- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Imaging and Fluorescence Quantification in Caenorhabditis elegans with Flow Vermimetry and Automated Microscopy
Published: Vol 11, Iss 10, May 20, 2021 DOI: 10.21769/BioProtoc.4024 Views: 5156
Reviewed by: Alexandros AlexandratosManish ChamoliDURAI SELLEGOUNDER
Original research article
The authors used this protocol in:

Jun 2020


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Gene activation and cellular biomarkers are commonly monitored using fluorescent signals from transgenic reporters or dyes. These quantifiable markers are critical for biological research and serve as an incredibly powerful tool, even more so when combined with high-throughput screening. Caenorhabditis elegans is a particularly useful model in this regard, as it is inexpensive to grow in vast numbers, has a rapid generation time, is optically transparent, and can readily fit within 384-well plates. However, fluorescence quantification in worms is often cumbersome. Quantification is frequently performed using laborious, low-throughput, bias-prone methods that measure fluorescence in a comparatively small number of individual worms. Here we describe two methods, flow vermimetry using a COPAS BioSorter and an automated imaging platform and analysis pipeline using a Cytation5 multimode plate reader and image analysis software, that enable high-throughput, high-content screening in C. elegans. Flow vermimetry provides a better signal-to-noise ratio with fewer processing steps, while the Cytation5 provides a convenient platform to image samples across time. Fluorescence values from the two methods show strong correlation. Either method can be easily extended to include other parameters, such as the measurement of various metabolites, worm viability, and other aspects of cell physiology. This broadens the utility of the system and allows it to be used for a wide range of molecular biological purposes.
Keywords: C. elegansBackground
Since its introduction in the 1960s, the nematode Caenorhabditis elegans has become a popular model across all of biology. There are multiple reasons for its popularity: it is small, simple, inexpensive, and easy to maintain, has substantial genomic conservation with human, very powerful and ubiquitous genetic tools, and is optically transparent. These characteristics make it particularly appropriate for high-throughput screening (HTS), where a comparatively simple assay is repeated thousands of times across a library of interest (often small molecules or genome-wide RNAi). A more powerful extension of HTS is high-content HTS, where complex or multiple phenotype readouts are simultaneously ascertained (e.g., the combination of survival and expression of a particular reporter, perhaps restricted to a particular tissue or subcellular localization). Live, whole organism screens are becoming increasingly popular, and are attractive methods to test the effects of gene knockdown or small molecule treatment on physiology, to identify bioactive and antimicrobial compounds, and to study genetic interactions. Indeed, many labs looking to identify small molecules and drugs to study aging (Lucanic et al., 2018), neurological diseases (Gosai et al., 2010), and host-pathogen interactions (Moy et al., 2009; Conery et al., 2014; Kirienko et al., 2016), have used worms for their first steps, and HTS have been developed for different types and purposes of experiments, in both agar- (Burns et al., 2006; Squiban et al., 2012; Lucanic et al., 2018) and liquid-based (Moy et al., 2009; Anderson et al., 2018) systems.
As more and more of these screens are conducted in C. elegans labs, output analysis has become a major bottleneck. Fluorescence quantification of images obtained from microscopy for phenotype-based screens often requires time-consuming and expensive manual labor that is prone to inadvertent bias. Additionally, the clustering, crossing, and/or tangling that often occur with C. elegans complicate measurement of individual animals in the images. Automated microscopy platforms and complex software algorithms have been developed to resolve these issues. ArrayScan VTI and SpotDetector BioApplication are examples of such software, and are able to recognize worms and quantify their sizes and fluorescence (Gosai et al., 2010; Maglioni et al., 2015). The Worm-align/Worm_CP workflow in CellProfiler also provides a cost-effective method to assess individual worms in images (Raviv et al., 2010; Wählby et al., 2012; Okkenhaug et al., 2020).
Unfortunately, while these pipelines are very useful for the measurement of phenotypes in individual animals, their computational demands still limit the sample sizes. Optimal numbers of worms in a well for a reliable count by using the above method is between 50 and 100, depending on the life stage of the worms (Maglioni et al., 2015). Conventional understanding is that this is low-throughput [defined as 1-500 individuals vs medium (500-10,000), high (10,000-100,000) or ultra-high (>100,000) throughput (Wildey et al., 2017)]. Many experiments may require measurements of individual animals from larger numbers of samples, such as searching for small effects or rare phenomena. For example, C. elegans at the L2 stage have a more oxidizing redox environment and increased variability between individuals compared to young adults (Bazopoulou et al., 2019), and measurement of the redox environment in a larger population provides a more accurate representation of the biological state of the organism. As another example, analysis of a large population may be necessary to identify rare crossover events between closely linked alleles in a mutant screen. Finally, experimental assays using a large number of individuals may be required to overcome substantial inter-individual variability (Angstman et al., 2015 and 2016).
Our lab routinely monitors gene activation via fluorescent transgenes and measures mitochondrial parameters using fluorescent dyes. The protocols we described (Tjahjono and Kirienko, 2017; Kang et al., 2018; Revtovich et al., 2019; Tjahjono et al., 2020) are invaluable for this purpose. Here we describe the imaging and fluorescence quantification protocols we routinely use. We commonly use a COPAS FlowPilot BioSorter (Union Biometrica) to obtain automated, unbiased, high-throughput measurements of fluorescence. This is analogous to flow cytometry, so we refer to it as flow vermimetry (from the Latin vermis for worm). This technique has been previously described for use in RNAi screens (Squiban et al., 2012). We also use the Cytation5 multimode plate reader/imager (BioTek) to obtain images and measure phenotypic changes over time. We have optimized the cellular analysis pipeline from Gen5 software for worm identification and fluorescence measurement. The Data Analysis section shows that the measurement values from these techniques demonstrate a high correlation. Advantages and limitations of each approach are also discussed.
Materials and Reagents
Bacterial Strains
Escherichia coli OP50
E. coli HT115 (RNAi-competent), obtained from Ahringer or Vidal RNAi library (Kamath et al., 2003; Rual et al., 2004)
Worm Strains
N2 Bristol (wild type)
WY703 (fdIs2 {3XESRE::GFP; pFF4[rol-6(su1006)]}) (Kuzmanov et al., 2014)
SLR115 {dvIs67 [Ptbb-6::GFP + Pmyo-3::dsRed]} (Munkácsy et al., 2016)
NVK93 (houIs002 {pJY323[Phsp-16.1::GFP]; pRF4 [rol-6(gf)]}) (Kuzmanov et al., 2014)
WY756 (fdEx139 {pJY312 [Phsp-16.1(dd)::GFP]; pRF4}) (Kuzmanov et al., 2014)
Reagents (store at room temperature)
LB Broth Miller (Luria-Bertani broth) (USBiological Life Science, catalog number: L1520)
Agar (USBiological Life Science, catalog number: A0930)
NaCl (USBiological Life Science, catalog number: S5000)
Peptone (USBiological Life Science, catalog number: P3300)
CaCl2 (Sigma, catalog number: C1016-500G)
MgSO4 (Fisher Scientific, catalog number: M63-500)
Cholesterol (Sigma, catalog number: C8667-5G)
KH2PO4 (ACROS Organics, catalog number: 7778-77-0)
K2HPO4 (USBiological Life Science, catalog number: P5100)
5% sodium hypochlorite solution (RICCA, catalog number: 7495.5-32)
Sodium hydroxide (NaOH) pellets (VWR, catalog number: 97064-476)
N-acetyl cysteine (ACROS Organics, catalog number: AC160280250)
Reagents (store at -20°C)
Carbenicillin (Fisher Scientific, catalog number: 50-213-247)
Tetracycline (Sigma, catalog number: 87128)
IPTG (GoldBio, catalog number: I2481C50)
Rotenone (Sigma, catalog number: R8875)
Buffers and Media (also see Recipes)
Nematode growth media (NGM) plates
Worm bleach solution
S Basal
Phosphate buffer
Other Materials
Cell culture microplate, 96-well, half area, black (Greiner Bio-One, catalog number: 675090)
Microplate, 96-well, non-treated polystyrene (Fisher Scientific, catalog number: 07-000-124)
15 ml conical sterile polypropylene centrifuge tubes (Fisher Scientific, catalog number: 12-565-268)
50 ml conical sterile polypropylene centrifuge tubes (Fisher Scientific, catalog number: 12-565-270)
0.6 ml microcentrifuge tubes (Fisher Scientific, catalog number: 05-408-120)
1.5 ml microcentrifuge tubes (Fisher Scientific, catalog number: 05-408-129)
2.0 ml microcentrifuge tubes (Fisher Scientific, catalog number: 05-408-138)
Petri dish, polystyrene, vented, 60 × 15 mm (Fisher Scientific, catalog number: 07-000-339)
Petri dish, polystyrene, vented, 94 × 16 mm (Fisher Scientific, catalog number: 07-000-323)
Pipette tips, 200 µl (VWR, catalog number: 89140-886)
Pipette tips, 1,000 µl (VWR, catalog number: 89140-918)
Pipette tips, 10 µl (VWR, catalog number: 89425-650)
Serological pipettes, 10 ml (Thermo Scientific, catalog number: 170356)
Serological pipettes, 25 ml (Thermo Scientific, catalog number: 170357)
Equipment
COPAS FlowPilot BioSorter (Union Biometrica)
Large Particle (LP) Sampler (Union Biometrica)
Cytation5 Cell Imaging Multi-Mode Reader (BioTek)
EL406 Washer Dispenser (BioTek)
Tube revolver/rotator (Fisher Scientific, catalog number: 11-676-341)
Centrifuge, MegafugeTM 8 Small Benchtop (Thermo Scientific, catalog number: 75007210)
Software
FlowPilot software for COPAS FP BioSorter instrument is obtained with a purchase of the equipment from Union Biometrica (https://www.unionbio.com/copas/software.aspx)
Gen5 software, version 3.0, for Cytation5 Cell Imaging Multi-Mode Reader is obtained with a purchase of the equipment from BioTek (https://www.biotek.com/products/software-robotics-software/gen5-software-features-for-imaging-microscopy/overview/)
Microsoft Excel is used for COPAS FlowPilot BioSorter output analysis
R (https://cran.r-project.org/) and RStudio (https://rstudio.com/products/rstudio/) are used for data visualization and statistical analysis
Procedure
Maintenance of C. elegans
Isolate eggs from gravid hermaphrodites using the hypochlorite method as follows:
Wash gravid hermaphrodites from 2-4 10 cm NGM plates with 15 ml of S Basal.
Pour worms in S Basal into a 15 ml conical tube.
Centrifuge the tube(s) at 1,000 × g for 30 s.
Aspirate the supernatant, leaving the worm pellet (volume: ~0.1-0.5 ml).
Add 3.5 ml of Worm Bleach Solution (see Recipes), shake vigorously, and wait for one minute.
Centrifuge the tube at 1,000 × g for 30 s.
Remove the supernatant by pouring it off the tubes or by using an aspirator.
Add 3.5 ml of Worm Bleach Solution (see Recipes) and shake vigorously.
Monitor the lysis of the worm cuticles by using a dissecting microscope. Shake vigorously periodically. Most worms will dissolve in four to five minutes, leaving the eggs, which are protected by the eggshells. Do not bleach the worms for more than seven minutes, as the eggs will start to die.
Centrifuge the tube at 1,000 × g for 30 s, remove supernatant.
Wash eggs with 15 ml of S Basal three times to remove all traces of bleach.
Add 5-6 ml of S Basal into the tube.
Rotate the tubes on a rotisserie at room temperature for 16-24 h to let the eggs hatch to obtain a synchronous L1 population.
Count the concentration of worms in the tube from Step A1 as follows:
Put 80 µl of S Basal into a 0.6 ml microcentrifuge tube.
Gently shake the tube containing L1 larvae from Step A1, take 20 µl of these L1 larvae and add it into the 0.6 ml microcentrifuge tube (5 times dilution).
Pipette up and down several times to disperse the worms in S Basal equally.
Take 10 µl from the microcentrifuge tube and spot it on a blank NGM plate in a shape of a line.
Count the number of L1 larvae on three different lines and average the obtained numbers.
Divide the averaged number by 2 to obtain worm count per µl in the original tube (or divide number by 10 and multiply by 5 to compensate for dilution).
To calculate the volume of worms needed for seeding, divide the desired number of worms by the worm count per µl. For example, to transfer 6,000 of L1 larva from a tube containing 30 worms per µl, use 200 µl (6,000/30) of the L1 larvae.
Transfer ~6,000 synchronized L1 larvae onto 2-4 10 cm NGM plates seeded with E. coli OP50.
Grow worms at 20°C for 72 h for the next cycle of egg isolation. If working with temperature-sterile worms, incubate them at 15°C for ~120 h instead.
Preparation of NGM-OP50 plates
Grow E. coli OP50 in LB broth for 16 h in a shaker-incubator at 37°C with 225 rpm of agitation to a saturated culture.
Centrifuge bacterial culture at 3,260 × g for 30 min at 4°C.
Concentrate bacteria by removing supernatant and adding S Basal (40 ml S Basal for each liter of the culture, achieving 25-fold concentration).
Seed 2 ml of concentrated bacteria onto each 10 cm NGM plate. Allow the OP50 lawn to dry before using. These plates can be stored in a closed container at 4°C for several weeks.
Preparation of RNAi-expressing bacteria
Grow RNAi-expressing E. coli in LB supplemented with 12.5 μg/ml tetracycline and 25 μg/ml carbenicillin for 16 h in a shaker-incubator at 37°C with 225 rpm of agitation.
Subculture with 100-fold dilution in LB supplemented with 25 μg/ml carbenicillin and grow for another 6 h in a shaking incubator at 37°C with 225 rpm of agitation.
Centrifuge bacterial culture at 3,260 × g for 20 min at room temperature.
Concentrate bacteria by removing supernatant and adding S Basal (40 ml S Basal for each liter culture, achieving 25-fold concentration).
Seed bacteria onto NGM plates supplemented with 25 μg/ml carbenicillin and 1 mM IPTG (2 ml of concentrated culture for each 10 cm plate and 0.5 ml of concentrated culture for each 6 cm plate).
For double RNAi experiment, measure optical densities (OD600) of each culture with a spectrophotometer. Dilute the cultures so that the final mix of cultures is equivalent and seed as described in Step C5.
Growing C. elegans for experimental setup
Using E. coli OP50
Pipette ~6,000 L1 worms onto 10 cm NGM plates seeded with E. coli OP50.
Incubate worms-containing plates in a 20°C incubator for 46 h.
Using RNAi-expressing E. coli
Pipette ~2,000 or ~6,000 L1 worms onto prepared 6 cm or 10 cm RNAi plates, respectively.
Incubate plates at 20°C for 46 h.
For either food sourse
Wash synchronized young adult worms from NGM plates into a 15 ml conical tube with S Basal.
Centrifuge tubes at 1,000 × g for 30 s.
Remove liquid and add 14 ml S Basal.
Repeat Steps D2a and D2b two more times to remove excess bacteria.
Remove liquid and add 1.8 ml (if 6 cm plate was used) or 5 ml (if 10 cm plate was used) S Basal to achieve density of ~1 worm/µl.
Pipette 100 µl of worms (~100 worms) into each well of a 96-well plate (half-area, black, clear bottom).
Image worms by using a Cytation5 (Procedure F) and/or measure fluorescence using LP Sampler and COPAS FP BioSorter (Procedure G).
C. elegans exposure to mitochondrial inhibitors
Wash synchronized L4/young adult worms from NGM plates into a 15 ml conical tube with 15 ml of S Basal.
Centrifuge tubes at 1,000 × g for 30 s.
Remove liquid and add 5 ml S Basal.
Pipette 100 µl of worms (~100 worms) into each well of a 96-well plate (half area, black, clear bottom).
Aspirate liquid with an EL406 Washer Dispenser (volume of remaining liquid: ~40 µl).
Add 60 µl of S Basal supplemented OP50 (final OD: 0.03) and compound of interest.
Note: In this study, we used a combination of 0, 25, 37.5, or 50 µM rotenone (Sigma) and 0, 1, 2, 3, 4, or 5 mM N-acetyl cysteine (NAC, ACROS Organics).
Image worms every two hours for twenty hours by using Cytation5 (Procedure F) and/or measure fluorescence by using LP Sampler and COPAS FP BioSorter (Procedure G).
Imaging with automated microscope (Cytation5)
Prepare worm-containing plates as described above (Procedures D and E).
Run Gen5 software.
Set up for kinetics experiment or take images at a certain time point. Imaging should be performed with identical settings for the biological replicates for the same experiment.
Analyze images with Gen5 cellular analysis pipeline:
Select an image.
Click on ‘Process’, select ‘Image Preprocessing’.
Apply Image preprocessing for fluorescence images, click ‘OK’.
On the transformed image, click on ‘Process’, select ‘Digital Phase Contrast’, click ‘OK’.
On the digital phase contrast and transformed image, click on ‘Analyze’.
Choose ‘Cellular Analysis’ on the ‘Analysis’ panel.
Click on ‘Edit options’ and adjust values (threshold, background, minimum and maximum object sizes, etc.) as optimized.
Click on ‘Apply’ and then ‘Apply changes’.
Export values into Microsoft Excel for downstream processing.
Fluorescence measurement with flow vermimetry (COPAS FP BioSorter and LP Sampler)
For operation with COPAS FP BioSorter
Prepare worm-containing plates as described above (Procedures D and E)
Wash worms into a 50 ml conical tube (from RNAi plates or from treatment with compounds).
Let worms settle to the bottom of the tube via gravity, aspirate liquid, and add 50 ml S Basal.
Once worms are re-settled, aspirate liquid and add 5 ml S Basal for every 5,000 worms (aim for a final concentration of 1 worm per µl).
Proceed to fluorescence measurement with COPAS FP BioSorter machine. Measurement should be performed with identical settings for the biological replicates for the same experiment.
Analyze data obtained from COPAS FP BioSorter.
For operation with LP Sampler and COPAS FP BioSorter
Prepare worm-containing plates as described above (Procedures D and E).
At the appropriate time point, wash the 96-well plate three times to remove any remaining drugs or dyes by using the EL406 machine. Allow worms to settle via gravity between washes.
Proceed to fluorescence measurement with LP Sampler and COPAS FP BioSorter machine. Measurement should be performed with identical settings within the same experiment.
Analyze data obtained from COPAS FP BioSorter.
Analyzing output data from COPAS FP BioSorter
Open the output file (delimited, .txt) in Microsoft Excel, R, or another statistical package.
Filter data based on ‘In Regions’ column, ‘0’ indicates samples that are detected outside of region of interest and ‘1’ indicates samples that lie within the region of interest.
Normalize fluorescence value (‘Green’, ‘Yellow’, or ‘Red’) with TOF.
Perform relevant statistical testing using normalized values.
Data analysis
Representative results
Based on the protocols described above, we routinely perform automated fluorescence quantification for medium-throughput, high-content assays to determine expression levels for several reporter strains. We compared the two pipelines for fluorescence measurement described above (flow vermimetry and automated image analysis). Flow vermimetry is a remarkably sensitive fluorescence detection method that allows rapid measurement of individual phenotypes within a population. It is worth noting that worms exhibiting a desirable phenotype can also be selected away from the general population and retrieved for further use, much like fluorescence-activated cell sorting.
The region of interest was first determined by comparing samples containing worms alone, compounds alone, and a combination of both. Time-of-flight (ToF) and extinction (optical density) values were used to separate worms from other particles, such as insoluble compounds that tend to aggregate over time (Figure 1A). Fluorophores were simultaneously excited with both solid-state lasers, and fluorescence values were determined using bandpass filters (Figure 1B). This system enables users to accurately and effectively determine the fluorescence of a large number of samples from a population. This has many applications, including mutant screening, drug discovery, and basic science research. In addition, phenotypic measurement of different worm populations can be performed by coupling the FP platform with an automated sampler and simple software scripting that allows worms to be sampled from 96-well (or other format) plates rather than 50 mL conical tubes. The system stores measured parameters in tab-delimited files [e.g., ‘Source well’, ‘In Regions’, ‘TOF’, ‘Extinction’, and fluorescence values: ‘Green’, ‘Yellow‘, and ‘Red’ (Figure 1C)] that can be accessed and analyzed by many downstream packages, including standard office productivity suites or more custom-generated scripting languages.
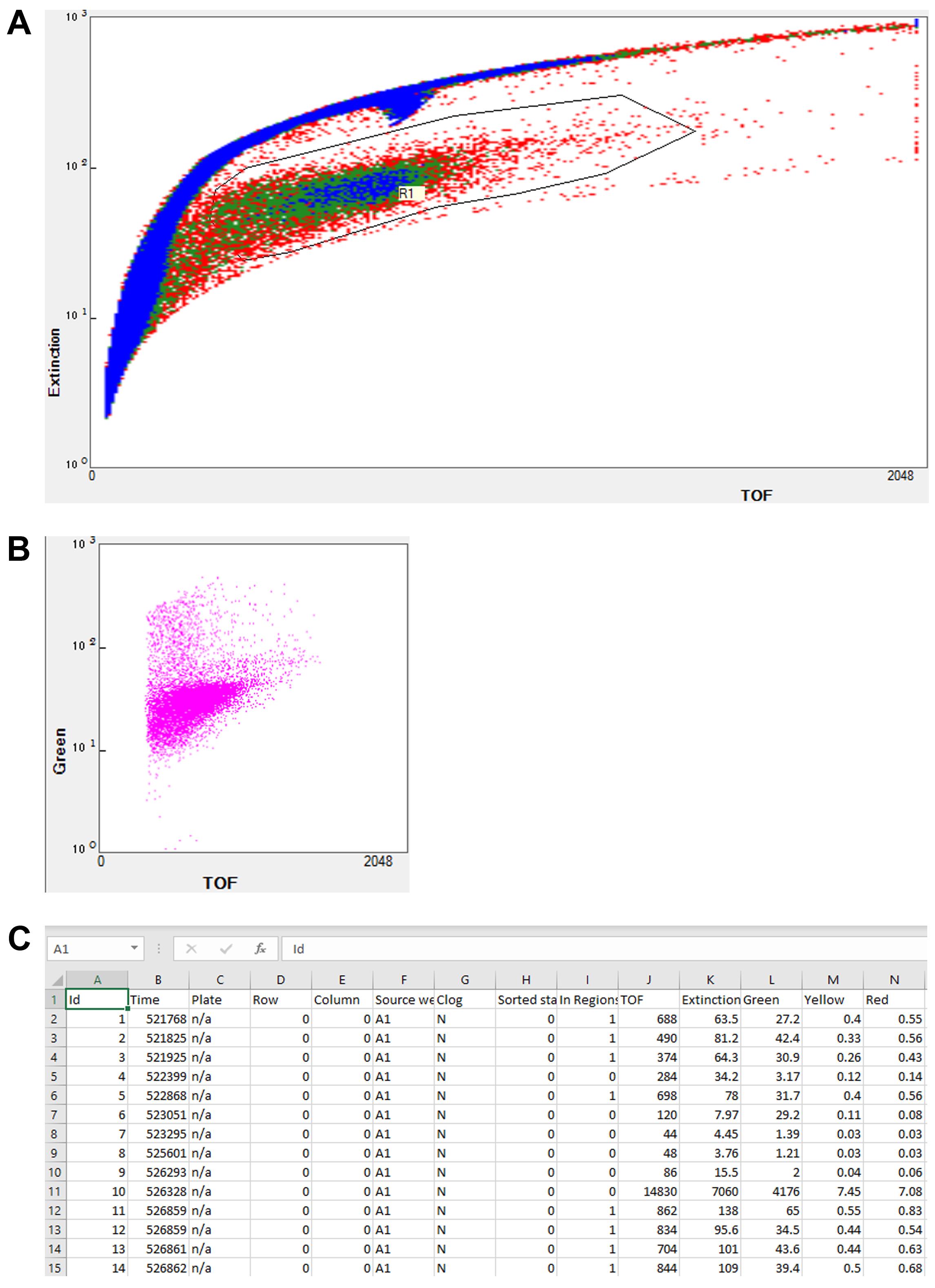
Figure 1. Flow vermimetry (with COPAS FP BioSorter) fluorescence measurement method. The FlowPilot software display consists of (A) a customizable plot for sample detection and selection, including customizable regions of interest (R1) and (B) customizable output plots. (C) Example of FlowPilot output opened in Microsoft Excel.While flow vermimetry provides measurements for each individual worm in a large population, the Cytation5 allows imaging in bright field and fluorescence channels for whole populations (Figure 2A). Representative images of worms from a fluorescent reporter strain that was treated with different mitochondrial stressors) are shown (Figure 2A). We have optimized the fluorescence quantification pipeline to differentiate worms from background in the acquired images by performing several crucial steps (based on a pipeline from Kirienko et al., 2013) (Figure 2B). First, fluorescent image backgrounds are flattened to improve the signal-to-noise ratio. Second, bright field images are converted into digital phase-contrast versions to minimize uneven illumination and improve contrast to optimize worm detection. Finally, cellular analysis is performed by specifying several parameters suitable for worms: threshold for object detection, minimum and maximum object size, and a smoothening algorithm to enhance object identification. This pipeline efficiently and effectively identifies worms and discards background particles, including insoluble compounds (Figure 2C). Fluorescence values are then calculated and exported into a file with a single click (Figure 2D).
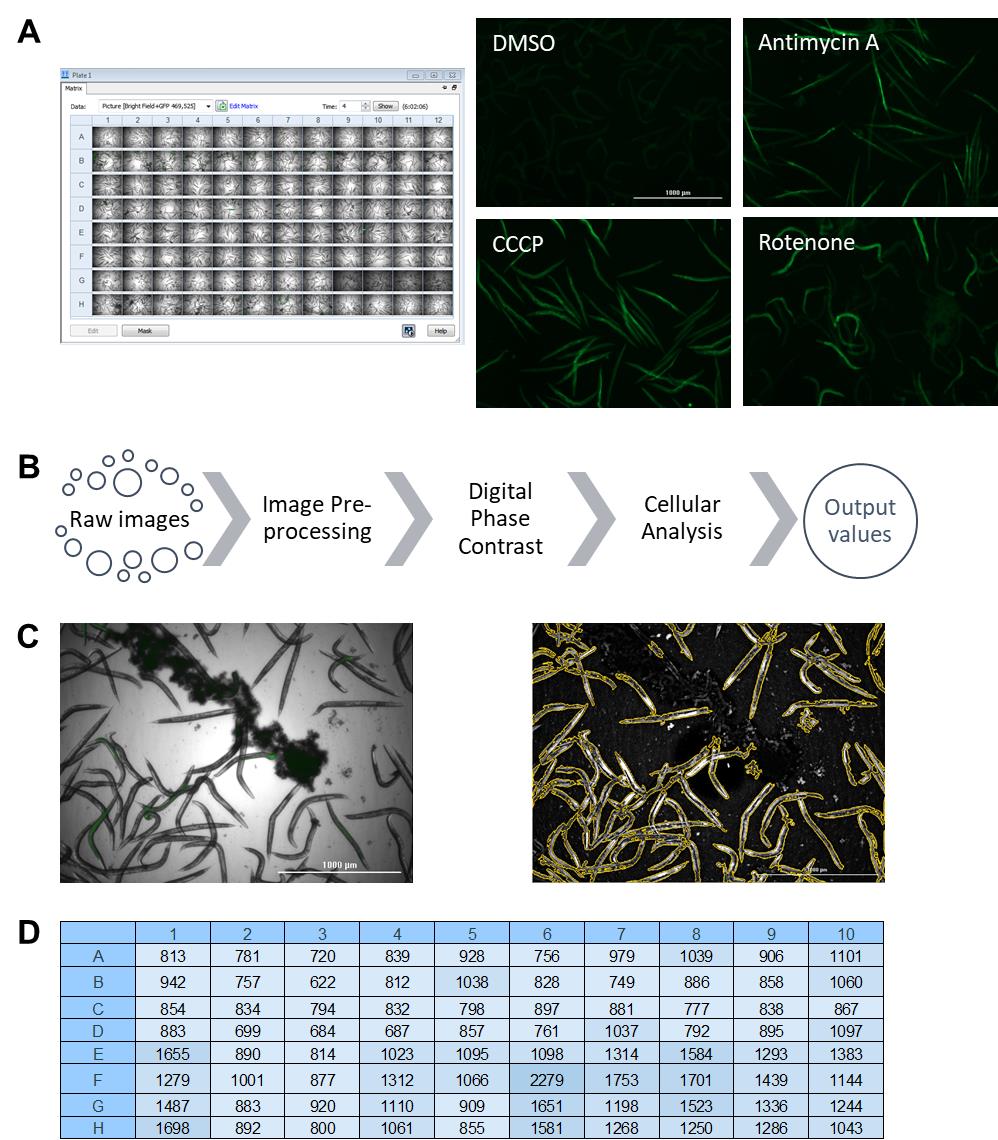
Figure 2. Pipeline for image acquisition and fluorescence quantification with Cytation5 and Gen5 software (BioTek). (A) Raw images obtained after imaging a 96-well plate, including representative fluorescence images of C. elegans reporter strain responsive to mitochondria-targeting drugs. (B) A pipeline for fluorescence quantification from raw images. (C) Example of worm identification before (left) and after (right) quantification pipeline. (D) Example of obtained fluorescence values from pipeline analysis after exporting into Microsoft Excel.To evaluate the consistency of these two approaches to produce reliable fluorescent values, we induced transgene expression in a C. elegans reporter strain carrying three tandem repeats of the Ethanol and Stress Response Element (ESRE) as a minimal promoter construct to drive expression of GFP (3XESRE::GFP) (Kirienko and Fay, 2010). This strain was exposed to rotenone (an inhibitor of Complex I of mitochondrial electron transport chain) and N-acetyl-L-cysteine (NAC, a canonical antioxidant and scavenger of reactive oxygen species). This combination potently activates the expression of the ESRE network, which is involved in the detection of reductive stress (Tjahjono et al., 2020). The concentrations of rotenone and NAC were varied to create a gradient of reporter expression. Worms were then split into two populations, one measured with flow vermimetry and the other imaged with the Cytation5. Normalized expression values from the Cytation5 machine (x-axis) were compared with values obtained from flow vermimetry (y-axis). The fluorescence values obtained strongly correlated (Figure 3).
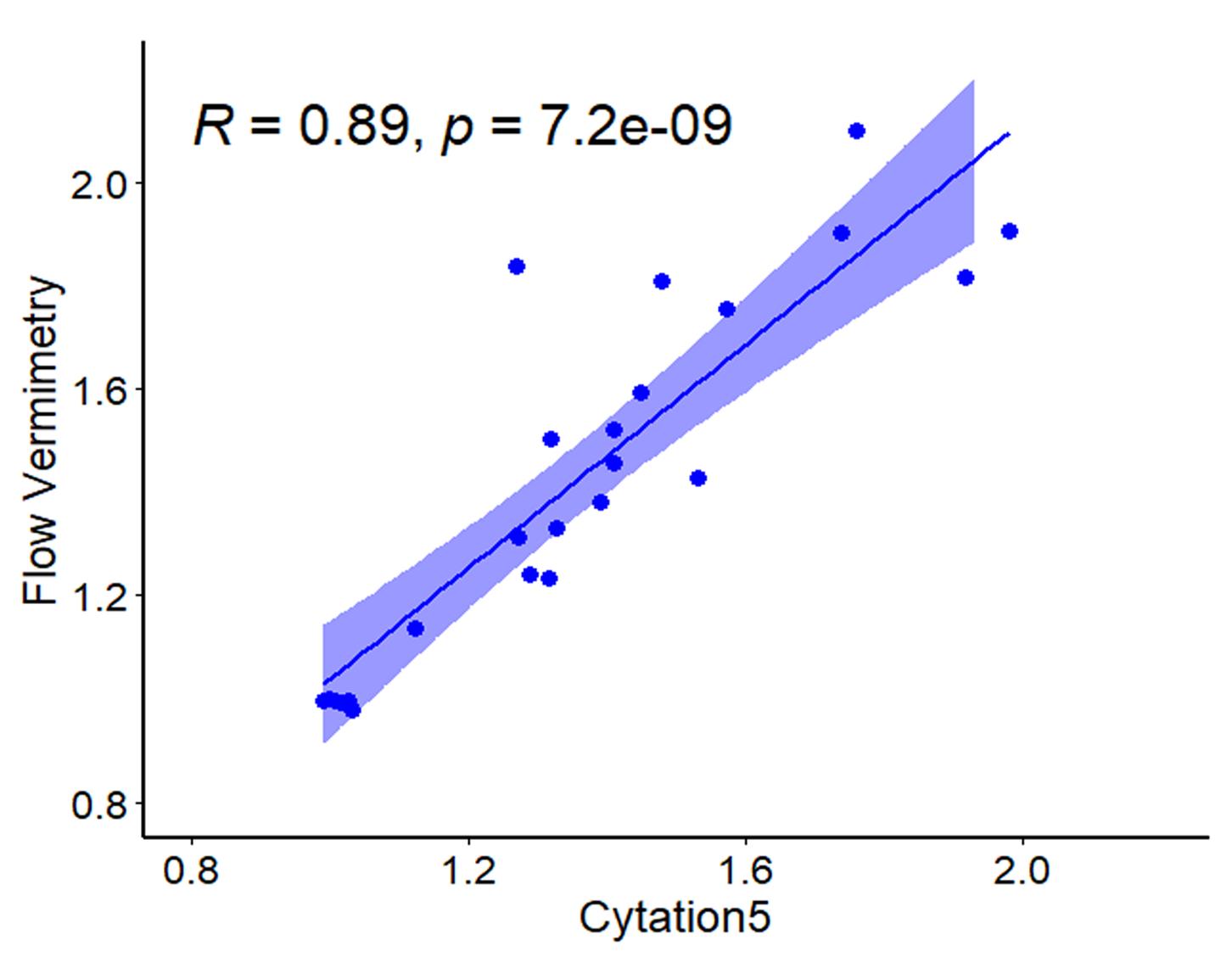
Figure 3. Fluorescence values obtained from flow vermimetry and Cytation5 are highly correlated. Fluorescence of the reporter strain carrying a 3XESRE::GFP was measured with flow vermimetry (y-axis) and imaged with Cytation5 (x-axis) after treatment with a gradient of rotenone (0, 25, 37.5, and 50 µM) and NAC (0, 1, 2, 3, 4, and 5 mM).Additionally, we assessed the ability of these approaches to quantify strong and weak fluorescence signals. Previously, we and others found that the loss of the transcription factor ATFS-1 in an spg-7(RNAi) mutant activates expression of the chaperone hsp-16.1 (Pellegrino et al., 2014; Munkácsy et al., 2016; Tjahjono et al., 2020). Expression of hsp-16.1 requires the presence of a pair of ESRE motifs in the gene’s promoter region. On this basis, we measured GFP signals for Phsp-16.1::GFP and Phsp-16.1(dd)::GFP (the latter of which lacks both ESRE motifs) upon exposure to empty vector (EV), atfs-1(RNAi), spg-7(RNAi), or spg-7(RNAi);atfs-1(RNAi). Flow vermimetry was able to recognize a wider range of fluorescence intensities but had somewhat higher variability (Figure 4A). As such, care needs to be taken to ensure that replicates are performed and that conditions are kept as consistent as possible.
Finally, variations (resulting in noise) are introduced both from the detector and optical setup of any fluorescence-based tool (Haider et al., 2016), as well as from the experiments and the samples’ inter-individual variability. Thus, we tested whether varying sample sizes dramatically altered the fluorescence output. We measured fluorescence expression of Ptbb-6::GFP reporter worms reared on E. coli expressing empty vector (EV), atfs-1(RNAi), spg-7(RNAi), or spg-7(RNAi);atfs-1(RNAi) via flow vermimetry. Interestingly, measurement of a sample size as low as 200 worms yielded very similar fluorescence values as measurement with 2000 worms, whether expression level was low or high (Figure 4B).
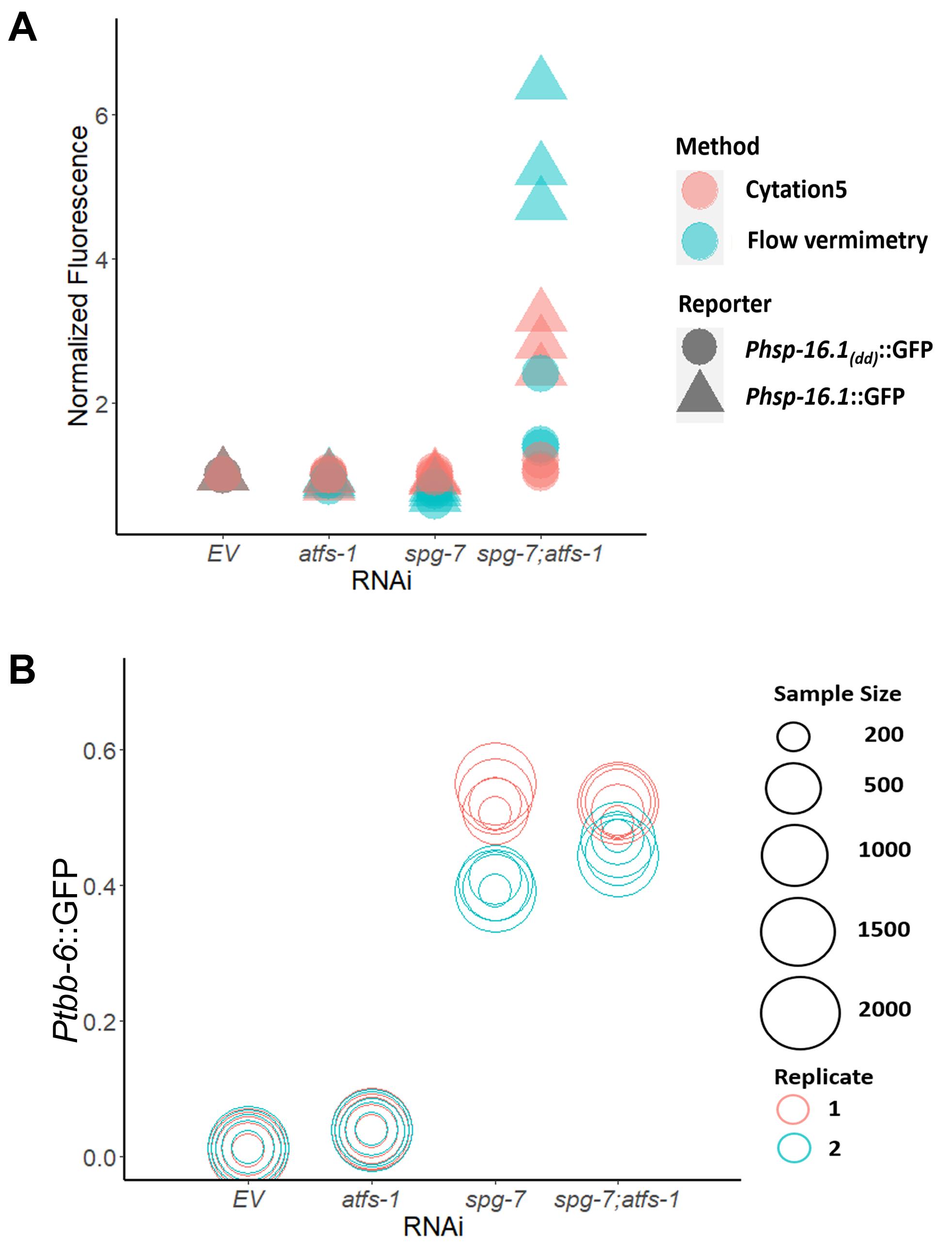
Figure 4. Flow vermimetry detected wider range of fluorescence signals. The fluorescence of (A) Phsp-16.1::GFP and Phsp-16.1(dd)::GFP, or (B) Ptbb-6::GFP worm reporter strains reared on E. coli expressing empty vector (EV), atfs-1(RNAi), spg-7(RNAi), or spg-7(RNAi);atfs-1(RNAi) was measured with (A) flow vermimetry and imaged with Cytation5 or (B) with flow vermimetry alone. ToF (Time of Flight) corresponds to worm length. At least 400 worms/replicate were used in (A). Sample sizes were varied in (B).The use of these tools for automated and high-content fluorescence quantification is invaluable to increase lab productivity. Either approach can efficiently measure the activation of multiple biomarkers from transgenic reporters or fluorescent dyes. Flow vermimetry provides records of individual phenotypes, allows data regarding population response to be obtained, rare outcomes to be screened for, and is an efficient and powerful method to obtain precisely targeted subpopulations for further analysis. It is also resistant to quantification problems that arise during overcrowding that can be problematic for microscopy-based techniques. On the other hand, the Cytation5 provides publication-quality images using a reliable pipeline for fluorescence quantification. This latter technique is also amenable to repeated imaging and analysis, allowing longer-term experiments during exposure to chemicals, pathogens, or even developmental events. Although these instruments have some degree of overlap, their combination allows for complex and nuanced study of biological events of interest.
Data visualization and statistical analysis
Correlation coefficient, p-value, and plot in Figure 3 were calculated and generated with the R package ggpubr (version 0.4.0). Figure 4A and 4B were generated with the R package ggplot2 (version 3.3.0). At least three biological replicates were analyzed, with each replicate comprised of ~400 worms (Figure 3 and Figure 4A). Sample sizes were indicated in graph for Figure 4B.
Notes
When analyzing flow vermimetry data, do not forget to filter out region ‘0’ from the ‘In Regions’ field. This is needed to ensure that the signals being analyzed originate from worms instead of debris.
Before performing imaging with Cytation5, ensure each channel is in focus. This can be done by performing an autofocus check and noting the focus position. If necessary, a small offset between channels may be used.
Recipes
Nematode Growth Media (NGM) (1 L)
18 g of agar
3 g of NaCl
2.5 g of peptone
Dissolved in 975 ml of Milli-Q water
Sterilize by autoclaving and wait until media cooled down to ~60°C before adding: 1 ml of CaCl2 (1 M), 1 ml of MgSO4 (1 M), 1 ml of cholesterol (5 mg/ml), and 25 ml of phosphate buffer
Pour into Petri dishes
Store at 4°C until ready to use
Worm Bleach Solution (100 ml)
10 ml of 5 M NaOH solution
20 ml of 5% sodium hypochlorite solution
70 ml of sterile Milli-Q water
Store at room temperature. Do not keep for more than a week
S Basal (1 L)
5.85 g of NaCl
1 g of K2HPO4
6 g of KH2PO4
Dissolved in 1 liter of Milli-Q water
Sterilize by autoclaving
Store at room temperature
Phosphate Buffer (1 L)
132 ml of K2HPO4 (1 M)
868 ml of KH2PO4 (1 M)
Store at room temperature
Acknowledgments
N.V.K., a CPRIT Scholar in Cancer Research, thanks the Cancer Prevention and Research Institute of Texas (CPRIT) for their generous support, CPRIT grant RR150044.This study was also funded by R35GM129294 (NIH NIGMS) and John S. Dunn Foundation Award. The funders had no role in study design, data collection or analysis, decision to publish or preparation of the manuscript. C. elegans strains used were provided by David Fay or obtained from the Caenorhabditis Genetics Center (CGC). CGC is supported by the NIH P40 OD010440 award. Techniques described were previously used in Kirienko lab, however data presented in this manuscript have not been published before.
Competing interests
The authors have declared that no competing interests exist.
References
- Anderson, Q. L., Revtovich, A. V. and Kirienko, N. V. (2018). A High-throughput, High-content, Liquid-based C. elegans Pathosystem. J Vis Exp(137).
- Angstman, N. B., Frank, H. G. and Schmitz, C. (2016). Advanced Behavioral Analyses Show that the Presence of Food Causes Subtle Changes in C. elegans Movement. Front Behav Neurosci 10: 60.
- Angstman, N. B., Kiessling, M. C., Frank, H. G. and Schmitz, C. (2015). High interindividual variability in dose-dependent reduction in speed of movement after exposing C. elegans to shock waves. Front Behav Neurosci 9: 12.
- Bazopoulou, D., Knoefler, D., Zheng, Y., Ulrich, K., Oleson, B. J., Xie, L., Kim, M., Kaufmann, A., Lee, Y. T., Dou, Y., Chen, Y., Quan, S. and Jakob, U. (2019). Developmental ROS individualizes organismal stress resistance and lifespan. Nature 576(7786): 301-305.
- Burns, A. R., Kwok, T. C., Howard, A., Houston, E., Johanson, K., Chan, A., Cutler, S. R., McCourt, P. and Roy, P. J. (2006). High-throughput screening of small molecules for bioactivity and target identification in Caenorhabditis elegans. Nat Protoc 1(4): 1906-1914.
- Conery, A. L., Larkins-Ford, J., Ausubel, F. M. and Kirienko, N. V. (2014). High-throughput screening for novel anti-infectives using a C. elegans pathogenesis model. Curr Protoc Chem Biol 6(1): 25-37.
- Gosai, S. J., Kwak, J. H., Luke, C. J., Long, O. S., King, D. E., Kovatch, K. J., Johnston, P. A., Shun, T. Y., Lazo, J. S., Perlmutter, D. H., Silverman, G. A. and Pak, S. C. (2010). Automated high-content live animal drug screening using C. elegans expressing the aggregation prone serpin α1-antitrypsin Z. PLoS One 5(11): e15460.
- Haider, S. A., Cameron, A., Siva, P., Lui, D., Shafiee, M. J., Boroomand, A., Haider, N. and Wong, A. (2016). Fluorescence microscopy image noise reduction using a stochastically-connected random field model.Sci Rep 6: 20640.
- Kamath, R. S., Fraser, A. G., Dong, Y., Poulin, G., Durbin, R., Gotta, M., Kanapin, A., Le Bot, N., Moreno, S., Sohrmann, M., Welchman, D. P., Zipperlen, P. and Ahringer, J. (2003). Systematic functional analysis of the Caenorhabditis elegans genome using RNAi.Nature 421(6920): 231-237.
- Kang, D., Kirienko, D. R., Webster, P., Fisher, A. L. and Kirienko, N. V. (2018). Pyoverdine, a siderophore from Pseudomonas aeruginosa, translocates into C. elegans, removes iron, and activates a distinct host response. Virulence 9(1): 804-817.
- Kirienko, D. R., Revtovich, A. V. and Kirienko, N. V. (2016). A High-Content, Phenotypic Screen Identifies Fluorouridine as an Inhibitor of Pyoverdine Biosynthesis and Pseudomonas aeruginosa Virulence. mSphere 1(4).
- Kirienko, N. V. and Fay, D. S. (2010). SLR-2 and JMJC-1 regulate an evolutionarily conserved stress-response network. EMBO J 29(4): 727-739.
- Kirienko, N. V., Kirienko, D. R., Larkins-Ford, J., Wahlby, C., Ruvkun, G. and Ausubel, F. M. (2013). Pseudomonas aeruginosa disrupts Caenorhabditis elegans iron homeostasis, causing a hypoxic response and death. Cell Host Microbe 13(4): 406-416.
- Kuzmanov, A., Karina, E. I., Kirienko, N. V. and Fay, D. S. (2014). The conserved PBAF nucleosome-remodeling complex mediates the response to stress in Caenorhabditis elegans. Mol Cell Biol 34(6): 1121-1135.
- Lucanic, M., Garrett, T., Gill, M. S. and Lithgow, G. J. (2018). A Simple Method for High throughput Chemical Screening in Caenorhabditis elegans. J Vis Exp(133).
- Maglioni, S., Arsalan, N., Franchi, L., Hurd, A., Opipari, A. W., Glick, G. D. and Ventura, N. (2015). An automated phenotype-based microscopy screen to identify pro-longevity interventions acting through mitochondria in C. elegans. Biochim Biophys Acta 1847(11): 1469-1478.
- Moy, T. I., Conery, A. L., Larkins-Ford, J., Wu, G., Mazitschek, R., Casadei, G., Lewis, K., Carpenter, A. E. and Ausubel, F. M. (2009). High-throughput screen for novel antimicrobials using a whole animal infection model.ACS Chem Biol 4(7): 527-533.
- Munkácsy, E., Khan, M. H., Lane, R. K., Borror, M. B., Park, J. H., Bokov, A. F., Fisher, A. L., Link, C. D. and Rea, S. L. (2016). DLK-1, SEK-3 and PMK-3 Are Required for the Life Extension Induced by Mitochondrial Bioenergetic Disruption in C. elegans. PLoS Genet 12(7): e1006133.
- Okkenhaug, H., Chauve, L., Masoudzadeh, F., Okkenhaug, L. and Casanueva, O. (2020). Worm-align and Worm_CP, Two Open-Source Pipelines for Straightening and Quantification of Fluorescence Image Data Obtained from Caenorhabditis elegans.J Vis Exp(159).
- Pellegrino, M. W., Nargund, A. M., Kirienko, N. V., Gillis, R., Fiorese, C. J. and Haynes, C. M. (2014). Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature 516(7531): 414-417.
- Raviv, T. R., Ljosa, V., Conery, A. L., Ausubel, F. M., Carpenter, A. E., Golland, P. and Wahlby, C. (2010). Morphology-guided graph search for untangling objects: C. elegans analysis. Med Image Comput Comput Assist Interv 13(Pt 3): 634-641.
- Revtovich, A., Lee, R. and Kirienko, N. (2019). Interplay between mitochondria and diet mediates pathogen and stress resistance in Caenorhabditis elegans.PLoS Genet 15(3): e1008011.
- Rual, J. F., Ceron, J., Koreth, J., Hao, T., Nicot, A. S., Hirozane-Kishikawa, T., Vandenhaute, J., Orkin, S. H., Hill, D. E., van den Heuvel, S. and Vidal, M. (2004). Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res 14(10B): 2162-2168.
- Squiban, B., Belougne, J., Ewbank, J. and Zugasti, O. (2012). Quantitative and automated high-throughput genome-wide RNAi screens in C. elegans. J Vis Exp(60).
- Tjahjono, E. and Kirienko, N. V. (2017). A conserved mitochondrial surveillance pathway is required for defense against Pseudomonas aeruginosa.PLoS Genet 13(6): e1006876.
- Tjahjono, E., McAnena, A. P. and Kirienko, N. V. (2020). The evolutionarily conserved ESRE stress response network is activated by ROS and mitochondrial damage.BMC Biol 18(1): 74.
- Wählby, C., Kamentsky, L., Liu, Z. H., Riklin-Raviv, T., Conery, A. L., O'Rourke, E. J., Sokolnicki, K. L., Visvikis, O., Ljosa, V., Irazoqui, J. E., Golland, P., Ruvkun, G., Ausubel, F. M. and Carpenter, A. E. (2012). An image analysis toolbox for high-throughput C. elegans assays.Nat Methods 9(7): 714-716.
- Wildey, M. J., Haunso, A., Tudor, M., Webb, M. and Connick, J. H. (2017). Chapter Five - High-Throughput Screening. In: Goodnow, R. A. (Ed.). Annual Reports in Medicinal Chemistry. 50: Academic Press. p. 149-95.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Tjahjono, E., Revtovich, A. V. and Kirienko, N. V. (2021). Imaging and Fluorescence Quantification in Caenorhabditis elegans with Flow Vermimetry and Automated Microscopy. Bio-protocol 11(10): e4024. DOI: 10.21769/BioProtoc.4024.
Category
Developmental Biology > Cell signaling > Stress response
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.