- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Identification of R-loop-forming Sequences in Drosophila melanogaster Embryos and Tissue Culture Cells Using DRIP-seq
Published: Vol 11, Iss 9, May 5, 2021 DOI: 10.21769/BioProtoc.4011 Views: 7146
Reviewed by: Qin TangAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
R-loops are non-canonical nucleic structures composed of an RNA–DNA hybrid and a displaced ssDNA. Originally identified as a source of genomic instability, R-loops have been shown over the last decade to be involved in the targeting of proteins and to be associated with different histone modifications, suggesting a regulatory function. In addition, R-loops have been demonstrated to form differentially during the development of different tissues in plants and to be associated with diseases in mammals. Here, we provide a single-strand DRIP-seq protocol to identify R-loop-forming sequences in Drosophila melanogaster embryos and tissue culture cells. This protocol differs from earlier DRIP protocols in the fragmentation step. Sonication, unlike restriction enzymes, generates a homogeneous and highly reproducible nucleic acid fragment pool. In addition, it allows the use of this protocol in any organism with minimal optimization. This protocol integrates several steps from published protocols to identify R-loop-forming sequences with high stringency, suitable for de novo characterization.
Graphic abstract:
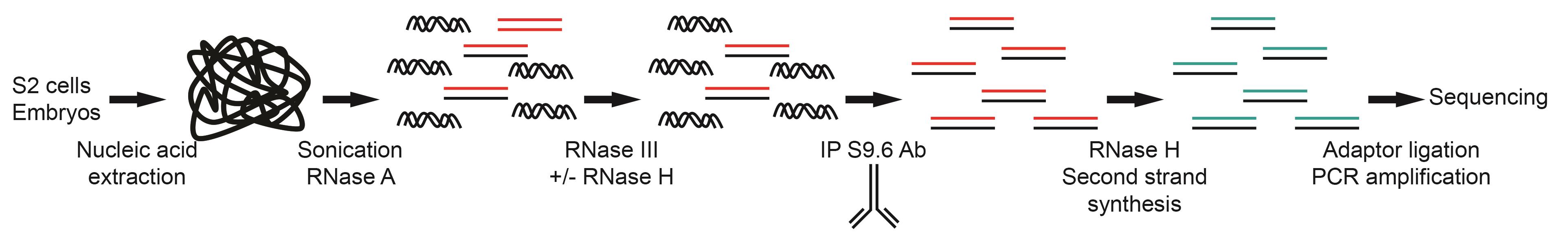
Figure 1. Overview of the strand-specific DRIP-seq protocol
Background
Overview
R-loops are triple-stranded nucleic acid structures that form when an RNA hybridizes with a complementary ssDNA, leading to displacement of the second DNA strand. R-loops were first described as a by-product of transcription and a source of genomic instability that needed to be resolved. However, research over the last decade has shown that R-loops can be associated with specific histone modifications and transcriptional status, induce the targeting of proteins, or act as promoters (Chedin and Benham, 2020; Niehrs and Luke, 2020). To study R-loop function, protocols have been developed to map these structures in multiple organisms. Differences have been observed between species: R-loop-forming sequences in mammals have a GC skew; those in Saccharomyces cerevisiae have an AT skew; and in plants, both GC and AT skews have been reported (Sanz et al., 2016; Wahba et al., 2016; Xu et al., 2017; Hartono et al., 2018). Differences in the properties and localization of R-loop-forming sequences between species and the association of R-loops with different diseases and gene misexpression highlight the importance of developing standard R-loop mapping protocols to perform rigorous comparisons.
R-loop maps in plants have shown that some R-loops can form differentially during development (Fang et al., 2019; Xu et al., 2020) and may potentially be involved in changes in transcriptional status. In mammals, active enhancers have been associated with R-loop formation, and these R-loops can act as promoters (Tan-Wong et al., 2019). The study of R-loops in the context of development in intact organisms with available genetic tools to evaluate their formation, resolution, and function, will allow a better understanding of R-loop biology.
Here, we provide a detailed strand-specific DRIP-seq protocol to identify R-loop-forming sequences in Drosophila melanogaster embryos and tissue culture cells (Figure 1). Similar to other DRIP protocols, our protocol relies on the S9.6 antibody to identify R-loop-forming sequences but also considers that the weak affinity of the S9.6 antibody for dsRNA can generate false positive signals (Phillips et al., 2013; Hartono et al., 2018; König et al., 2017). This single-strand DRIP-seq protocol in association with the genetic tools available for Drosophila melanogaster should be a powerful system to study R-loop function in the whole organism, during development, and in different tissues (Figure 2, Table 1). DRIP-seq can be combined with genetic manipulation to overexpress or knockdown genes with a view to identifying their effect on R-loop formation, or with cell synchronization to follow R-loop formation during the cell cycle. Finally, the use of sonication rather than restriction enzymes to fragment the genomic DNA means that this protocol could potentially be adapted to other organisms with minimal optimization.
Limitations
The protocol relies on the S9.6 antibody to immunoprecipitate RNA–DNA hybrid-containing fragments. This antibody is not a perfect tool: it shows differential binding affinity based on the sequence of the R-loop with no correlation with the GC content and a preference for longer RNA–DNA hybrids. It can bind dsRNA, albeit with an affinity 5-times lower than that for RNA–DNA hybrids (Phillips et al., 2013; König et al., 2017). The latter two limitations can be overcome during the preparation of nucleic acids. After nucleic acid extraction, ssRNA and dsRNA are removed by incubating the nucleic acids with RNase A in the presence of 0.5 M NaCl to avoid the degradation of RNA in the RNA–DNA hybrids. This first digestion step is followed by an incubation with RNase III to degrade any dsRNA that were not degraded by RNase A. By using two different RNases before immunoprecipitation, we removed all ssRNA and dsRNA but also potentially some R-loops, which can be sensitive to these RNases. Thus, these steps make the experiment more stringent and limit false positives but could be modified to balance stringency with sensitivity, since some R-loops may be sensitive to these treatments. Removal of RNA that can interact with S9.6 is most relevant when the RNA component of the R-loop is to be sequenced; although, we have observed that the RNA–DNA hybrid pulldown efficiency is increased when stringent RNase treatment is used. It is also essential to have an RNase H-treated negative control to evaluate the specificity of the pulldown (Figure 3). We found that commercially available E. coli RNase H does not consistently completely digest RNA–DNA hybrids in nucleic acid preparations. This can be solved by producing and using highly active human RNase H1 and RNase H2. This negative control is especially important during optimization of the DRIP protocol; it ensures that the technique is specific. It is also used to call peaks when DRIP-seq is performed (Figures 2, 4, and 5).
To avoid bias toward longer fragments during the immunoprecipitation step, we fragment the nucleic acids by sonication (Figure 4A). Sonication allows us to obtain a homogenous population of fragments with an average size of 300 bp. Sonication is performed after RNase A treatment to avoid the possibility of generating new RNA–DNA hybrids during the DRIP procedure. Sonication is reported to reduce the recovery of some RNA–DNA hybrids as compared with restriction digestion (Crossley et al., 2020).
Advantages
Advantages compared with other DRIP protocols
This protocol uses a gentle lysis step to extract the nucleic acids. Cell lysis can be performed on tissue culture cells, whole embryos, or dissected tissues from larval, pupal, or adult Drosophila melanogaster. Although the ~200 mg Drosophila embryos needed to perform one DRIP-seq experiment is relatively high, the possibility to freeze tissues and pool them for a single nucleic acid extraction makes it feasible. By dissecting Drosophila melanogaster and performing DRIP on discs, organs, or sorted cell populations, it should be possible to identify cell- or tissue-specific R-loops.
This DRIP protocol, contrary to several protocols developed for mammalian cells (e.g., Sanz et al., 2016), does not use a restriction enzyme cocktail to fragment the nucleic acids. Instead, sonication is used, which leads to the fragmentation of nucleic acids at an average size of 300 bp (Figure 4A). These fragments have a homogenous size and the sonication is highly reproducible. An increase in resolution by using sonication, as compared with restriction digestion, in mammalian cells has recently been demonstrated (Crossley et al., 2020). By using sonication, the protocol can easily be adapted to other organisms with little optimization; the only step that may require optimization is lysis. Sonication has another advantage: it leads to disruption of the displaced single-strand DNA of the R-loop, which makes it possible to prepare strand-specific sequencing libraries using either the DNA or the RNA moiety of the RNA–DNA hybrid (Wahba et al., 2016).
Advantages compared with other methods
Another method to identify R-loop-forming sequences relies on a catalytically inactive form of the RNase H1 enzyme (dRNase H1) (Ginno et al., 2012; Chen et al., 2017). The use of dRNase H1 in cells presents several potential problems, which may explain the differences in the identification of R-loop-forming sequences with this method versus the S9.6 antibody. Firstly, RNase H1 is not the only regulator of R-loops in cells; it has been suggested that topoisomerases are the main enzymes responsible for the resolution of R-loops that form co-transcriptionally in human cells (Manzo et al., 2018; Zhang et al., 2019), while RNase H1 and H2 target R-loops once they are formed. Secondly, a recent article by Lockhart et al., 2019 demonstrated that RNase H1 is activated upon stress, while RNase H2 displays the main activity under physiological conditions in S. cerevisiae. Thirdly, with dRNase H1, R-loop identification may be limited to R-loops that are accessible, protein-free, and normally degraded by RNase H enzymes. Fourthly, expression of dRNase H1 may stabilize R-loops. While this may allow detection of transient R-loops, it could also skew interpretation of where stable R-loops exist. Finally, RNase H1 has two RNA–DNA hybrid binding domains, both of which need to bind to induce degradation of the RNA moiety of the hybrid (Nowotny et al., 2008). This may prevent or limit the detection of smaller RNA–DNA hybrids. Thus, while dRNase H1 may be a useful tool in some contexts, the interpretation of DRIP results may be more straightforward. Comparison of results from both methods could also yield complementary information.
Similar to DRIP, dRNase H1 has been used to isolate RNA–DNA hybrids after extraction of nucleic acids from tissue culture cells; however, bias of the enzyme toward longer hybrids and its weaker affinity as compared with the S9.6 antibody make it less efficient (Ginno et al., 2012).
Finally, native bisulfite sequencing is an alternative high-resolution method for identifying R-loops. Bisulfite converts cytosine to uracil in single-stranded DNA (Yu et al., 2003); thus, this method does not detect RNA–DNA hybrids but the ssDNA strand that is displaced when they form. However, the presence of methyl-cytosine in the genome blocks modification of the ssDNA, and ssDNA can be displaced by the formation of other non-canonical DNA structures such as G-quadruplexes and I-motifs, which could lead to false negative or false positive results, respectively.
The single-strand DRIP-seq protocol presented here has been optimized to identify R-loop-forming sequences in Drosophila melanogaster embryos and tissue culture cells, and can potentially be used in other organisms with minimal optimization. This protocol could be a standardized means to evaluate R-loops across developmental stages or in different tissues or organisms.
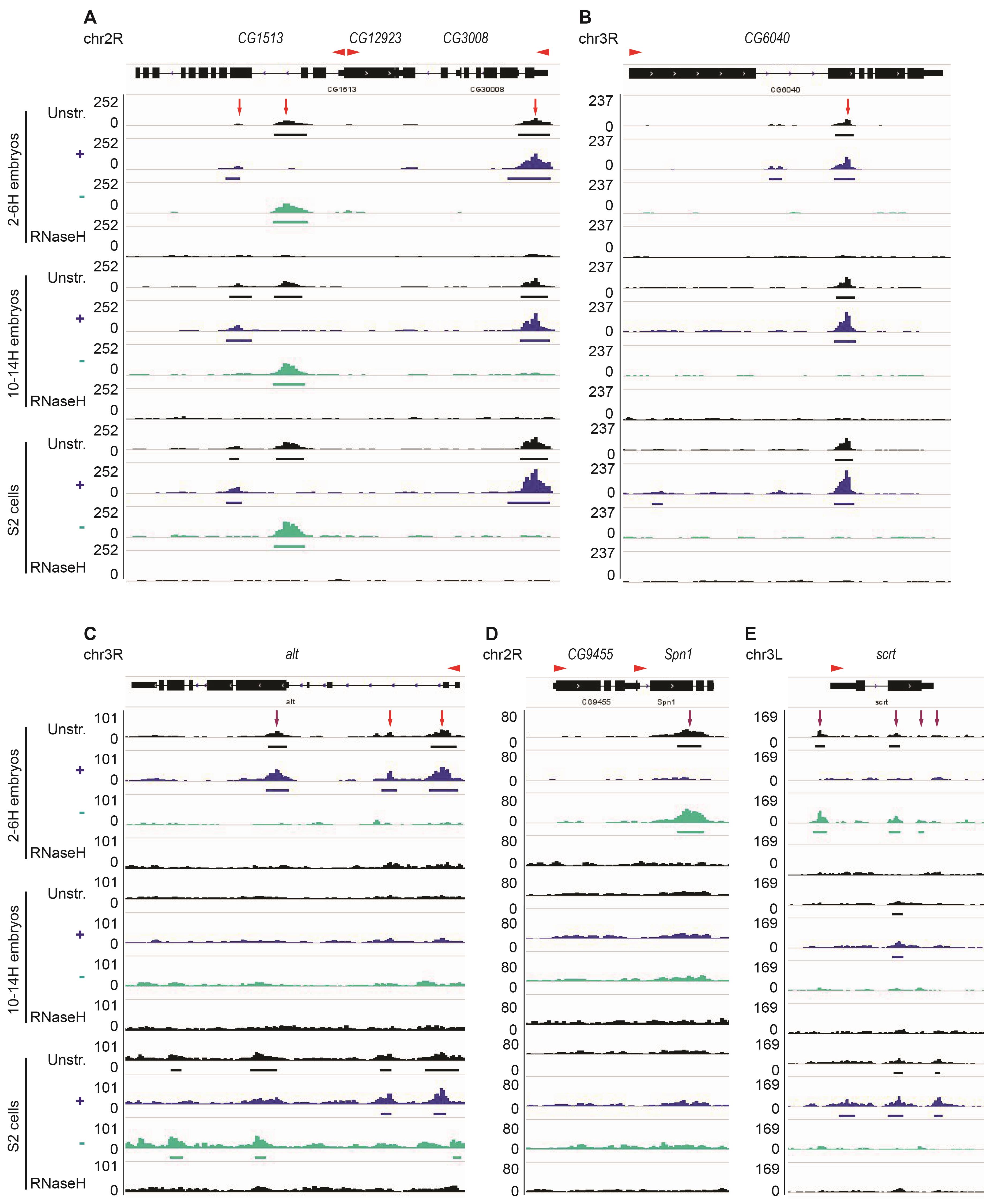
Figure 2. Examples of shared and distinct R-loops among S2 cells and early and later stage embryos. A-B. R-loops form over the CG1513, CG3008, and CG6040 genes in S2 cells and 2-6 H and 10-14 H embryos (red arrow). No R-loop is formed over the CG12923 gene. C. At the alt gene, three R-loops are detected in 2-6 H but not in 10-14 H embryos. Two of these R-loops (red arrows) are also present in S2 cells, while one of them is replaced by an R-loop on the opposite strand (purple arrow). D. R-loop formed over Spn1 is only present in 2-6 H embryos (purple arrows). E. R-loops form over scrt on the + strand in 2-6 H embryos and the - strand in 10-14 H embryos and in S2 cells. “Unstr” corresponds to unstranded DRIP-seq data, “+” and “-” indicate the strand-specific track, and RNase H-treated sample acts as a negative control. The red arrowhead indicates the orientation of the transcript. Note that the “+” and “-” strands refer to the DNA strand such that “+” strand R-loops should arise from transcription in the leftward direction. Thus, in A, the R-loop in the CG3008 gene is in the expected orientation to have arisen from gene transcription, while that in the gene CG1513 would be derived from antisense transcription.
Table 1. Data availability
Materials and Reagents
RNase H1 and H2 expression and purification
Amicon® ultra centrifugal filter, 0.5 ml 10K (EMD Millipore, catalog number: UFC5010BK)
Amicon® ultra centrifugal filter, 0.5 ml 3K (EMD Millipore, catalog number: UFC5003BK)
Econo-column (2.5*20 cm) (Bio-Rad, catalog number: 7374252)
E. coli RosettaTM 2(DE3)pLysS SinglesTM Competent Cells – Novagen (Sigma, catalog number: 71401)
Ni-NTA agarose (Qiagen, catalog number: 1018244)
Glutathione-superflow resin (Takara, catalog number: 635607)
PreScission protease plus (Homemade; commercial enzyme could also be used)
Ampicillin (Bioshop, catalog number: AMP201.100)
LB Broth Miller (Bioshop, catalog number: LBL407.500)
Albumin, bovine serum (Bioshop, catalog number: ALB001.100)
Lysozyme (Bioshop, catalog number: LYS702.5)
Imidazole (OmniPur) (Millipore, catalog number: 5710-OP)
Glycerol (Bioshop, catalog number: GLY001.4)
IPTG (Bioshop, catalog number: IPT002.5)
RNase H2 extraction buffer (see Recipes)
RNase H1 lysis buffer (see Recipes)
RNase H1 wash buffer (see Recipes)
RNase H1 elution buffer (see Recipes)
RNase H size column buffer (see Recipes)
RNase H storage buffer (see Recipes)
Protease inhibitors (see Recipes)
Protease inhibitors and additives
TLCK (Sigma, catalog number: T7254)
Benzamidine (Bioshop, catalog number: BEN601.25)
Pepstatin A (Bioshop, catalog number: PEP605.25)
1,10-Phenanthroline (Sigma, catalog number: 131377-5G)
PMSF (Fisher Scientific, catalog number: 19538125)
Aprotinin (Bioshop, catalog number: APR200)
Leupeptin (Bioshop, catalog number: LEU011.50)
NP40 (Nonidet P40 substitute, Fluka catalog number: 74385) (can substitute Sigma, catalog number: 74385)
DTT (Bioshop, catalog number: DTT002.100)
Activity testing of RNase H1 and H2
rNTP (NEB, catalog number: N0450S)
UTP α-P32 (Perkin Elmer, catalog number: BLU007H250UC)
T7 RNA polymerase (NEB, catalog number: M0251L)
SSC 20× (see Recipes)
Tri-sodium citrate (Bioshop, catalog number: CIT001.205)
Drosophila melanogaster embryo collection
Fly bottles
Oregon R flies (Dr Éric Lécuyer lab; available through Bloomington Drosophila stock center)
Agar A (Bioshop, catalog number: FB0010)
Sugar (RedPath)
Apple juice
TEGOSEPT, 1 KG (Nipagin) (Diamed.ca, catalog number: GEN20-258)
Homemade sieve made with NITEX (can be obtained from https://flystuff.com/)
Funnel
Fly cages (fly cages, food, and bottles can be obtained from https://flystuff.com/)
Fly food (prepared in-house)
Methanol (Bioshop, catalog number: MET302.4)
Embryo lysis buffer (see Recipes)
Apple juice plate (see Recipes)
1× PBS (see Recipes)
1× PBT (see Recipes)
Cell culture
S2 cells (Invitrogen, catalog number: R69007)
GibcoTM Schneider's Drosophila Sterile Medium (Thermo Fisher Scientific, catalog number: 21720-024)
FBS (Thermo Fisher Scientific, catalog number: 16140-089)
Nucleic acid extraction and preparation
Proteinase K (Biobasic, catalog number: PB0451)
Phase lock gel, heavy (VWR, catalog number: 10847-802)
DNase I (RNase-free) (NEB, catalog number: M0303L)
AmbionTM RNase III (Thermo Fisher Scientific, catalog number: AM2290)
RNase A (Qiagen, catalog number: 19101)
UltraPureTM DNase/RNase-free distilled water (Thermo Fisher Scientific, catalog number: 10977015)
Covaris microTUBE AFA fiber pre-split snap-cap 6*16 mm (Covaris, catalog number: 520045 )
Phenol-chloroform isoamyalcohol (Bioshop, catalog number: PHE512.400)
Chloroform (Bioshop, catalog number: CCL402.1)
Reagent alcohol (Sigma, catalog number: 277649-1)
Tris (Bioshop, catalog number: TRS001.10)
Acetic acid, glacial (Thermo Fisher Scientific, catalog number: 351271-212)
EDTA (Bioshop, catalog number: EDT002.500)
Sodium acetate (Bioshop, catalog number: SAA304.5)
Sodium chloride (Bioshop, catalog number: SOD002.10)
1× RNase H buffer (see Recipes)
TE (see Recipes)
DRIP
Anti-DNA–RNA Hybrid [S9.6] antibody (Kerafast, catalog number: ENH002, hybridomas are available through ATCC, HB-8730)
DynabeadsTM Protein G for Immunoprecipitation (Thermo Fisher Scientific, catalog number: 10004D)
NucleoSpin® Gel and PCR (Macherey-Nagel, catalog number: 740609.250)
DNA Clean & ConcentratorTM (Zymo Research, catalog number: D4014)
BSA, molecular biology grade (NEB, catalog number: B9000S)
10× DRIP binding buffer (see Recipes)
1× DRIP binding buffer (see Recipes)
DRIP elution buffer (see Recipes)
Library preparation and qPCR
NEBNext® Multiplex Oligos for Illumina® (Index Primers Set 1) (NEB, catalog number: E7735S)
NEBNext® UltraTM II Directional RNA Library Prep Kit for Illumina® (NEB, catalog number: E7760S)
RNase H (New England Biolabs (NEB), catalog number: M0297L)
PowerUp SYBR Green PCR master mix (Thermo Fisher Scientific, catalog number: A25741)
Agarose and SDS-PAGE gel preparation and staining
SYBRTM Gold Nucleic Acid Gel Stain (Thermo Fisher Scientific, catalog number: S11494)
SYPRO Ruby Solution (Lonza, catalog number: 50562)
Glycine (Bioshop, catalog number: GLN001.10)
0.5× TBE (see Recipes)
10× DNA loading buffer (see Recipes)
SDS-PAGE running buffer (see Recipes)
Protein loading buffer (see Recipes)
Plasmids
pHis-MBP-hRNaseH1 (Alecki et al., 2020)
pGEX6P1-hsRNASEH2BCA (Addgene, catalog number: 108693)
vg-pETBlue (Alecki et al., 2020)
Equipment
Typhoon Imager (GE Healthcare) for in-gel fluorescence imaging and phosphorimaging
Viaa7 PCR system (Thermo Fisher Scientific) for real-time PCR
Covaris E220 (Covaris) for nucleic acid sonication
Centrifuge Avanti® J-E (Beckman Coulter)
AKTA FPLC (or equivalent chromatography system)
HiLoad 20/60 Superdex 200 column (GE Healthcare, catalog number: GE28-9893-36) (available from Sigma)
Superdex 200 10/300 GL size exclusion column (GE Healthcare, catalog number: 28990944)
Sonicator (Sonics, Vibra cellsTM) (probe sonicator for cell lysis)
Nutator rotating platform to gently mix tubes
Bacterial shaker
10 cm Petri dish
Liquid nitrogen for flash freezing
-80°C Freezer
Nanodrop spectrophotometer
Water bath
24°C Incubator
27°C Shaking incubator for S2 cells; S2 cells can also be grown on plates at room temperature
Procedure
RNase H1 and H2 expression and purification
RNase H1 expression and purification (Figures 3A-3D)
Transform E. coli Rosetta cells with pHis-MBP-hRNaseH1. The next day, inoculate 2 ml LB-ampicillin (100 ng/µl) with a single colony and allow it to grow O.N. at 37°C in a shaking incubator.
Add 1 ml culture from (a) to 200 ml LB-ampicillin and allow it to grow O.N. at 37°C in a shaking incubator.
Add 100 ml culture from (b) to 1 L LB-ampicillin and induce RNase H1 expression with 0.1 mM IPTG for 4 h at 37°C.
Harvest the cells by centrifuging for 20 min at 4,000 × g, 4°C.
Discard the supernatant, freeze the cell pellet in liquid nitrogen, and store for at least a few hours at -80°C.
Thaw the cell pellet on ice and add 20 ml RNase H1 lysis buffer containing 0.1 mg/ml lysozyme, 100 µg RNase A, 100 µg DNase I, and protease inhibitors.
Incubate for 30 min on ice.
Keep the cells on ice and sonicate 12 times for 15 s ON, 15 s OFF at an amplitude of 60%.
Centrifuge for 20 min at 20,000 × g, 4°C.
Keep the supernatant as the crude extract. The supernatant can be frozen in liquid nitrogen and stored at -80°C for future purification.
Wash 3 ml Ni-NTA beads 3 times with 15 ml RNase H1 lysis buffer in a 50-ml tube by centrifuging for 2 min at 207 × g.
Add the crude extract to the beads and incubate O.N. at 4°C on a nutator. Starting from this step, all the washes and elutions must be performed at 4°C to ensure minimal protein degradation.
Wash an Econo column with RNase H1 lysis buffer and transfer the beads and crude extract into the column.
Save the flowthrough (if all the RNase H1 protein did not bind to the beads, the flowthrough can be incubated with the Ni-NTA resin a second time).
Wash 4 times with 15 ml RNase H1 wash buffer supplemented with protease inhibitors and collect each wash.
Elute the protein 5 times with 3 ml RNase H1 elution buffer containing protease inhibitors; collect 1-ml fractions.
Load 2.5 µl each fraction on a 10% SDS-PAGE gel and stain with SYPRO Ruby following the manufacturer’s instructions (Figure 3B).
Pool the elutions with similar concentrations and dialyze them against 1 L chilled RNase H storage buffer.
Equilibrate a HiLoad 20/60 Superdex 200 size exclusion column on an AKTA FPLC with 2-3 CV 20 mM Tris-HCl pH 7.0 containing 150 mM NaCl and 0.2 mM PMSF at 4°C (equilibration can be performed overnight).
Concentrate the sample to ≤ 5 ml if necessary. Centrifuge at ≥ 10,000 × g for 10 min at 4°C to remove any precipitate and load on the Superdex 200 column.
Run the Superdex 200 column at 4°C. Collect 1.5-ml fractions.
Run 5 µl each fraction on a 10% SDS-PAGE gel and stain with SYPRO Ruby.
Pool the fractions containing RNase H1 protein without degradation products and dialyze them twice O.N. at 4°C against 2 L chilled RNase H storage buffer.
Concentrate the protein 2–3-fold with an Amicon ultra column 10,000 MCWO following the manufacturer’s instructions to obtain a protein concentration of 0.5–1 µg/µl.
Aliquot, freeze in liquid nitrogen, and store at -80°C.
Run 2.5 µl on a 10% SDS-PAGE gel and stain with SYPRO Ruby to confirm the quality of the preparation (Figure 3D).
Determine the protein concentration on a NanoDrop using an extinction coefficient = 81,050 M-1cm-1 and a MW = 32 kDa. For 1 L of culture, ~9 mg purified RNase H1 is expected.
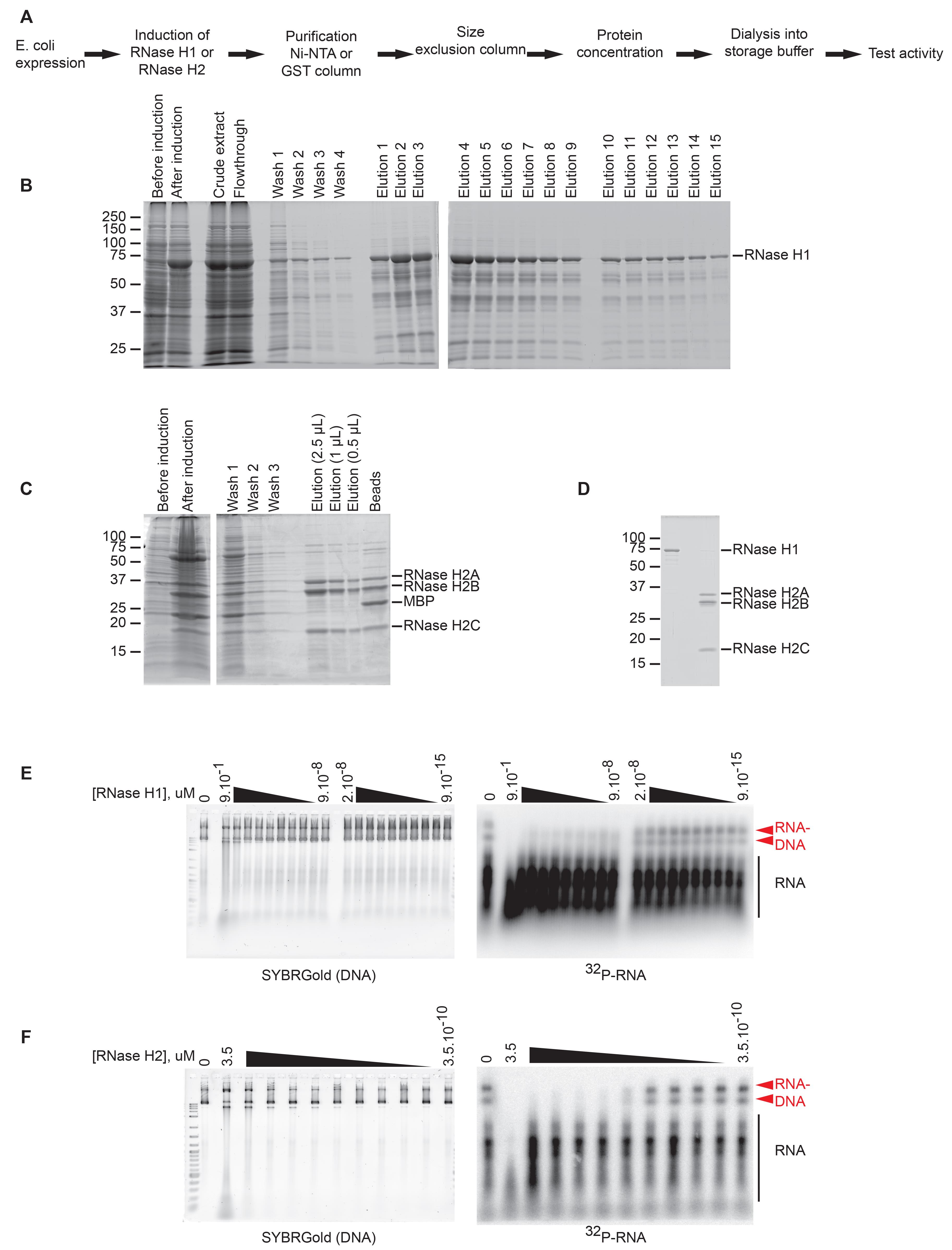
Figure 3. Preparation of human RNase H1 and H2 proteins. A. Overview of RNase H1 and H2 expression and purification. B-C. SDS-PAGE gels of RNase H1 (B) and RNase H2, and (C) induction and purification on an Ni-NTA or GST column. D. SDS-PAGE gel of RNase H1 and H2 protein after purification and dialysis, stained with SYPRO Ruby. E-F. Agarose gel of transcribed DNA after incubation with RNase H1 (E) or RNase H2 (F), showing that both enzymes are active and degrade RNA–DNA hybrids.RNase H2 expression and purification (Figures 3A-3D)
Transform E. coli Rosetta cells with pGEX6P1-hsRNASEH2BCA. Inoculate two 5-ml starter cultures of LB-ampicillin (100 ng/µl) with a single colony and grow O.N. at 37°C with shaking.
The next day, inoculate 1 L LB-ampicillin with the 10-ml culture and allow to grow for 4 h at 37°C on a nutator.
Allow the culture to sit for 1 h at RT to cool. Save 500 µl to run on an SDS-PAGE gel as the uninduced fraction.
Induce with 0.1 mM IPTG O.N. at 20°C with shaking.
Harvest the cells by centrifuging for 20 min at 15,000 × g, 4°C.
Resuspend the pellet in 15 ml 1× PBS containing 0.2% Tween-20, 10 mM MgCl2, and protease inhibitors.
Freeze in liquid nitrogen and thaw at 37°C (3 times) to facilitate cell lysis.
Sonicate on ice 30 times for 30 s ON, 30 s OFF at an amplitude of 60%.
Centrifuge for 20 min at 20,000 × g, 4°C to pellet the cell debris.
Save the supernatant as the crude extract and run 10 µl on a 12% SDS-PAGE gel to verify the induction. The crude extract can be frozen in liquid nitrogen and stored at -80°C for future purification.
Wash 1.5 ml glutathione superflow beads 3 times with 1× RNase H2 extraction buffer.
Add the crude extract to the beads and incubate O.N. at 4°C on a nutator. All the remaining steps should be carried out at 4°C.
Wash the beads twice with 10 ml 1× RNase H2 extraction buffer containing protease inhibitors and once with 4 ml 1× RNase H2 extraction buffer without protease inhibitors.
Resuspend the beads in 2 ml 1× extraction buffer supplemented with 25 µg PreScission protease plus (we prepare our own PreScission; commercial enzyme could also be used).
Incubate O.N. at 4°C on a nutator.
Collect the flowthrough containing the RNase H2 released by PreScission cleavage.
Repeat the incubation with 1× RNase H2 extraction buffer and PreScission protease.
Analyze 10 µl input and washes, 1 µl each elution, and 10 µl beads on a 12% SDS-PAGE gel.
Stain the gel with SYPRO Ruby following the manufacturer’s instructions (Figure 3C).
Pool the elutions containing protein and concentrate them 5-fold with an Amicon ultra column 3,000 MCWO following the manufacturer’s instructions. The final concentration should be ~30 mg/ml in a volume of ~750 µl.
Equilibrate a 24-ml Superdex 200 10/300 GL size exclusion column with 2 CV 20 mM Tris-HCl pH 7.0 containing 150 mM NaCl and 0.2 mM PMSF.
Load the RNase H2 on the column and collect 500-µl fractions.
Run 5 µl each fraction on a 12% SDS-PAGE gel and stain with SYPRO Ruby.
Pool all the fractions containing the RNase H2 complex and dialyze them O.N. at 4°C against 1 L chilled RNase H storage buffer.
Aliquot, freeze in liquid nitrogen, and store at -80°C.
Run 0.1, 0.5, and 2.5 µl on a 12% SDS-PAGE gel next to a standard dilution of BSA and stain with SYPRO Ruby to determine the protein concentration (Figure 3D).
For 2 L of culture, ~5 mg purified RNase H2 trimer is expected.
Activity testing of RNase H1 and H2 (Figures 3E-3F)
A plasmid containing a sequence prone to forming R-loops is transcribed and used as a template to measure the ability of RNase H1 or RNase H2 to degrade R-loops. We use a plasmid with the vg PRE cloned into pETBlue (Alecki et al., 2020).
Assemble on ice (Table 2).
Table 2. In vitro transcription assayFinal concentration Volume (µl) Tris-HCl 1 M pH 8.0 40 mM 2 MgCl2 100 mM 8 mM 4 NaCl 1 M 25 mM 1.26 Spermidine 100 mM 2 mM 1 DTT 1M 30 mM 1.5 ATP 1 mM 40 nM 2 CTP 1 mM 40 nM 2 GTP 1 mM 40 nM 2 UTP 1 mM 8 nM 0.4 UTP α-P32 2.6 nM 0.2 DNA(vgcorePRE-pET) 100 ng/µl 5 T7 RNA polymerase (NEB) 0.125 H2O DNase/RNase-free 30.3 Final volume 50 Incubate for 30 min at 30°C.
Stop the reaction by heating the sample for 10 min at 65°C.
Titrate RNase H1 or RNase H2.
Assemble on ice (Table 3).
Table 3. Activity testing of purified RNase H1 or RNase H2Per reaction (µl) H2O DNase/RNase-free 5 10× RNase H buffer 1 Transcribed vgcorePRE-pET 3 RNase H1 or RNase H2 1 Final volume 10 Prepare serial dilutions of RNase H1 (10-fold steps) or RNase H2 (5-fold steps) in storage buffer.
Incubate for 1 h at 37°C.
Add 1 µl 10× DNA loading buffer.
Load on a 1% agarose 0.5× TBE gel.
Stain with SYBRGold and image.
Incubate the gel for 15 min in H2O.
Incubate the gel for 20 min in 75 mM NaOH.
Incubate the gel for 15 min in 0.5 M Tris-HCl 1.5 M NaCl.
Incubate the gel for 30 min in 6× SSC.
Transfer the gel O.N. to a HYBOND membrane (downward transfer) with the wick in 20× SSC.
Expose the membrane to a phosphorimager screen.
Scan on a Typhoon or equivalent phosphorimager (Figures 3E-3F).
Embryo collection
Note: This site (https://openspim.org/Drosophila_embryo_sample_preparation) provides a more detailed explanation of how to collect embryos.
Transfer 5 bottles of flies into a cage containing a 10-cm apple juice plate smeared with yeast paste to allow the flies to lay eggs. To collect enough embryos, 2–4 cages are used.
Allow the flies to lay eggs on the apple juice plates for 1 h before starting a timed collection.
Change the apple juice plates and allow the flies to lay eggs for 4 h.
Change the apple juice plates and allow the embryos to age in the incubator on the plate for 2 h or 10 h in order to collect 2-6 h or 10-14 h Drosophila embryos.
Remove the yeast, add household bleach diluted 1:2 with H2O to the plate, and incubate the embryos for 2 min to dechorionate them.
Transfer the embryos to a homemade sieve (we cut a large hole in the cap of a 50-ml tube and glue nylon mesh over it (a cell strainer (40-100 µm mesh size) can also be used) and wash with water.
Dry excess liquid on paper towel and transfer the embryos to a pre-weighed 1.7-ml tube.
Record the weight of the embryos, freeze in liquid nitrogen, and store at -80°C.
Nucleic acid extraction from Drosophila melanogaster embryos
Notes:
To have enough material for one DRIP-seq experiment, start with 500 μl or 200 mg embryos
Before they are washed with methanol, embryos stick to plastic. To minimize the loss of material, only glass vials and glass pipettes are used until the methanol wash; 8-ml glass scintillation vials work well for this step. For all steps involved in nucleic acid preparation, low-binding tubes and tips are used.
Transfer 500 μl or 200 mg embryos to a glass vial.
Wash embryos with 4 ml 1× PBS.
Remove the PBS and wash the embryos with 4 ml 1× PBT.
Transfer the embryos to a bottle containing 3 ml 1× PBS and 3 ml N-heptane.
Shake for a few seconds to mix the 2 phases.
Remove the lower phase (PBS) and leave the interphase intact.
Add 3 ml methanol and shake vigorously for 1 min.
Remove the top and interphase.
Wash the embryos with 3 ml methanol.
Transfer the embryos to a 15-ml tube and wash with 4 ml 1× PBS.
Resuspend the embryos in 4 ml embryo lysis buffer and incubate for 2 h at 50°C. Every 15 min, invert the tube to mix.
Centrifuge at 4,000 × g for 15 min. Transfer the supernatant to a 50-ml tube.
Add 4 ml phenol/chloroform/isoamyl alcohol. Incubate for 1 h on a nutator at 4°C.
Centrifuge at 4,000 × g for 15 min. Transfer the upper phase to a 50-ml tube.
Add 4 ml phenol/chloroform/isoamyl alcohol. Incubate for 1 h on a nutator at 4°C.
Centrifuge at 4,000 × g for 10 min. Transfer the upper phase to a clean 50-ml tube.
Add 4 ml chloroform/isoamyl alcohol. Incubate for 1 h on a nutator at 4°C.
Centrifuge at 4,000 × g for 10 min. Transfer the upper phase to a clean 50-ml tube.
To precipitate the nucleic acids, add 200 μl 3 M KOAc pH 5.2 and 2.8 ml isopropanol. Incubate for 30 min on a nutator at 4°C.
Gently transfer the white filaments to a 1.7-ml microfuge tube containing 1 ml 70% ethanol. Use a 1-ml pipette tip or cut the end off a 200-µl pipet tip to transfer the filaments without breaking.
Wash 3 times with 1 ml 70% ethanol without centrifugation.
Remove as much ethanol as possible.
Centrifuge for 1 min at 1,000 × g and remove the ethanol. This step can be repeated to remove the residual ethanol.
Air-dry the nucleic acid pellet for 1–4 h depending on the size of the pellet (until it becomes transparent).
Resuspend in 1 ml TE O.N. at 4°C on a nutator. At this step, the nucleic acid is viscous. Nucleic acid can be stored for a few weeks at -20°C before nuclease digestion and purification for DRIP.
Nucleic acid extraction from Drosophila S2 cells
Drosophila S2 cells are grown at room temperature in Schneider’s media containing 10% FBS.
Transfer 2 ×107 cells to a 50-ml tube.
Pellet the cells at 500 × g for 5 min.
Remove the supernatant and wash the cell pellet with 10 ml 1× PBS.
Pellet the cells at 500 × g for 5 min.
Resuspend the cells in 3 ml TE and transfer to three 1.7-ml tubes.
Add 26 μl 20% SDS and 60 μg proteinase K to each tube. Mix gently by inverting the tube several times.
Incubate O.N. at 37°C.
Transfer to 2-ml phase lock tubes.
Add 1 volume phenol/chloroform/isoamyl alcohol. Shake and centrifuge at 14,000 × g for 5 min.
Transfer the supernatant to a 50-ml tube containing 2.4 volumes of 100% ethanol and a 1/10 volume of 3 M NaOAc pH 5.2.
Note: It should be possible to substitute the KOAc used for Drosophila embryos for NaOAc.
Invert the tube gently to precipitate the nucleic acids.
Transfer the nucleic acids to a 1.7-ml tube containing 1 ml 70% ethanol.
Wash 5 times with 1 ml 70% ethanol by removing as much ethanol as possible without centrifugation.
Remove all the ethanol.
Centrifuge for 1 min at 1,000 × g and remove all the ethanol. This step can be repeated to remove the residual ethanol.
Air-dry the nucleic acid pellet for a few hours until the pellet becomes transparent.
Resuspend in 1 ml TE O.N. at 4°C on a nutator. At this step, the nucleic acid solution is viscous. Nucleic acid can be stored for a few weeks at -20°C before nuclease digestion and purification for DRIP.
Nuclease digestion and sonication of nucleic acids
Note: All the measurements on the NanoDrop are carried out using dsDNA parameters. The samples collected to measure the concentration on the NanoDrop and run on a gel are: 1) gDNA prior and 2) after RNase A treatment; 3) after sonication; 4, 5) after RNase III ± RNase H treatment.
Quantitate nucleic acids on a NanoDrop (concentration should be around 2 μg/μl and A260/A280 ≥ 2.0).
Run 1 μl nucleic acids on a 1% agarose 1× TAE gel to verify that the gDNA is intact. Stain the gel with SYBR Gold. gDNA should be above 10 kb and a smear of RNA should be visible between 100 bp and 1.5 kb.
RNAase A digestion: Incubate 250 mg nucleic acids in the presence of 0.5 M NaCl and 0.1 mg/ml RNase A in a final volume of 1.5 ml for 3 h at 37°C.
Transfer to 2-ml phase lock tubes and add 1 volume phenol/chloroform/isoamyl alcohol.
Mix vigorously and centrifuge at 14,000 × g for 5 min.
Transfer 500 μl upper phase to a 1.7-ml tube containing 1 ml 100% ethanol and 50 μl 3 M NaOAc pH 5.2.
Invert gently to precipitate the nucleic acids.
Transfer the nucleic acids to a 1.7-ml tube containing 400 μl 70% ethanol.
Remove all the ethanol.
Dry the nucleic acids for 10–30 min depending on the size of the pellet.
Resuspend in 500 μl TE by pipetting gently up and down and incubate on ice for at least 30 min.
Measure the concentration of nucleic acids on a NanoDrop and adjust the volume to a concentration below 40 ng/μl (A260/A280 should be 1.8–2.0). Save 1 μl to run on an agarose gel to verify complete degradation of free RNA.
Sonication using a Covaris E220: Split the sample into 130-µl aliquots (~5 µg) for sonication in a 130-µl Covaris microtube. Sonicate using the following parameters (Table 4):
Table 4. Parameters for the sonication using a Covaris E220Peak incident power (W) 140 Duty factor 10% Cycles per burst 200 Treatment (s) 80 Pool the sonicated nucleic acids.
Save 1 μl to load on the gel to verify sonication. From a band above 10 kb before sonication, a smear should be observed with an average size of 300–400 bp after sonication.
RNase H and RNase III digestion: Split the nucleic acids from step 14 into two aliquots. To both aliquots, add 100 µl 10× RNase H buffer and 2 units RNase III. Bring the volume to 1 ml. To one of the aliquots (RNase H-treated), add 10 μg RNase H1 and 10 μg RNase H2.
Incubate both digests O.N. at 37°C.
Transfer to 2-ml phase lock tubes and add 1 volume phenol/chloroform/isoamyl alcohol.
Mix vigorously and centrifuge for 5 min at 14,000 × g.
Transfer 500 μl upper phase to a 1.7-ml tube containing 1 ml 100% ethanol and 50 μl 3 M NaOAc pH 5.2.
Invert gently to precipitate the nucleic acids.
Centrifuge for 15 min at 16,000 × g.
Remove the supernatant and wash the pellet with 400 μl 70% ethanol.
Centrifuge for 5 min at 16,000 × g and remove all the ethanol.
Allow the nucleic acid pellet to dry for approximately 10 min.
Resuspend in 400 μl TE by gently pipetting up and down and incubate on ice for at least 30 min.
Measure the concentration on a NanoDrop and save 1 μl to run on a gel. This measurement is used to calculate the volume needed for the 4.4 µg used for DRIP (DRIP Step F2a).
Analyze the test aliquots from each step of the procedure (Steps E1-E5) on a 1% agarose 1× TAE gel and stain with SYBR Gold (Figure 4A). Nucleic acids can be stored for a few days at -20°C before DRIP.
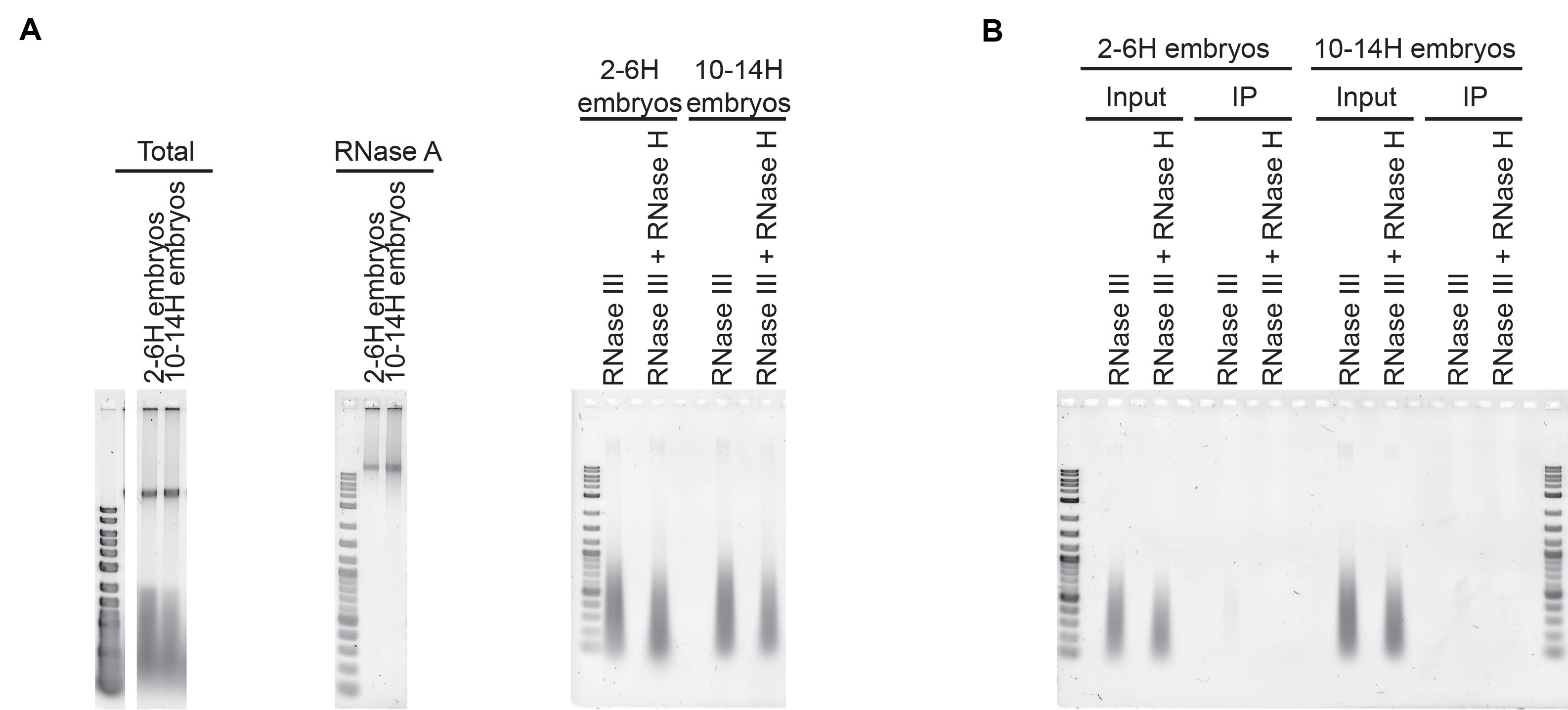
Figure 4. DRIP and nucleic acid preparation. A. Nucleic acid preparation from Drosophila melanogaster embryos. Total nucleic acids after extraction from embryos (left). Nucleic acids after RNase A digestion and before sonication (middle). Nucleic acids after sonication and RNase III +/- RNase H digestion (right). B. Input and elution from a DRIP experiment performed on 2-6 h and 10-14 h Drosophila melanogaster embryos. A faint smear is observed in the elution but not in the elution of the RNase H-treated sample.
DRIP
Notes:
For DRIP, all the steps are carried out using low-retention tubes and pipette tips.
For sequencing, we perform 3 DRIP experiments as described above in parallel and pool the elutions after the final purification.
Bead preparation
Wash 40 μl Protein G Dynabeads twice with 1× PBS containing 5 mg/ml BSA.
Resuspend the beads in 2 volumes 1× PBS containing 5 mg/ml BSA and add 10 μg S9.6 antibody.
Incubate O.N. at 4°C on a nutator.
Wash the beads once with 1× PBS containing 5 mg/ml BSA.
Keep the antibody-bound beads on ice or at 4°C.
DRIP
For each DRIP, dilute 4.4 μg previously purified DNA (Step E27) based on the NanoDrop measurement in 440 μl TE and add 50 μl 10× DRIP binding buffer.
Save 50 μl for input and incubate the remaining sample with the S9.6-Dynabeads O.N. at 4°C with rotation.
Capture the beads for 30 s using a magnetic rack, remove the supernatant, resuspend the beads in 700 μl 1× DRIP binding buffer, and incubate for 10 min at RT with rotation.
Repeat the washes twice, for a total of 3 washes.
Resuspend the beads in 250 μl DRIP elution buffer and 140 μg proteinase K.
Incubate for 45 min at 50°C; invert the tube every 5 min.
Collect the supernatant (IP/elution).
Input and IP purification
Note: The nucleic acids are purified on two successive columns, the first of which is used to eliminate SDS from the samples.
The input and IP are purified on Macherey-Nagel (MN) PCR cleanup columns using NTB buffer following the kit instructions. The elution is performed with 50 μl MN elution buffer.
The input and elution are purified on Zymoresearch DNA purification columns following the kit instructions. Elute the input and IP with 50 μl 10 mM Tris-HCl pH 8.0. For sequencing, elute the IP with 8.5 μl 10 mM Tris-HCl pH 8.0 and pool the 3 IPs together.
Run 2 μl each sample on a 1.5% agarose 1× TAE gel and stain for at least 1 h with SYBR Gold. A smear should be visible in the IP but not in the RNase H-treated IP (Figure 4B). We store the samples overnight at -20°C and prepare the libraries the next day. It should also be possible to store the samples at -80°C for a few weeks until library preparation.
- Library preparation and qPCR
Library preparation using an NEBNext® UltraTM II Directional RNA Library Prep Kit for Illumina®.
Notes:
The DNA moiety of the RNA–DNA hybrid is sequenced by starting with the second strand synthesis; the RNA moiety is removed in the second strand synthesis by RNase H digestion.
The number of PCR cycles is determined following the manufacturer’s instructions based on the amount of nucleic acid (Step G1a).
Estimate the amount of material in the IP and input on a bioanalyzer. The amount of nucleic acid should be above 1 ng to make the library (Figure 5A).
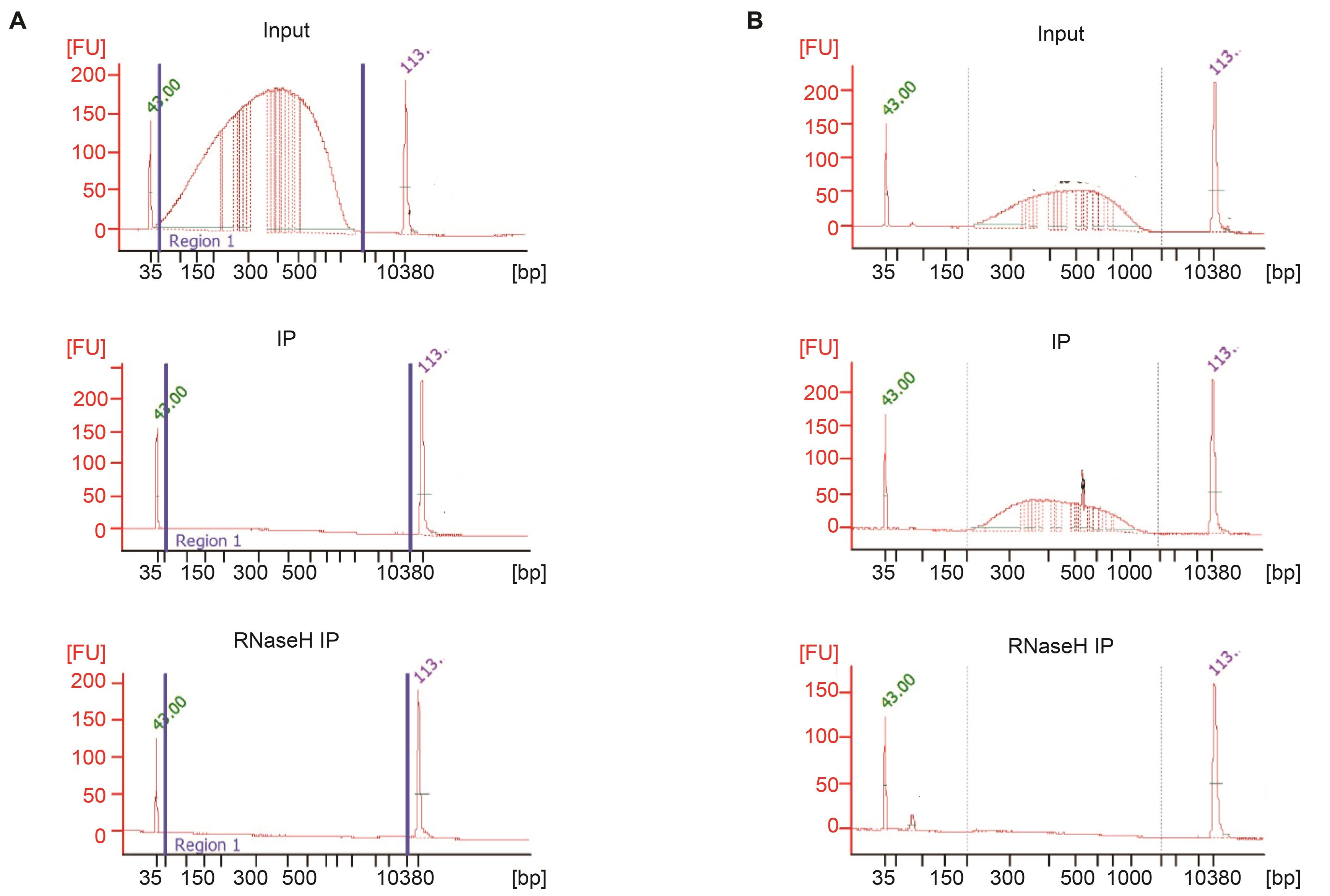
Figure 5. DRIP library preparation. Input and elution from 10-14 h embryos before (A) and after (B) the library preparation. The elution is quantitated by a bioanalyzer. The same amount of input and RNase H elution is used to prepare the libraries. The entire immunoprecipitated sample of the RNase H-treated nucleic acid is used to prepare the library since we are unable to determine the amount of nucleic acid present.Adjust the amount of input to the IP. For the RNase H-treated IP, the amount of material recovered cannot be quantitated. We use the whole sample and are able to generate a high complexity library, although the number of reads is typically 2–5× lower.
Prepare the library using NEB RNA Ultra II for Illumina, start at the second strand cDNA synthesis step (Table 5).
Table 5. Modified second strand synthesis reactionSecond strand synthesis reaction Volume Input and IP 20 μl NEBNext second strand synthesis reaction buffer with dUTP mix (10×) 8 μl NEBNext second strand synthesis enzyme mix 4 μl Random primer 1 μl RNase H (NEB), 1.6 U 0.3 μl Nuclease-free water 46.7 μl Assemble on ice and incubate for 1 h at 16°C.
For the next steps, follow the kit instructions.
Quality control of the library
Using qPCR, verify enrichment of R-loops at 3 positive sites and their absence at 2 negative sites (Figure 6; primer sequences in Table 6).
Dilute 1 μl each library in 10 μl 0.1× TE.
Set up the qPCR plate with a standard curve using gDNA from S2 cells. This gDNA can be prepared as described in section D, digested with RNase A, and stored at -80°C. We prepare a dilution series in 10-fold steps from 25 to 0.025 ng/μl and use 2 μl each standard for qPCR with each primer set (standard curve is 50-5-0.5-0.05 ng total gDNA).
qPCR reactions (5 μl total volume, run in a 384-well plate) consist of 2.5 μl Power Up SYBR Green master mix, 2 μl diluted library or standard genomic DNA from S2 cells, and 0.25 μl each primer diluted to 1 μM in water.
qPCR is run on a Viaa7 instrument with 40 cycles, Tm = 60°C, and extension time = 1 min.
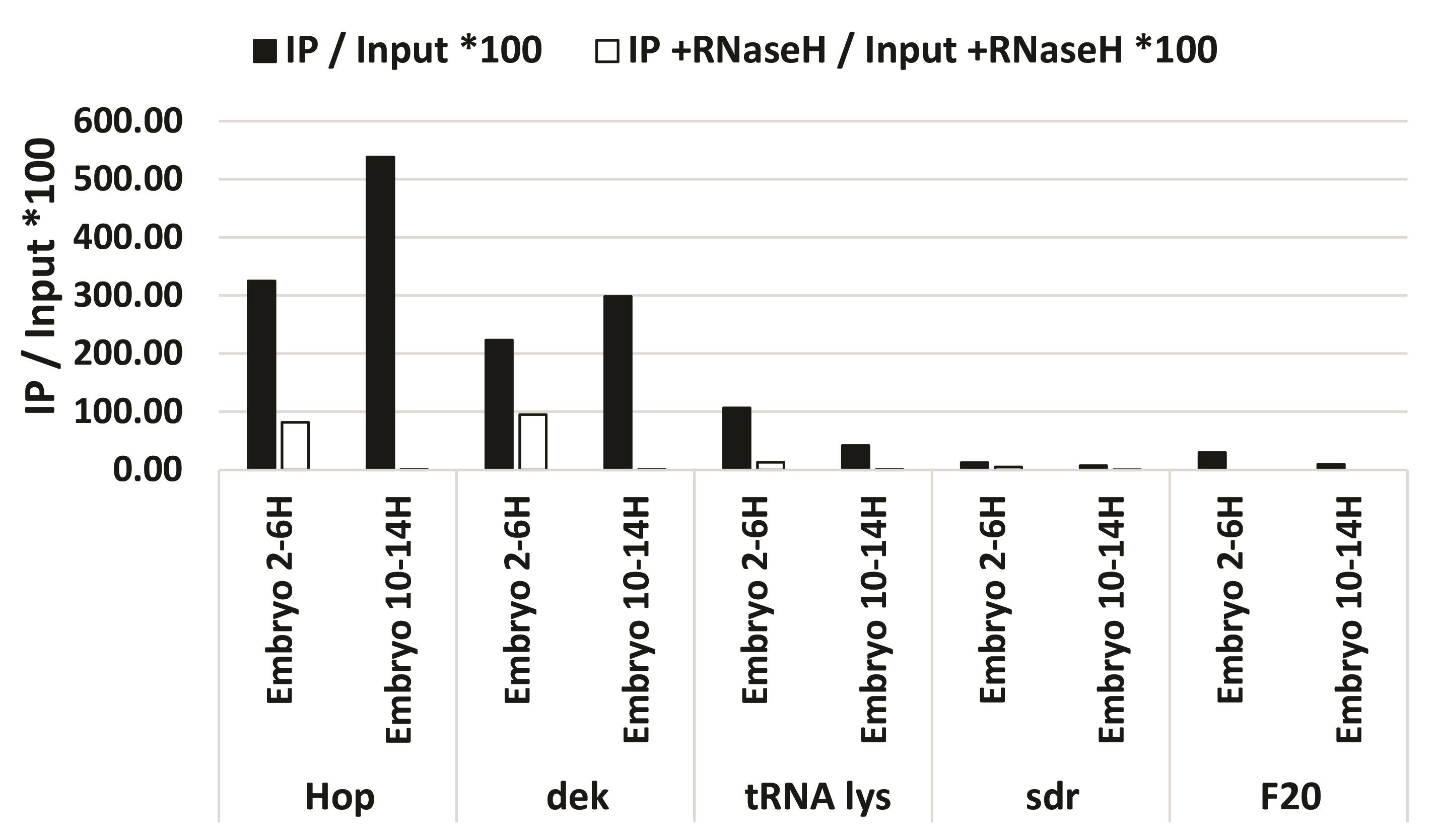
Figure 6. qPCR after library preparation. 3 positive and 2 negative sites confirmed the DRIP and library preparation of the input, IP, and RNase H-treated IP by qPCR.Table 6. qPCR primers
Primer name Sequence Hop F CTACAAGCAGGCGAAGGTTT R-loop (positive) Hop R CTTGATCTCAGGGGTGCGAT dek F GCGATGAGCCAGAAGATGAG R-loop (positive) dek R CTTGGACTCATCAGTGGCAT tRNA lys F GCCAAGCTCATTTTCTACGATCT R-loop (highly transcribed gene, positive) tRNA lys R GTCCGACAACGCCGATGATA Sdr F ACAGCTGATGTCGCTCACAT No R-loop (negative) Sdr R CGCTGAATGATCACCAGGTGA F20 F (CG12754) TCAAGCCGAACCCTCTAAAAT No R-loop (negative) F20 R AACGCCAACAAACAGAAAATG Using the bioanalyzer
Run 1 μl each library on a bioanalyzer (Figure 5B).
The average size of the fragments should be 400 bp.
Sequence the library on a HiSeqTM sequencing system with a depth of 50 million reads per sample. Paired end sequencing was used here, with a read length of 50 bases.
Data analysis
The work-flow for DRIP-seq analysis as described in Alecki et al. (2020) is similar to standard ChIP-seq analysis. The basic work-flow is trimming of the adaptors, quality control of the FASTQ files, alignment to the genome, removal of PCR duplicates, and calling of the peaks. The protocol diverges from ChIP-seq in the steps of splitting data by strand before peak calling and calling peaks against both the input and the RNase H-treated samples. All the analysis steps can be carried out on a Galaxy (usegalaxy.org) or using simple bash scripts. This workflow identifies a stringent set of reproducible peaks that meet strict criteria for being R-loops (i.e., they are sensitive to RNase H).
Notes:
Others have described the use of a two-fold reduction in read counts in RNase H-treated samples to filter DRIP-seq peaks (Crossley et al., 2020) rather than comparing peaks called in DRIP versus RNase H-treated DRIP. This approach may be more flexible to accommodate sequencing experiments in which the RNase H digestion is suboptimal and should be considered, particularly if RNase H filtering removes a large number of peaks (>10%).
We call peaks from both strands together or after separating the bam files into forward (F) and reverse (R) strands. The strand-specific analysis is more informative since it makes it possible to infer the orientation of transcription that produced the RNA. A fraction of peaks have an R-loop signal on both strands (~10% of peaks); we do not know whether these are technical artifacts (Alecki et al., 2020). Some, particularly in the case of embryo analysis, may be the result of mixed cell populations. It may be desirable to remove them for downstream analysis (Crossley et al., 2020), which can be done using bedtools to intersect the F and R strands.
Crossley et al. (2020) recently described a synthetic spike-in strategy that can be used to normalize DRIP-seq data, allowing quantitative comparisons across conditions. This could be especially valuable when comparing Drosophila of different genotypes.
Trim the adaptors and remove low-quality reads using Trimmomatic (Bolger et al., 2014) or fastp (v0.20.0) (Chen et al., 2018).
Align the reads to the Drosophila genome using Bowtie2 (v 2.3.1) (Langmead and Salzberg, 2012) (--fr –no-mixed–no-unal).
Use Samtools (v. 1.4.1) (Li et al., 2009) to convert the sam files generated by Bowtie2 to bam files, sort the bam files, and create a bam index for each file.
Use Picard (http://broadinstitute.github.io/picard). Mark duplicates to filter PCR duplicates. Sambamba (v0.7.1) (Tarasov et al., 2015) can also be used for this step.
Generate strand-specific bam files (for all samples: DRIP, RNase H DRIP, and Input) based on samflags using the samtools view as follows:
Forward strand: samtools view –f 99; samtools view –f 147, followed by samtools merge.
Reverse strand: samtools view –f 83; samtools view –f 163, followed by samtools merge.
To carry out this procedure in Galaxy, use the samtools view and select “A filtered/subsampled selection of reads”; then set the correct combination of “Require that these flags are set.”
For example, for the forward strand, first output the –f99 reads with these flags set:
–read is paired –read is mapped in a proper pair =mate strand =read is the first in a pair;
then output –f147 reads with these flags set;
–read is paired –read is mapped in a proper pair –read strand =read is the second in a pair;
samtools merge is used as above to merge f99 and f147 (F strand) and f83 and f163 (R strand).
Use MACS2 (v2.1.1) (Zhang et al., 2008) to call peaks of DRIP versus Input using broad peak settings, with DRIP as the treatment and Input as the control. For example, for peak calling with paired-end bam files aligned to the dm6 version of the Drosophila genome:
(macs2 callpeak –t DRIP.bam –c DRIP_input.bam –f BAMPE –g 1.4e+08 –n DRIP –outdir DRIP_peaks –broad).
Use MACS2 to call peaks of DRIP versus RNase H-treated DRIP with broad peak settings, with DRIP as the treatment and RNase H-treated as the control.
Use the BEDTools (Quinlan and Hall, 2010) intersect to retain peaks present in IP versus input and IP versus RNase H IP (bedtools intersect –a IP_versus_IN_peaks.bed –b IP_versus_RNaseHIP_peaks.bed –u >filtered_peaks.bed).
Use the BEDtools intersect to retain only peaks present in both replicates.
Notes
This DRIP protocol consists of 4 steps, with the most critical being the preparation of the nucleic acids for immunoprecipitation. We encourage the users to perform DRIP-qPCR several times to ensure reproducibility before performing sequencing.
Time-line (Table 7).
Table 7. Time-line of DRIP-seq on Drosophila melanogaster embryos and tissue culture cells
Day 1 Embryo collection Tissue culture cell lysis Day 2 Embryo lysis and nucleic acid extraction Nucleic acid extraction Day 3 Nucleic acid preparation I + bead preparation Day 4 Nucleic acid preparation II + immunoprecipitation Day 5 Bead washing + elution + purification Day 6 Library preparation and qPCR Embryo collection
Note that embryos can be collected and stored at -80°C for a few months. This protocol can be performed in one week but it is also possible to stop at several steps. The nucleic acids can be stored at -20°C for a few days after extraction from tissue culture cells or embryos, after the preparation of the nucleic acids (before immunoprecipitation) and before the qPCR or library preparation.
Nucleic acid extraction and preparation
For DRIP protocols performed on protein-free nucleic acids after cell lysis, it is essential that the cell lysis and purification be performed gently to avoid breaking R-loops or creating new RNA–DNA hybrid by spurious annealing of RNA and complementary DNA. Thus, from extraction until sonication, do not centrifuge the nucleic acids at high speed. To resuspend the nucleic acids, do not vortex; instead pipette gently up and down.
DRIP
Nucleic acids from other species can be included before immunoprecipitation as a spike-in control to normalize the results between different samples or experimental conditions (Chen et al., 2016).
Recipes
Embryo lysis buffer
50 mM Tris-HCl pH 8.0
100 mM EDTA
100 mM NaCl
0.5% SDS
5 mg/ml proteinase K
1× RNase H buffer
50 mM Tris-HCl
75 mM KCl
3 mM MgCl2
10 mM DTT
pH 8.3
10× DRIP binding buffer
100 mM NaPO4 pH 7.0
1.4 mM NaCl
0.5% Triton X-100
1× DRIP binding buffer
10× DRIP binding buffer diluted in TE
DRIP elution buffer
50 mM Tris-HCl pH 8.0
10 mM EDTA
0.5% SDS
TE
10 mM Tris pH 8.0
1 mM EDTA
Apple juice plate
3.5% agar
4% sugar
40% apple juice
0.30% nipagin diluted in ethanol (2.5% of the final volume)
RNase H2 extraction buffer
1× PBS
0.2% Tween-20
10 mM MgCl2
RNase H1 lysis buffer
25 mM Tris-HCl
300 mM NaCl
5 mM imidazole
pH 8.0 adjusted with NaOH
RNase H1 wash buffer
50 mM Tris-HCl
300 mM NaCl
20 mM imidazole
pH 8.0 adjusted with NaOH
RNase H1 elution buffer
50 mM Tris-HCl
300 mM NaCl
250 mM imidazole
pH 8.0 adjusted with NaOH
RNase H size column buffer
20 mM Tris-HCl pH 7.0
150 mM NaCl
Add 0.1 mM PMSF immediately before use
RNase H storage buffer
20 mM Tris-HCl pH 7.5
50 mM NaCl
1 mM DTT
0.1 mM EDTA
20% glycerol
Add 0.1 mM PMSF immediately before use
Protease inhibitors and additives (Table 8)
Note: The buffers used to prepare RNase H1 and H2 proteins contain freshly added protease inhibitors, NP40, and DTT, except for the size column buffers, which have only PMSF and DTT added.
Table 8. List of protease inhibitors and additivesFinal concentration TLCK 13.5 µM Benzamidine 100 µM Pepstatin 3 µM Phenanthroline 55 µM PMSF 100 µM Aprotinin 1.5 µM Leupeptin 23 µM NP40 0.05% DTT 1 mM 0.5× TBE
44.5 mM Tris
44.5 mM Boric acid
1 mM EDTA pH 8.0
10× DNA loading buffer
20% glycerol
0.1 M EDTA pH 8.0
1% SDS
0.25% Bromophenol Blue
0.25% xylene cyanol
SSC 20×
3 M NaCl
0.3 sodium citrate
pH 7.0 adjusted with HCl
1× TAE
40 mM Tris-HCl
20 mM Acetate
1 mM EDTA
1× PBS
137 mM NaCl
2.7 mM KCl
10 mM Na2HPO4
1.8 mM KH2HPO4
1× PBT
1× PBS
0.1% Triton X-100
3 M KOAc pH 5.2
3 M potassium acetate
pH 5.2 adjusted with glacial acetic acid
3 M NaOAc pH 5.2
3 M sodium acetate
pH 5.2 adjusted with glacial acetic acid
SDS-PAGE running buffer
25 mM Tris-HCl
192 mM Glycine
0.1% SDS
Protein loading buffer
6× SDS-sample buffer, added to samples at a final concentration of 1×
0.35 M Tris, pH 6.8
30% glycerol
1% SDS
0.0001% Bromophenol Blue
Acknowledgments
This protocol was adapted from Ginno et al. (2012) and published previously in Alecki et al. (2020). Work in the authors’ lab is funded by the Canadian Institutes for Health Research (CIHR).
Competing interests
The authors declare no competing interests.
References
- Alecki, C., Chiwara, V., Sanz, L. A., Grau, D., Arias Perez, O., Boulier, E. L., Armache, K. J., Chedin, F. and Francis, N. J. (2020). RNA-DNA strand exchange by the Drosophila Polycomb complex PRC2. Nat Commun 11(1): 1781.
- Bolger, A. M., Lohse, M. and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15): 2114-2120.
- Chedin, F. and Benham, C. J. (2020). Emerging roles for R-loop structures in the management of topological stress. J Biol Chem 295(14): 4684-4695.
- Chen, K., Hu, Z., Xia, Z., Zhao, D., Li, W. and Tyler, J. K. (2015). The Overlooked Fact: Fundamental Need for Spike-In Control for Virtually All Genome-Wide Analyses. Mol Cell Biol 36(5): 662-667.
- Chen, L., Chen, J. Y., Zhang, X., Gu, Y., Xiao, R., Shao, C., Tang, P., Qian, H., Luo, D., Li, H., Zhou, Y., Zhang, D. E. and Fu, X. D. (2017). R-ChIP Using Inactive RNase H Reveals Dynamic Coupling of R-loops with Transcriptional Pausing at Gene Promoters. Mol Cell 68(4):745-757.e5.
- Chen, S., Zhou, Y., Chen, Y. and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34(17): i884-i890.
- Crossley, M. P., Bocek, M. J., Hamperl, S., Swigut, T. and Cimprich, K. A. (2020). qDRIP: a method to quantitatively assess RNA-DNA hybrid formation genome-wide. Nucleic Acids Res 48(14): e84.
- Fang, Y., Chen, L., Lin, K., Feng, Y., Zhang, P., Pan, X., Sanders, J., Wu, Y., Wang, X. E., Su, Z., Chen, C., Wei, H. and Zhang, W. (2019). Characterization of functional relationships of R-loops with gene transcription and epigenetic modifications in rice. Genome Res 29(8): 1287-1297.
- Ginno, P. A., Lott, P. L., Christensen, H. C., Korf, I. and Chédin, F. (2012). R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol Cell 45(6): 814–825.
- Hartono, S. R., Malapert, A., Legros, P., Bernard, P., Chedin, F. and Vanoosthuyse, V. (2018). The Affinity of the S9.6 Antibody for Double-Stranded RNAs Impacts the Accurate Mapping of R-Loops in Fission Yeast. J Mol Biol 430(3): 272-284.
- König, F., Schubert, T. and Langst, G. (2017). The monoclonal S9.6 antibody exhibits highly variable binding affinities towards different R-loop sequences. PLoS One 12(6): e0178875.
- Langmead, B. and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4): 357-359.
- Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16): 2078-2079.
- Lockhart, A., Pires, V. B., Bento, F., Kellner, V., Luke-Glaser, S., Yakoub, G., Ulrich, H. D. and Luke, B. (2019). RNase H1 and H2 Are Differentially Regulated to Process RNA-DNA Hybrids. Cell Rep 29(9): 2890-2900 e2895.
- Manzo, S. G., Hartono, S. R., Sanz, L. A., Marinello, J., De Biasi, S., Cossarizza, A., Capranico, G. and Chedin, F. (2018). DNA Topoisomerase I differentially modulates R-loops across the human genome. Genome Biol 19(1): 100.
- Niehrs, C., and Luke, B. (2020). Regulatory R-loops as facilitators of gene expression and genome stability. Nat Rev Mol Cell Biol 21(3): 167-178.
- Nowotny, M., Cerritelli, S. M., Ghirlando, R., Gaidamakov, S. A., Crouch, R. J. and Yang, W. (2008). Specific recognition of RNA/DNA hybrid and enhancement of human RNase H1 activity by HBD. EMBO J 27(7): 1172-1181.
- Phillips, D. D., Garboczi, D. N., Singh, K., Hu, Z., Leppla, S. H. and Leysath, C. E. (2013). The sub-nanomolar binding of DNA-RNA hybrids by the single-chain Fv fragment of antibody S9.6. J Mol Recognit 26(8): 376-381.
- Quinlan, A. R. and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26(6): 841-842.
- Sanz, L. A., Hartono, S. R., Lim, Y. W., Steyaert, S., Rajpurkar, A., Ginno, P. A., Xu, X. and Chedin, F. (2016). Prevalent, Dynamic, and Conserved R-Loop Structures Associate with Specific Epigenomic Signatures in Mammals. Mol Cell 63(1): 167-178.
- Tan-Wong, S. M., Dhir, S. and Proudfoot, N. J. (2019). R-Loops Promote Antisense Transcription across the Mammalian Genome. Mol Cell 76(4): 600-616 e606.
- Tarasov, A., Vilella, A. J., Cuppen, E., Nijman, I. J. and Prins, P. (2015). Sambamba: fast processing of NGS alignment formats. Bioinformatics 31(12): 2032-2034.
- Wahba, L., Costantino, L., Tan, F. J., Zimmer, A. and Koshland, D. (2016). S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes Dev 30(11): 1327-1338.
- Xu, W., Xu, H., Li, K., Fan, Y., Liu, Y., Yang, X., and Sun, Q. (2017). The R-loop is a common chromatin feature of the Arabidopsis genome. Nature Plants 3(9): 704-714.
- Xu, W., Li, K., Li, S., Hou, Q., Zhang, Y., Liu, K., and Sun, Q. (2020). The R-Loop Atlas of Arabidopsis Development and Responses to Environmental Stimuli. Plant Cell 32(4): 888–903.
- Yu, K., Chedin, F., Hsieh, C.-L., Wilson, T.E., and Lieber, M.R. (2003). R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat Immunol 4(5): 442-451.
- Zhang, T., Wallis, M., Petrovic, V., Challis, J., Kalitsis, P., and Hudson, D.F. (2019). Loss of TOP3B leads to increased R-loop formation and genome instability. Open Biol 9(12). DOI: https://doi.org/10.1098/rsob.190222.
- Zhang, Y., Liu, T., Meyer, C. A., Eeckhoute, J., Johnson, D. S., Bernstein, B. E., Nusbaum, C., Myers, R. M., Brown, M., Li, W. and Liu, X. S. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9(9): R137.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Alecki, C. and Francis, N. J. (2021). Identification of R-loop-forming Sequences in Drosophila melanogaster Embryos and Tissue Culture Cells Using DRIP-seq. Bio-protocol 11(9): e4011. DOI: 10.21769/BioProtoc.4011.
Category
Molecular Biology > RNA
Developmental Biology > Cell growth and fate > Differentiation
Systems Biology > Genomics > DRIP-seq
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.