang, G., Liu, W., Zhang, Q., Qian, W. and Sun, J. (2016). Characterization of differential pore-forming activities of ESAT-6 proteins from Mycobacterium tuberculosis and Mycobacterium smegmatis. FEBS Lett 590(4): 509-19.
- Ray, S., Vazquez Reyes, S., Xiao, C. and Sun, J. (2019). Effects of membrane lipid composition on Mycobacterium tuberculosis EsxA membrane insertion: A dual play of fluidity and charge. Tuberculosis (Edinb) 118: 101854.
- Ruiz-Argüello, M. B., Basanez, G., Goni, F. M. and Alonso, A. (1996). Different effects of enzyme-generated ceramides and diacylglycerols in phospholipid membrane fusion and leakage. J Biol Chem 271(43): 26616-21.
- Sun, J. and Collier, R. J. (2010). Disulfide bonds in the ectodomain of anthrax toxin receptor 2 are required for the receptor-bound protective-antigen pore to function. PLoS One 5(5): e10553.
- Sun, J., Lang, A. E., Aktories, K. and Collier, R. J. (2008). Phenylalanine-427 of anthrax protective antigen functions in both pore formation and protein translocation. Proc Natl Acad Sci U S A 105(11): 4346-51.
- Sun, J., Vernier, G., Wigelsworth, D. J. and Collier, R. J. (2007). Insertion of anthrax protective antigen into liposomal membranes: effects of a receptor. J Biol Chem 282(2): 1059-65.
- Vazquez-Reyes, S., Ray, S., Aguilera, J., Sun, J. 2020) Development of a new liposome model that enhances membrane permeabilizing activity of Mycobacterial tuberculosis EsxAB heterodimer.
- Zhang, Q., Wang, D., Jiang, G., Liu, W., Deng, Q., Li, X., Qian, W., Ouellet, H. and Sun, J. (2016). EsxA membrane-permeabilizing activity plays a key role in mycobacterial cytosolic translocation and virulence: effects of single-residue mutations at glutamine 5.Sci Rep 6: 32618.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Aguilera, J., Vazquez-Reyes, S. and Sun, J. (2021). A Fluorescence Dequenching-based Liposome Leakage Assay to Measure Membrane Permeabilization by Pore-forming Proteins. Bio-protocol 11(10): e4025. DOI: 10.21769/BioProtoc.4025.
- Aguilera, J., Karki, C. B., Li, L., Vazquez Reyes, S., Estevao, I., Grajeda, B. I., Zhang, Q., Arico, C. D., Ouellet, H. and Sun, J. (2020). Nα-Acetylation of the virulence factor EsxA is required for mycobacterial cytosolic translocation and virulence. J Biol Chem 295(17): 5785-5794.
Category
Biochemistry > Protein > Interaction > Protein-lipid interaction
Microbiology > Microbial proteomics > Membrane proteins
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.