- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Spin Labeling of RNA Using “Click” Chemistry for Coarse-grained Structure Determination via Pulsed Electron-electron Double Resonance Spectroscopy
(*contributed equally to this work) Published: Vol 11, Iss 9, May 5, 2021 DOI: 10.21769/BioProtoc.4004 Views: 6228
Reviewed by: Angeliki GiannoulisYINNIAN FENGAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Understanding the function of oligonucleotides on a molecular level requires methods for studying their structure, conformational changes, and internal dynamics. Various biophysical methods exist to achieve this, including the whole toolbox of Electron Paramagnetic Resonance (EPR or ESR) spectroscopy. An EPR method widely used in this regard is Pulsed Electron-Electron Double Resonance (PELDOR or DEER), which provides distances in the nanometer range between electron spins in biomolecules with Angstrom precision, without restriction to the size of the biomolecule, and in solution. Since oligonucleotides inherently do not contain unpaired electrons, these have to be introduced in the form of so-called spin labels. Firstly, this protocol describes how nitroxide spin labels can be site-specifically attached to oligonucleotides using “Click” chemistry. The reaction provides little byproducts, high yields, and is conveniently performed in aqueous solution. Secondly, the protocol details how to run the PELDOR experiment, analyze the data, and derive a coarse-grained structure. Here, emphasis is placed on the pitfalls, requirements for a good dataset, and limits of interpretation; thus, the protocol gives the user a guideline for the whole experiment i.e., from spin labeling, via the PELDOR measurement and data analysis, to the final coarse-grained structure.
Graphical abstract:

Schematic overview of the workflow described in this protocol: First, the spin-labeling of RNA is described, which is performed as a "Click"-reaction between the alkyne-functionalized RNA strand and the azide group of the spin label. Next, step-by-step instructions are given for setting up PELDOR/DEER distance measurements on the labeled RNA, and for data analysis. Finally, guidelines are provided for building a structural model from the previously analyzed data.
Background
The function of biomolecules is rooted in their three-dimensional structure, dynamics, and interaction with other molecules. For example, proteins and oligonucleotides adopt structures that provide interaction sites or binding pockets for metal ions, organic ligands, and other oligonucleotides or proteins. Upon complex formation, the structure of the biomacromolecule may change, and it is this structural change that is the basis for function; thus, methods are needed that enable resolution of the structures along the trajectory of the conformational change. X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, electron microscopy (EM), small-angle X-ray scattering (SAXS), atomic force microscopy (AFM), optical trap, and magnetic tweezers are powerful methods for obtaining the structures of biomolecules at atomic resolution (Salas et al., 2015; Lottspeich and Engels, 2018; Geffroy et al., 2018); yet, they also have their limitations. X-ray crystallography requires the biomolecule to be crystallized and is, as EM, limited in providing information on dynamics and conformational intermediates. NMR spectroscopy can follow the dynamics of biomolecules since it is performed in liquid solution, but is limited with respect to the size of the biomolecule. Complementary to these techniques, fluorescence microscopy (FM), Förster resonance energy transfer (FRET), and electron paramagnetic resonance (EPR) spectroscopy are biophysical methods that allow resolution of the dynamics of structural changes without size restriction and in solution (Yang et al., 2012; Lottspeich and Engels, 2018; Kuzhelev et al., 2018). In the toolbox of EPR spectroscopy (Schweiger et al., 2001; Goldfarb et al., 2018), a set of pulse sequences summarized under the term Pulsed Dipolar EPR Spectroscopy (PDS; Schiemann et al., 2007) measures the dipolar coupling between the spins of unpaired electrons. This coupling encodes the inter-spin distance, from which structural and dynamic information can be derived. Owing to the high sensitivity of PDS, measurements at biomolecular concentrations down to the nanomolar range are feasible (Fleck et al., 2020). Out of all PDS techniques, Pulsed Electron-Electron Double Resonance (PELDOR or DEER) is the most prominent and works in a distance range from 1.5 to 16 nm upon deuteration of the buffer and the whole protein (Schmidt et al., 2016) with Angstrom precision (Jeschke, 2012; Tsvetkov et al., 2019). PELDOR imposes no size restriction (Malygin et al., 2019), and can be performed on biomolecules in liquid (Yang et al., 2012) or frozen solution (Duss et al., 2014), in membranes (Dastvan et al., 2019), and within cells (Theillet et al., 2016). However, since PELDOR requires unpaired electrons and biomolecules are usually diamagnetic, techniques are needed for site-directed spin labeling (Shelke and Sigurdsson, 2014). Focusing on RNA oligonucleotides, there are two principal strategies for spin labeling (Ward and Schiemann, 2014): the first is the phosphoramidite approach, where a spin-labeled phosphoramidite is incorporated into the RNA strand during solid-phase synthesis (Beaucage et al., 1992; Lottspeich and Engels, 2018) and the second is the post-synthetic method, where a functionalized RNA strand is labeled after synthesis (Kerzhner et al., 2016). The major drawbacks of the phosphoramidite approach are the rather laborious synthesis of the spin-labeled phosphoramidite and the easy reduction of the label during RNA synthesis, leading to an EPR-silent state. For the post-synthetic strategy, RNA strands modified with a unique functional group can be obtained commercially and are then reacted with a spin label carrying the complementary functional group. However, labeling with high yields and efficient purification with minimal product loss can be challenging. Even though the length of commercially available RNA oligonucleotides is limited, there are well-established methods for obtaining longer RNA strands, e.g., via enzymatic ligation (Duss et al., 2014; Kerzhner et al., 2018).
In this protocol, built on the publications Kerzhner et al., 2018, Wuebben et al., 2019 and 2020, we first introduce a workflow for the post-synthetic spin labeling of RNA oligonucleotides based on the “Click” reaction. Here, the “Click” reaction is performed as a copper-(I)-catalyzed [2+3] cycloaddition between an alkyne-substituted 5’-uridine in the RNA strand and an azide group on a nitroxide label (Figure 1). The reaction is usually quantitative and fast, and the purification of the labeled RNA can be easily performed via reversed-phase High Performance Liquid Chromatography (HPLC). Secondly, the protocol outlines how to set up and perform the PELDOR measurement. Thirdly, it provides a workflow for data analysis and the transformation of the distances into structures. Importantly, possible pitfalls are highlighted and tips provided on how to overcome experimental difficulties, which are rarely found in the literature.
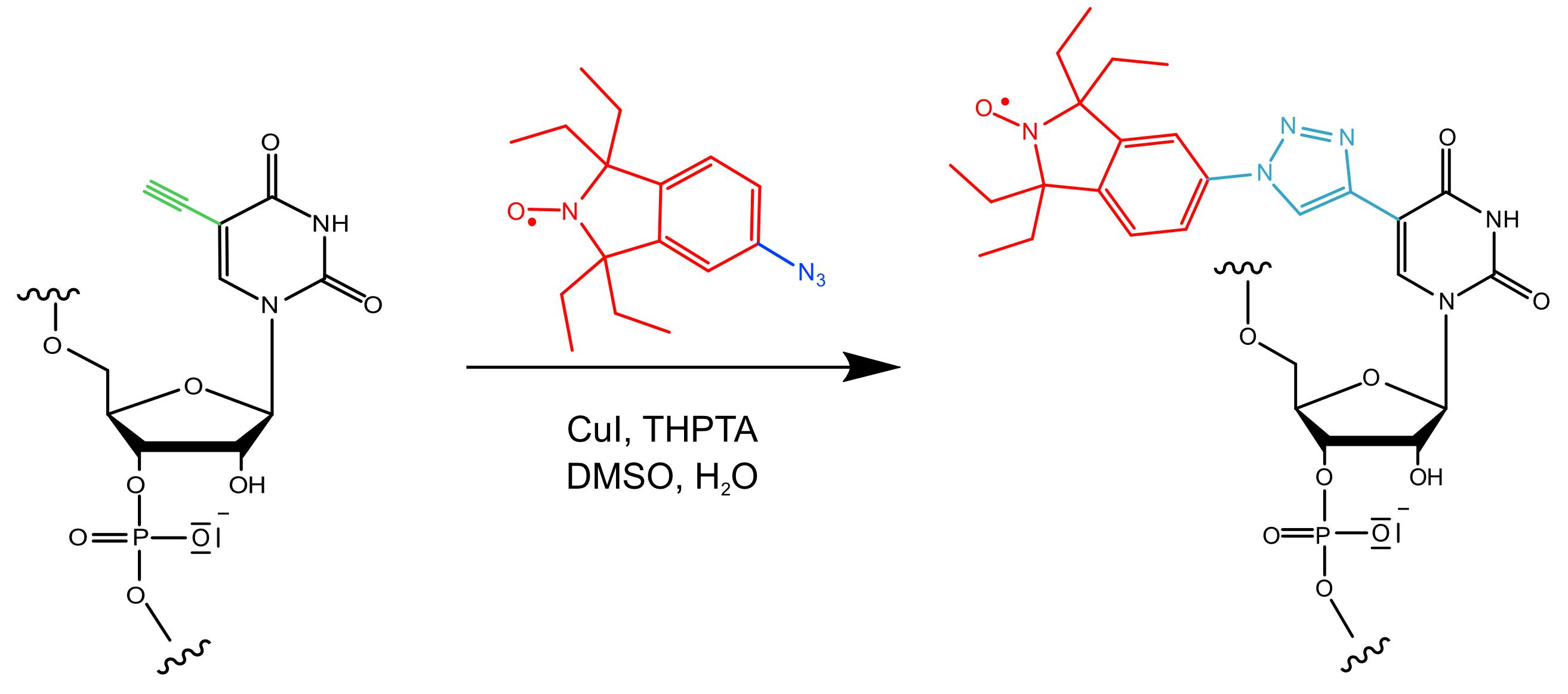
Figure 1. Reaction scheme of the “Click” reaction used in this protocol.The azide-functionalized spin label (azide group in blue; nitroxide moiety and 1,1,3,3-tetraethyl-5-azoisoindolin-2-oxyl backbone in red) is attached to a 5-ethynyl-2′-deoxyuridine nucleotide (ethynyl group in green) in the RNA strand (black). Copper, in its oxidation state (I), forms a complex with tris ((1-hydroxy-propyl-1H-1,2,3-triazol-4-yl) methyl) amine (THPTA), catalyzing this reaction in aqueous solution, which leads to the formation of a triazol linker (cyan) covalently connecting the nitroxide moiety to the RNA.
Beyond this application, labeling via “Click” chemistry is restricted neither to RNA nor to nitroxides or spin labels in general. It can be applied to e.g., DNA (El-Sagheer and Brown, 2010) and proteins (Nikić et al., 2015) and can be used for fluorescent labels (Liang et al., 2019). For protein labeling, one would make use of unnatural amino acids e.g., 4-ethynyl-L-phenylalanine (Widder et al., 2019). Note, however, that the reaction conditions will have to be adapted to the particular system under study. The workflow for the Q-band PELDOR experiment is universally applicable to measurements of nitroxide-labeled biomolecules. Likewise, the data analysis and distance-to-structure transformation can be adapted for other PDS techniques or other types of spin labels.
Materials and Reagents
Note: It is very important to set up an RNase-free environment. Therefore, use only autoclaved pipette tips, clean your bench and tools with 70% ethanol or RNase Away, and wear protective gloves.
RNase Away (Thermo Fisher, catalog number: 7003PK) or 70% Ethanol (Julius Hoesch GmbH, catalog number: 125900)
1.5 ml and 2 ml Eppendorf tubes
2.5 nmol dried RNA oligonucleotide with one or more 5-ethynyl-2’-deoxyuridine modifications (commercially synthesized and delivered in dried form e.g., from Metabion or Biomers, see procedure for more details), stored at -20°C (Note 1)
Diethylpyrocarbonate (DEPC, Carl Roth, catalog number: K028.3)-treated water
Copper(I)-iodide (Cu(I); Carl Roth, catalog number: 0305.1), stored at room temperature under ambient conditions in a regular container, as received
Dimethyl sulfoxide (DMSO, Carl Roth, catalog number: A994.2), stored at room temperature
3.6 µl 250 mM tris ((1-hydroxy-propyl-1H-1,2,3-triazol-4-yl) methyl) amine (THPTA, Sigma, catalog number: 762342-100MG) solution in DMSO, stored at -20°C, vortex the solution prior to use
2 µl 100 mM 1,1,3,3-tetraethyl-5-azoisoindolin-2-oxyl (Wuebben et al., 2019) or 1,1,3,3-tetramethyl-5-azoisoindolin-2-oxyl (Kerzhner et al., 2018) solution in DMSO, stored at -20°C, vortex the solution prior to use
Hamilton syringe, 50 µl
Buffer A, acetonitrile (Carl Roth, catalog number: 8825.2), stored at room temperature
Buffer B, 0.1 M triethylammonium acetate (TEAA) (1 M solution, Labomedic, catalog number: 2001741), stored at room temperature
For Liquid Chromatography-Mass Spectrometry (LC-MS): Buffer C, 10 mM triethylamine (Merck, catalog number: 90340-2.5L), stored at room temperature
For LC-MS, Buffer D, 0.1 M hexafluoroisopropanol (Merck, catalog number: 18127-50ML), stored at room temperature
For spin counting and determination of labeling efficiency: 10 µl capillaries (Hirschmann Laborgeräte, catalog number: L925.1)
Deuterated ethylene glycol (EG-d6) as a cryoprotectant (Merck, catalog number: 530549), stored at room temperature
Deuterated water (D2O) as a solvent (Deutero, order number: 00506), stored at room temperature
Pipette and matching elongated pipette tips (Sorenson BioScience, 200 µl MµltiFlex Round, catalog number: # 28480) to transfer the sample into the EPR tube
For PELDOR measurement: Q-band EPR sample tubes with 3 mm outer diameter made from clear-fused quartz (CFQ) (Wilmad LabGlass, catalog number: G542PN000001470)
Light-duty tissue wipers (VWR, European catalog number: 115-0202)
Liquid nitrogen (Air Liquide) for freezing the sample and liquid helium (Air Liquide) for cooling the sample in the resonator
DEPC-treated water (see Recipes)
Equipment
SpeedVac vacuum concentrator: Eppendorf Concentrator plus (Eppendorf, Rotor: F-45-48-11) or freeze dryer: Alpha 3-4 LSC basic (Martin Christ Gefriertrocknungsanlagen)
Thermomixer (Eppendorf)
Table-top centrifuge (Eppendorf)
Milli-Q Ultrapure Water System (Merck Millipore)
NanoDrop Spectrophotometer (Thermo Fisher)
High-Performance Liquid Chromatography (HPLC) system (Agilent, Series 1,200)
Reversed-phase C18 column, heatable (Zorbax 300SB-C18, 4.6 × 150 mm for RNA sequences shorter than 50 nucleotides or Zorbax 300SB-C18, 9.4 × 250 mm column for RNA sequences exceeding 50 nucleotides)
Amicon® Ultra-0.5 centrifugal filter device with a nominal molecular weight limit (NMWL) of 3,000 Da (Merck, catalog number: UFC500396)
Liquid Chromatography-Mass Spectrometry (LC-MS) system (HTC esquire from Bruker Daltonics in combination with an HPLC system). If you have no access to an LC-MS system, you can perform an analytical HPLC run and subsequently determine the exact mass via matrix-assisted laser desorption/ionization (MALDI) mass spectrometry or electrospray ionization (ESI) mass spectrometry
For spin counting and determination of labeling efficiency: continuous wave (cw) EPR spectrometer (Bruker BioSpin, model: EMXnano)
Pulsed EPR spectrometer (Bruker BioSpin, model: ELEXSYS E580) equipped with a Q-band microwave bridge (Bruker) and an ER5106-QT2 resonator (Bruker) for PELDOR measurements. When using an X-band resonator for continuous wave operation (e.g., Bruker ER4119HS), spin counting experiments can also be performed on this spectrometer
150 W travelling wave tube amplifier (TWT, Applied Systems Engineering, model: 187 Ka)
Liquid helium cryostat (Oxford Instruments, model: CF935)
Temperature controller (Oxford Instruments, model: iTC503S)
Helium transfer line (Oxford Instruments NanoScience, model: LLT600 or LLT650) and gas flow controller (Oxford Instruments)
Turbomolecular pump for evacuating the cryostat (Pfeiffer Vacuum, model: HiCUBE 80 Eco)
Membrane pump for maintaining a constant stream of cold helium gas (KNF Neuberger, model: PM26962–026.1.2)
Dewar vessels for shock-freezing and handling the EPR sample tubes at liquid nitrogen temperatures (KGW Isotherm, Type 00C, 13.5 cm in height; Type 3C, 26 cm in height)
100 L liquid helium tank (Cryo Anlagenbau, model CS 100 H)
Safety goggles and cold protection gloves
PC with Linux, as delivered with the spectrometer
Software
Matlab Version R2018a or later (The MathWorks Inc., www.mathworks.com). For DeerNet, the Deep Learning Toolbox and the Signal Processing Toolbox for Matlab should be installed
DeerAnalysis (e.g., DeerAnalysis 2019) toolbox for Matlab, available free of charge (G. Jeschke, https://epr.ethz.ch/software.html)
Software for data analysis and graphing, e.g., Origin (OriginLab Corporation, www.originlab.com), SciDAVIS (http://scidavis.sourceforge.net), or Excel (Microsoft Corporation, https://products.office.com/excel)
Xepr-software, as delivered with the EPR spectrometer, for hardware control and data acquisition (Bruker)
In silico spin labeling software, e.g., mtsslSuite (G. Hagelueken, www.mtsslsuite.isb.ukbonn.de; Hagelueken et al., 2015), MMM (G. Jeschke, https://epr.ethz.ch/software.html; Polyhach et al., 2011) or the GFN/FF-based CREST/MD (S. Grimme, https://github.com/grimme-lab; Spicher et al., 2020)
IDT OligoAnalyzer Tool (IDT, www.idtdna.com/pages/tools/oligoanalyzer)
SnrCalculator (D. Abdullin, https://github.com/dinarabdullin/SnrCalculator)
Procedure
RNA Labeling
Order your modified RNA oligonucleotide. For this protocol, the following sequence is used:
5′-GCG GGG ACG ACC CXG C-3′, with X = 5-ethynyl-2′-deoxyuridine
The labeling position should not disturb the overall RNA structure or its function; thus, avoid labeling at catalytically active sites or binding pockets. To minimize possible perturbations, design the modified oligonucleotide strands by exchanging a naturally occurring uridine nucleotide with the modified 5-ethynyl-2′-deoxyuridine. Moreover, where possible, it is preferable to integrate the modification into a double-stranded RNA region since this promotes narrow distance distributions.
Note: Establish an RNase-free environment: Clean your bench and tools e.g., pipettes with 70% ethanol or RNase Away. Wear protective gloves throughout this workflow.
Aliquot the RNA
Dissolve the RNA oligonucleotide in DEPC-treated water (see Recipes).
Aliquot 2.5 nmol RNA into 1.5-ml Eppendorf tubes (Note 1).
Dry the RNA oligonucleotide aliquots in a SpeedVac vacuum concentrator or a freeze dryer. Store the dried RNA oligonucleotide aliquots at -20°C.
Set up the RNA labeling reaction
Pre-heat the Thermomixer to 60°C.
Dissolve the dried RNA oligonucleotide in 4.4 µl DEPC-treated water (see Recipes).
Weigh 1-2 mg CuI (X) into a 2-ml Eppendorf tube and calculate the volume of DMSO (Y), to obtain a 50 mM Cu+ solution:
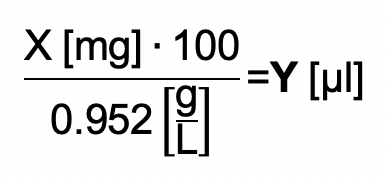
Read Note 2 before you continue!
Dissolve CuI in Y µl DMSO and immediately prepare the Cu+-complex by mixing together:
20 µl DMSO
8 µl 50 mM Cu+ solution from the previous step
3.6 µl 250 mM THPTA/DMSO solution
Vortex the mixture and incubate for 5 min at room temperature
Set up the labeling reaction by pipetting the following solutions into the 1.5-ml Eppendorf tube containing the dissolved RNA (Step A3b):
2 µl spin label/DMSO solution, taken from the 100 mM stock solution
4.6 µl THPTA-Cu+ solution (Step A3d)
Shortly vortex the reaction mixture and incubate the reaction at 300 rpm and 60°C for 30 min (Note 3)
Desalt the RNA oligonucleotide and remove Cu+ via an Amicon Ultra centrifugal filter device
Add 480 µl DEPC-treated water to each reaction tube and transfer each solution to an Amicon Ultra-0.5 filter, previously inserted into an Amicon collection tube.
Spin and concentrate for 30 min at 14,000 × g and room temperature.
Add 500 µl DEPC-treated water to the Amicon filter device and spin again for 30 min at room temperature and 14,000 × g, discard the flowthrough.
Repeat Step A4c one more time.
For RNA recovery, turn the Amicon filter device around and insert it into a clean Amicon collection tube. Spin for 2 min at room temperature and 14,000 × g.
Pipette 50 µl Milli-Q water into the Amicon filter device and wash the membrane by pipetting up and down or by vortexing. Then, place the filter device again upside down and spin for 2 min at room temperature and 14,000 × g.
Wash the filter membrane again, as described in Step A4f, two more times. The sample volume should now be around 200 µl.
Determine the amount of RNA oligonucleotide (nRNA) using the NanoDrop: Clean the NanoDrop measurement pedestal with 70% ethanol using a light-duty tissue wiper. Using a pipette, transfer 1 µl DEPC-treated water onto the measurement pedestal and run a blank measurement as a reference. Clean the pedestal with a light-duty tissue wiper, pipette 1 µl RNA solution onto the measurement pedestal and measure the absorbance at 260 nm (Abs260nm). Then, calculate nRNA with the extinction coefficient (εRNA), either provided by the vendor or calculated using an online tool such as the IDT OligoAnalizer Tool and the volume of your RNA oligonucleotide sample (VRNA) using:
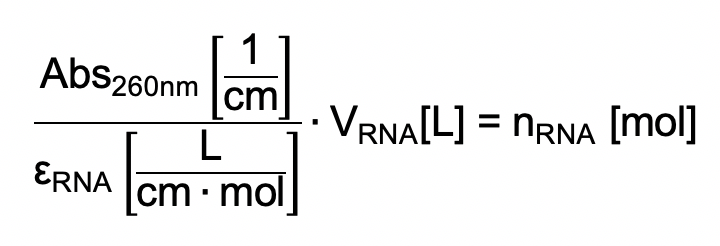
Evaporate the excess water from your RNA oligonucleotide sample, either with the SpeedVac vacuum concentrator or the freeze dryer, until you obtain a volume that does not exceed half of your HPLC-loop capacity. If using a 100 µl injection loop, e.g., inject a volume of 30-50 µl.
Reversed-phase HPLC purification
Start with the equilibration of your reversed-phase column. Therefore, re-buffer the column to 8% buffer A (acetonitrile) and 92% buffer B (0.1 M triethylammonium acetate).
Perform a blank run to check the purity of your column before you purify your sample: Load 30-50 µl Milli-Q water with a Hamilton syringe into the injection loop and start the HPLC run. The blank run uses the same operation parameters as the purification run of your RNA sample. In general, you will need to fine-tune the HPLC run for each new RNA sequence. However, you can use the following settings as a starting point for RNA sequences shorter than 50 nucleotides: Elute with a gradient of 8% → 25% buffer A for 20 min with a flow rate of 1.5 ml/min. Then, equilibrate your column back to 8% buffer A. For short RNA sequences, use the analytical Zorbax 300SB-C18, 4.6 × 150 mm column, but for sequences exceeding 50 nucleotides, use the preparative Zorbax 300SB-C18, 9.4 x 250 mm column. The elution profile for the preparative column is: Elute with 8% acetonitrile for 10 min and then apply a gradient of 8% → 23% buffer A for 55 min with a constant flow rate of 2 ml/min. Finally, equilibrate your column back to 8% buffer A. It is important to heat your HPLC column to avoid the formation of tertiary RNA structures (Figure 2). Heat the column to 60°C, also during the blank run, to ensure the same conditions.
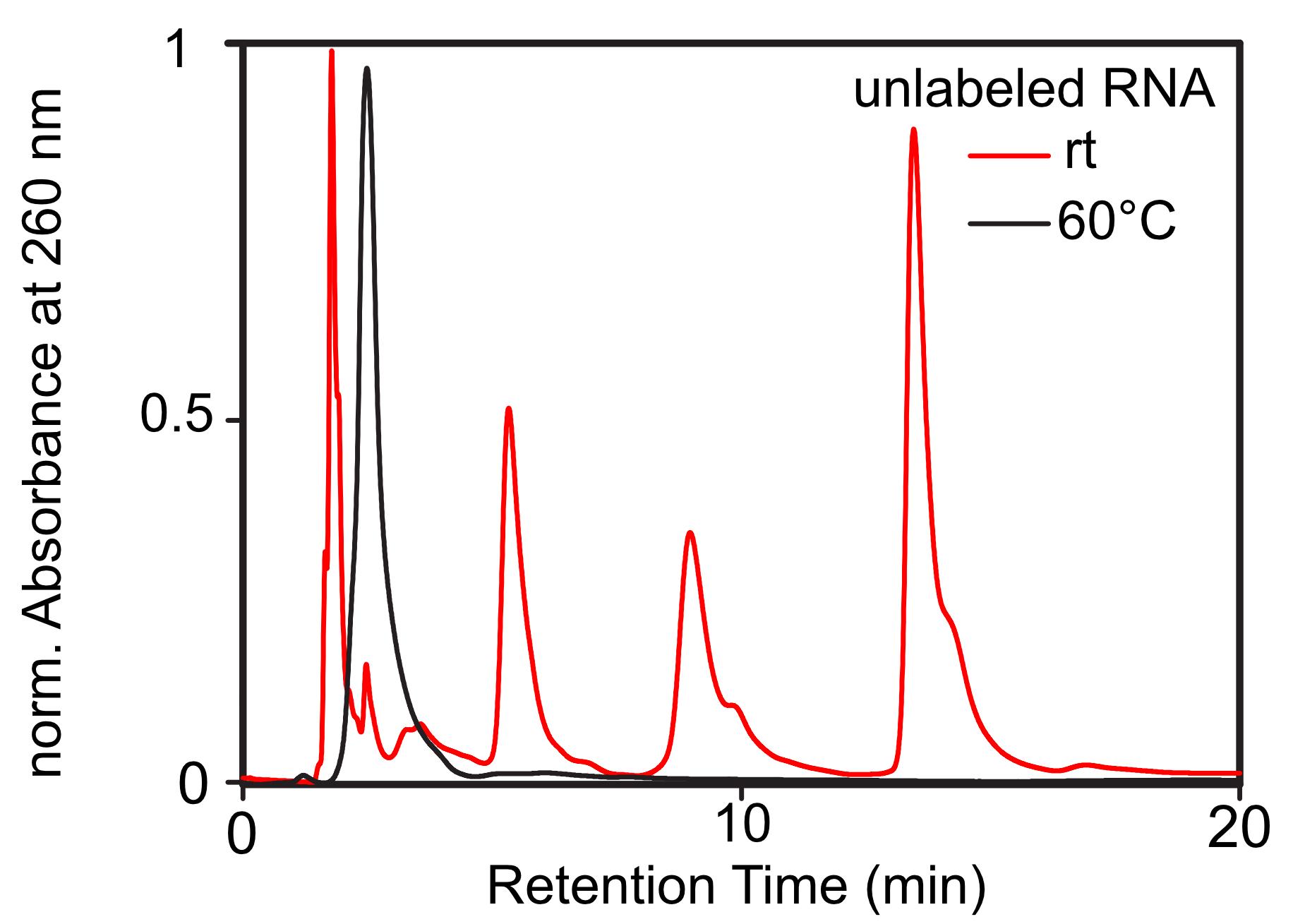
Figure 2. Exemplary normalized HPLC chromatograms of unlabeled RNA at different column temperatures. Upon column heating, the formation of tertiary structures is inhibited, which leads to a single peak (black chromatogram).Inject your RNA sample into the loop. Do not inject more than 2.5 nmol RNA to avoid column overloading. However, you should check the capacity and properties in the manufacturer’s instructions of the particular column you use.
Run the HPLC method.
Separately collect the fractions of unlabeled and labeled RNA. The collection can be performed manually or, if your HPLC is equipped with a fraction collector, automatically. If you use a fraction collector, you can set a constraint to collect everything that exceeds an absorbance of e.g., 15 mAU.
Pool each HPLC peak separately and concentrate the solutions in the SpeedVac vacuum concentrator until you obtain a volume of about 500 µl for each peak.
After the HPLC run has finished, proceed with the purification of your next sample or store the HPLC column. The storage of your column is achieved by rebuffering it to 80% buffer A.
Desalt the labeled RNA with an Amicon Ultra centrifugal filter device
Desalt the RNA samples using an Amicon Ultra centrifugal filter device, as already described above in Steps A4a-A4h.
After desalting your RNA samples, concentrate them in the SpeedVac vacuum concentrator until you reach the desired concentration. Yields of 50-70% with respect to the starting amount of RNA are usually obtained; the losses largely originate from the purification steps. The labeling itself is quantitative, as shown in the HPLC chromatograms (Figure 3).
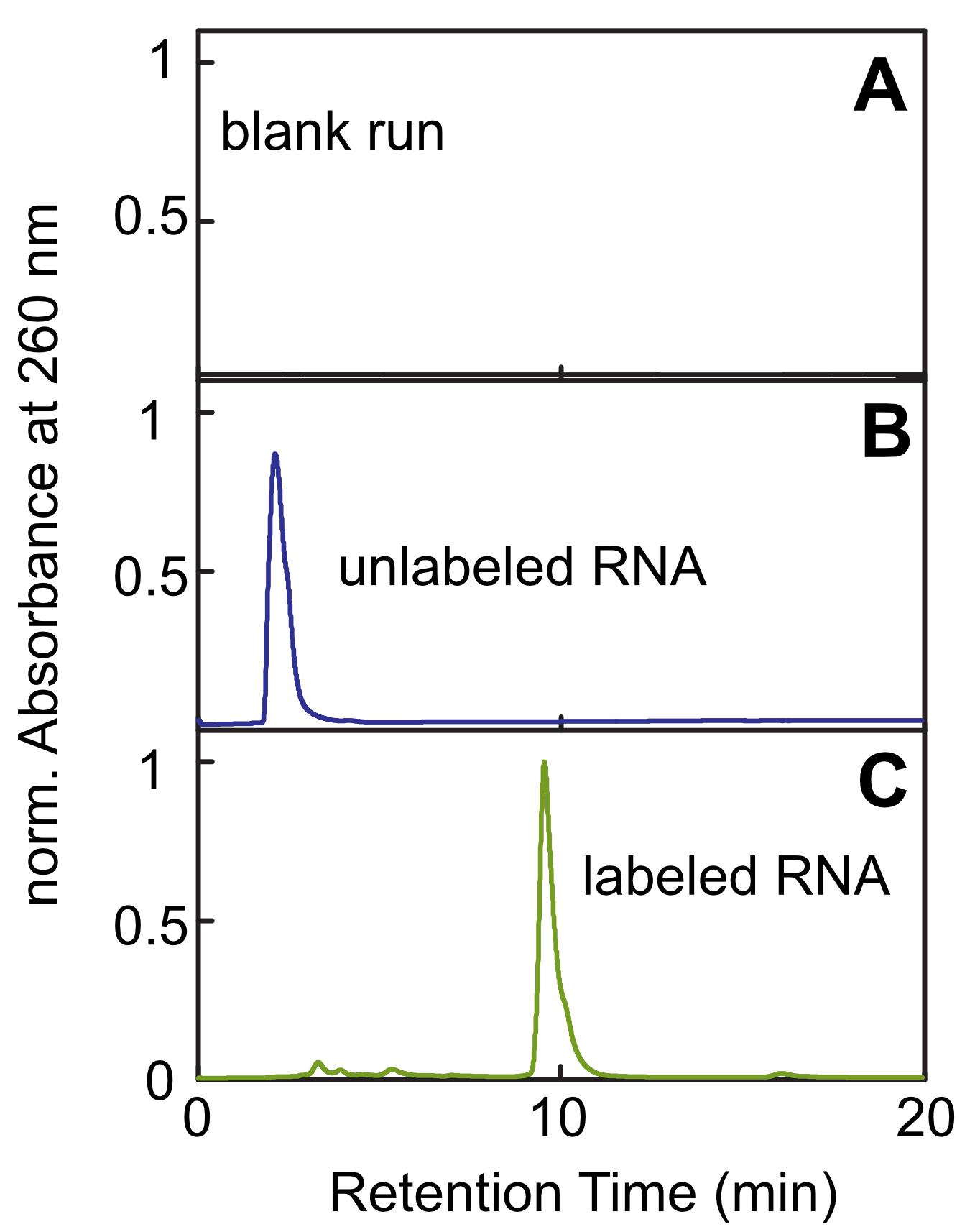
Figure 3. Normalized chromatograms of different HPLC runs. A. Chromatogram obtained by injection of water (blank run). B. Chromatogram obtained by injection of unlabeled RNA. C. Chromatogram obtained by injection of RNA from a labeling reaction. From the chromatogram of the labeled RNA (C), it can be inferred that there is almost no unlabeled RNA left in the reaction mixture, as only a negligible peak at a retention time of ~2 min is obtained. For further purification, only the peak at a retention time of 10 min has been collected and used.
Determine the purity of your labeled RNA sequence via LC-MS (or HPLC and mass spectrometry separately)
Perform an LC-MS measurement of the unlabeled RNA, as a reference. The procedure is analogous to that of an HPLC run (equilibration of the column with buffer C, injection, run, and storage). Use a gradient of 5% → 20% buffer D for 20 min with a constant flow rate of 0.4 ml/min.
Perform an LC-MS measurement of the labeled RNA sample; use the gradient specified in Step A7a. Successful labeling can be checked by determining the exact mass of the species present in a specific LC peak.
If you do not have access to an LC-MS system, perform an analytical HPLC run and subject the fractions containing RNA to ESI-MS or MALDI-MS to determine the exact mass.
Determine the spin labeling efficiency
Determine the concentration of labeled RNA(cRNA) using the NanoDrop (Step A4h).
Prepare 10 µl 25 µM RNA solution, if each RNA strand is singly labeled. If the RNA strand is doubly labeled, halve the RNA concentration.
Fill the solution into a 10 µl capillary.
Perform a cw EPR measurement as described in the user’s manual of the EMXnano spectrometer. Use the “spin count” routine from Bruker to determine the spin concentration(cspin). Another way to determine the spin concentration is to doubly integrate the cw EPR spectrum and to relate this double integral value to that of a reference sample of known spin concentration. The error margin of both spin quantitation methods is 10-20%.
Calculate the labeling efficiency. For singly labeled RNA sequences, the efficiency is given by:

For doubly labeled sequences, use:

Keep in mind that although LC-MS reveals almost quantitative labeling, the yield of labeled RNA with respect to the starting RNA is 50-70% due to losses during purification.
Prepare the PELDOR sample
For singly labeled RNA, transfer 2.0 nmol into an Eppendorf tube and dry it in the SpeedVac vacuum concentrator or freeze-dryer. Halve the amount for doubly labeled RNA.
Dissolve the RNA in 64 µl D2O. Add 16 µl EG-d6 as a cryoprotectant and mix the solution carefully in an Eppendorf tube. This yields a sample of 80 µl with a spin concentration of 25 µM containing 20% v/v EG-d6 (Note 4). During PELDOR measurements, the resonator should be completely filled with your sample; thus, even if your RNA amount is scarce, avoid preparing samples with a volume less than 60 µl.
Fill the entire 80 µl into a 3 mm outer diameter Q-band EPR tube with the Eppendorf pipette using the elongated pipette tips. Carefully shake the EPR tube such that the sample collects at the bottom.
Freeze the sample by carefully immersing the EPR tube into a Dewar vessel with liquid nitrogen. As EPR tubes may crack during freezing, wear safety goggles and use cold-protection gloves when handling liquid nitrogen. Keep the tube in the Dewar vessel with liquid nitrogen.
PELDOR Measurements
Prepare the ELEXSYS E580 Q-Band EPR spectrometer (Bruker BioSpin) for the PELDOR measurement:
As EPR spectrometers are sensitive devices that can be damaged upon operator errors, stick to this protocol and the instructions in the user manual of the instrument. In case of doubt, seek advice from a more experienced colleague.
Connect the turbomolecular pump to the cryostat, switch on the pump, and evacuate the cryostat to a reduced pressure of ~10-4 mbar. Evacuating overnight may be helpful to reach the required vacuum conditions.
Switch on the heat exchanger of the spectrometer and check the temperature of the inward and return flow. Depending on the ambient temperature, the former should be around 10-15°C and the latter should not surpass 25-30°C. If the spectrometer has a closed cooling circuit, check the water level in this system and refill if necessary.
Switch on the spectrometer console, the magnet power supply, and the TWT amplifier. It is important to keep the TWT in the Standby mode to prevent damage of the detection system. Give the system ~1 h to warm-up before starting measurements as this ensures electronic stability of the spectrometer.
Connect the overflow valve of the helium tank to the helium recovery system, if available in your facility. Make sure that the gasflow through the tubing is not blocked (e.g., due to bending) and open the overflow valve on the helium tank to prevent pressure build-up.
From this step on, wear cold protection gloves and safety goggles! Open the needle valve on the helium transfer line. Slowly (~3 min for the whole procedure) insert the transfer line into the helium tank. As the transfer line enters the liquid helium, the gas meter will indicate a gasflow and occasionally, a hiss-sound can be perceived. If helium gas exits the tank directly from the upper opening, first screw the nut encircling the transfer line hand-tight and then fix it with a spanner as soon as the transfer line is fully inserted into the tank.
Immerse the lance of the transfer line into ethanol and check for gasflow. Wipe the gas outlet with a tissue and connect the lance to the cryostat of the spectrometer.
Close the cryostat with an empty sample rod to prevent condensation of air within the cryostat.
Connect the membrane pump to the port on the transfer line. Open the needle valve on the transfer line approximately ¼ turn and switch on the membrane pump to maintain a stream of cold helium gas. Using the gasflow controller of the cryostat, adjust the helium gasflow to ~1 L/h. Switch on the iTC503S temperature controller and set the target temperature to 50K. Cooling down the cryostat from ambient temperatures to 50K usually takes 20-30 min (Note 5).
Take the EPR tube out of the liquid nitrogen Dewar vessel and quickly wipe it with a tissue to remove potential contaminants and ice. However, make sure not to keep the EPR tube outside the liquid nitrogen for too long to prevent the sample from warming up. If liquid nitrogen has condensed within the tube, warming up the tube might lead to ejection of the sample or even rupture of the tube.
Insert the EPR tube into a tightly fitting sample holder and adjust its position such that the sample will be located in the EPR-active zone of the resonator. For the ER5106-QT2 resonator, the center of the sample should be 38 mm below the lower end of the tube holder.
Screw the sample holder, with the EPR tube inserted, into a sample rod.
Ensure that the spectrometer is in the Standby mode. Stop the membrane pump, wait until the needle of the gasflow controller has dropped to zero, and remove the empty sample rod from the cryostat. Quickly, but also carefully and straight, insert the sample rod, with the EPR tube mounted, as far as it will go. Do not leave the cryostat open for a longer period to prevent air condensation (Note 6). Wait at least 20 min before you proceed to the next steps so that the sample is thermally equilibrated.
Spectrometer tuning and safety check
Familiarize yourself with the basic functions and operation windows of the Xepr-software by means of the spectrometer manual. In particular, look up keywords such as “Pulse Tables,” “PulseSPEL,” “SpecJet,” “FT EPR Parameters,” “FT Bridge,” “Microwave Bridge Tuning Dialog Box,” and “Acquisition Trigger.” If you need further explanations, ask a more experienced EPR colleague for advice. In the following steps, the buttons, input boxes, and menu bars in the Xepr-software that you should use at a particular step are set in quotation marks.
Connect the Xepr-software to the spectrometer by selecting “Connect to Spectrometer” in the “Acquisition”-menu and open the “Microwave Bridge Tuning Dialog Box.” Go into the “Tune”-mode and set the “Attenuation” to 10 dB. Set the microwave (MW) frequency to ~33.7 GHz.
Over-couple the resonator (Note 7). The ER5106-QT2 resonator has two screws to adjust the cavity length (right screw) and the cavity coupling (left screw). Adjust the cavity length such that the resonator tuning dip becomes visible in the MW Bridge Tuning Dialog Box. Note that there are two tuning dips for the ER5106-QT2 resonator, a “real” dip and a “fake” dip. These can be distinguished as the “fake” dip moves markedly when the coupling screw is rotated, whereas the “real” dip stays in place. Changing the cavity length moves both dips simultaneously.
Adjust the cavity coupling and the cavity length such that the “real” dip is centered in the Tuning Dialog Box and that the minimum of the “fake” dip is on the right side of the window.
Perform the safety check of the spectrometer’s detection system as described in the instrument manual. Attention: Do not attempt to switch the TWT into the Operate mode, if the defense pulses are absent, as this would damage the detection circuitry. In case of doubt, consult your spectrometer administrator.
Optimize a standing Hahn echo (Weber, 2005)
Adjust the “Center Field” B0 to the value corresponding to g ~2.00 at the current MW frequency ν (e.g., B0 ~11,980 G at ν ~33.7 GHz).
In the Pulse Tables, program the Hahn echo sequence (π/2–τ–π–τ–Echo) into the “+x”channel using pulse lengths of 12 ns and 24 ns for π/2 and π-pulses, respectively. Use τ = 200 ns for the interpulse delay. After entering pulse lengths and delays into the Pulse Tables, confirm each entry by pressing “Enter” on the keyboard.
Select the “Acquisition Trigger”-channel from the dropdown menu and set the acquisition trigger “Length” to 4 ns at the “Position” 0 ns. Set the “Integrator Time Base” to 1.0 ns, the “Shot Rep. Time” to 3,000 µs, and the number of “Shots Per Point” to 10.
Click “Start” in the Patterns window, “Run” in SpecJet, and decrease the MW “Attenuation” to observe the Hahn echo in SpecJet. Adjust the “Attenuation” to maximize the Hahn echo amplitude i.e., to obtain π/2 and π-pulses at the given pulse lengths (Note 8).
Set the “No. of Averages” in SpecJet to 1. Set the “Video Bandwidth” to 20 MHz and adjust the “Video Gain” amplification such that the echo is not clipped. Fine-adjust the MW phase such that the echo is fully detected in the real channel of the quadrature detector (green trace in SpecJet); the signal in the imaginary channel (yellow trace in SpecJet) should be zero on average. Slight changes in the “Center Field” may be helpful in this context to fully bring the sample on resonance.
Record the echo-detected field-swept EPR spectrum
Adjust the acquisition trigger “Position” in the Pulse Tables in such a way that the echo starts at the left of the SpecJet window and increase the acquisition trigger “Length” to ~120 ns, in order to cover the whole echo (Figure 4A). This maximizes the spectral resolution of the field-swept spectrum (Figure 4B).
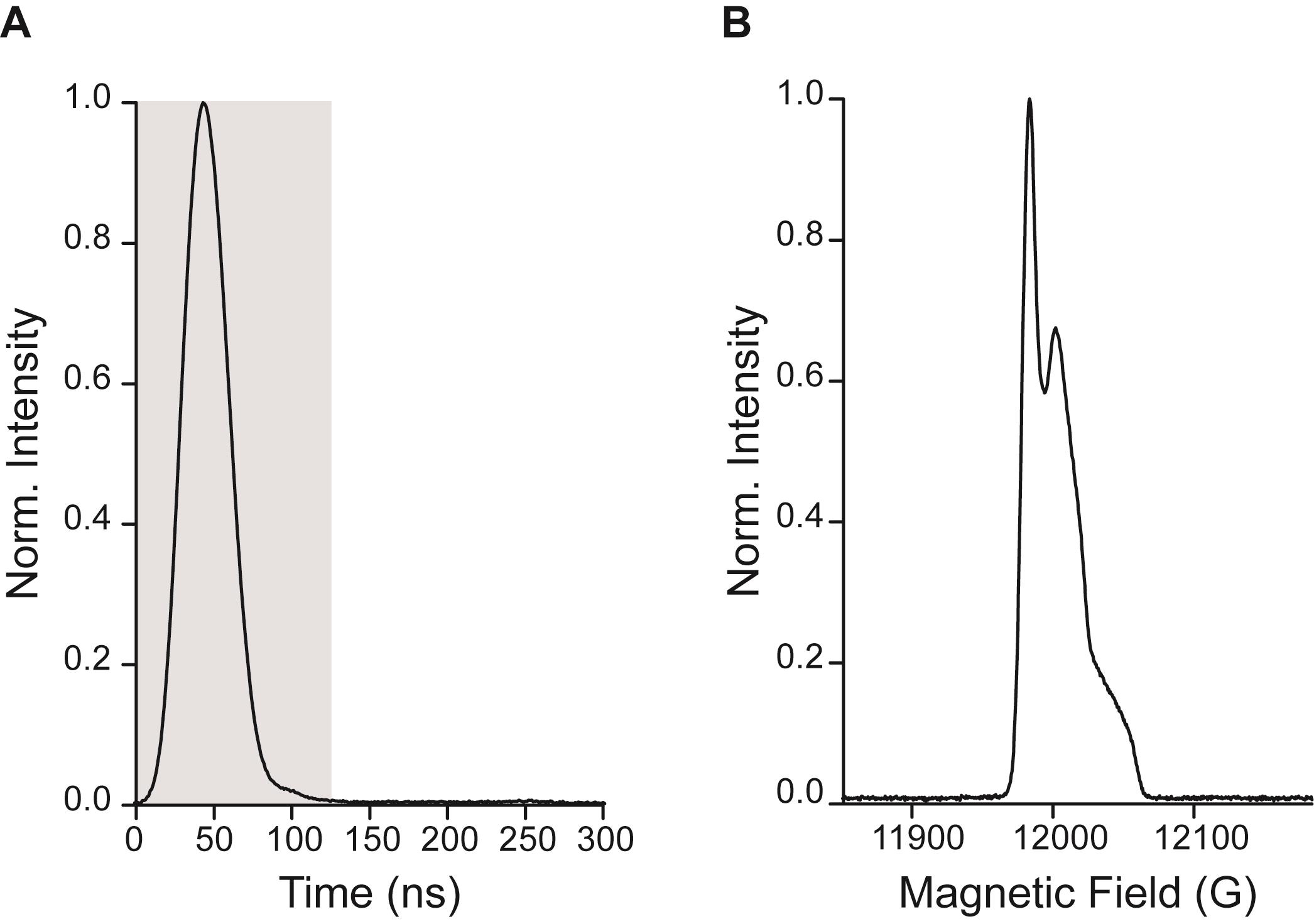
Figure 4. Initial steps for setting up pulsed EPR experiments. A. Hahn echo. B. Echo-detected field-swept EPR spectrum. The grey area in (A) marks the integration region to obtain the field-swept spectrum.In the “Field”-tab within the FT EPR Parameters window, set the “Sweep Width” to 400 G.
Switch to the “Acquisition”-tab to set the “X-Axis Size”, i.e., the number of points in the spectrum. Usually, 800 is an appropriate value, corresponding to a resolution of 0.5 G/point.
Press the “Run” button in the main window to record the spectrum (Figure 4B). Depending on the spin concentration, average multiple scans to obtain a good signal-to-noise ratio (SNR) and save the data to disk.
Read off the magnetic field value that yields the maximum signal intensity in the field-swept spectrum and write it down, you will need it later.
Perform the Two-Pulse Electron Spin Echo Envelope Modulation (Two-Pulse ESEEM) experiment to obtain information on transverse electron spin relaxation, which limits the dipolar evolution window in the PELDOR experiment. In the Two-Pulse ESEEM experiment, the Hahn echo amplitude is monitored as a function of the interpulse delay τ. The default PulseSPEL program provided by Bruker records it as a function of τ. Note, however, that some literature references show these Hahn echo decay curves as a function of 2τ.
Set the “Center Field” to the magnetic field value of the signal maximum in the field-swept spectrum.
In the “Acquisition”-tab, open “PulseSPEL” by clicking the corresponding button.
Load the standard PulseSPEL program for the Two-Pulse ESEEM experiment, which is by default located in the folder:
“xeprFiles/PulseSPEL/sharedPulseSPEL/Standard/Spel2009/ESEEM”, and the corresponding variable definitions “descrESEEM.def”.
Set the variables to the values shown in Table 1.
Table 1. Parameters for the Two-Pulse ESEEM experiment
Parameter Description Value p0 Length of π/2-pulse Use the values which you identified as optimal in section B, step 3d. p1 Length of π-pulse d1 Initial interpulse delay τ 200 ns d0 Acquisition trigger offset 0 ns (for setup)
Usually ~432 ns (for experiment)SRT Shot-Repetition-Time 3,000 * srtu (with srtu = 1.02 µs). h Number of Shots per Point 10 d30 Time increment 8 ns Click the buttons “Compile,” “Show Program,” and “Validate” in this order.
In the “Acquisition”-toolbar, choose the option “Run from PulseSPEL,” select the experiment “2P ESE Setup,” and press the “Run”-button in the main program window. Read off the time value at which the echo amplitude is maximal and set this value as the acquisition trigger delay parameter d0. Usually, this value amounts to d0 = 432 ns.
Select the “2P ESEEM” experiment with a two-step phase cycle, set the number of points for this experiment (parameter dim2 in the PulseSPEL program) to 1,024, and click the “Run” button in the main window to record the Hahn echo decay curve (Figure 5B).
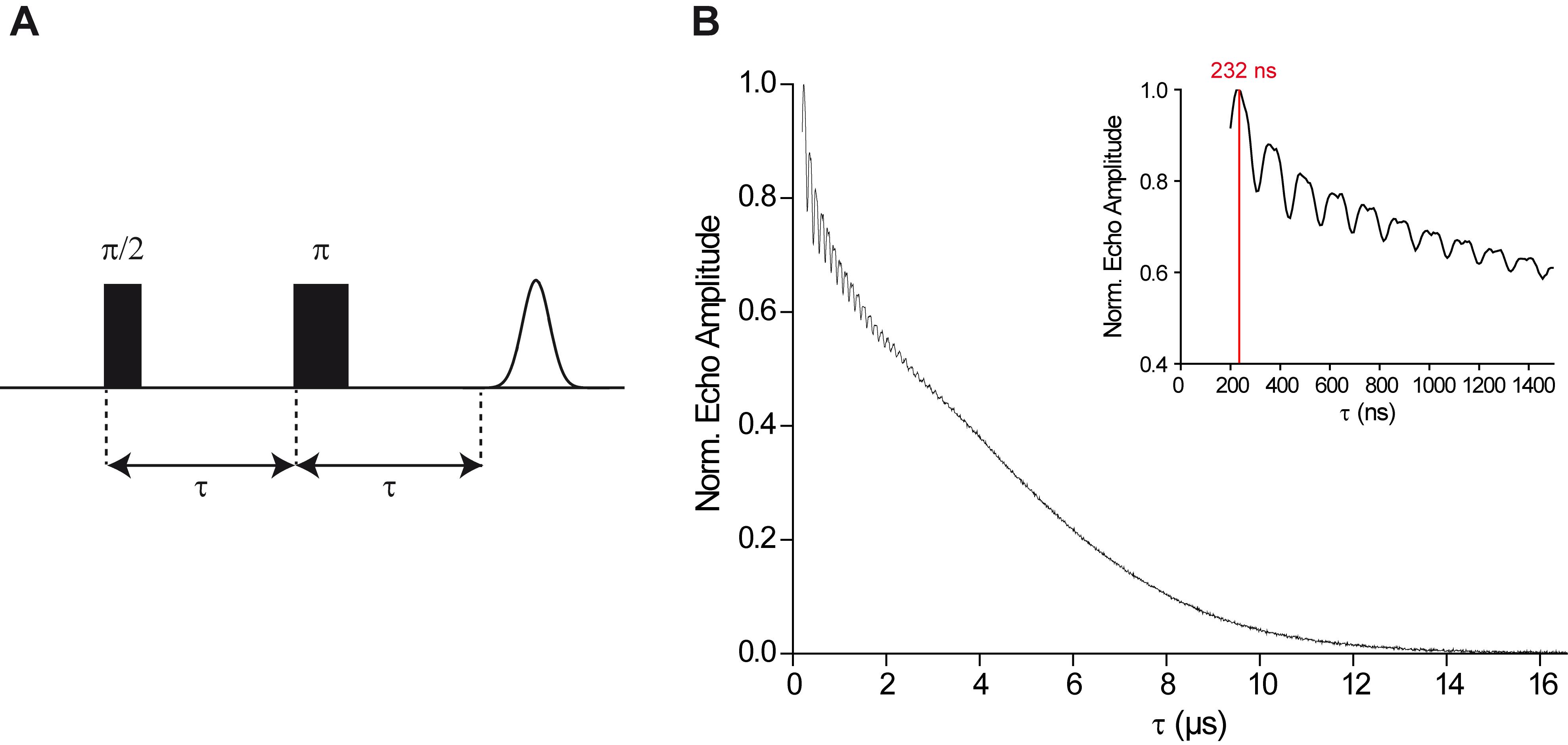
Figure 5. The Two-Pulse ESEEM experiment. A. Schematic representation of the Hahn-echo sequence. In the Two-Pulse ESEEM experiment, the interpulse delay τ is incremented and the echo amplitude is measured as a function of τ. B. Typical Hahn echo decay curve. The inset shows the initial 1.5 µs of the trace, the maximum at 232 ns is highlighted by a red line.If the curve does not reach the zero level at long interpulse delays τ, increase the number of points and/or the time step d30. Read off the time value at which the echo intensity has dropped to almost zero and write it down.
Setting up the PELDOR experiment
PELDOR is a double-frequency experiment that uses the probe frequency νprobe to create a signal on a spin A in a molecule and flips a spin B in the same molecule with the pump frequency νpump. The four-pulse PELDOR sequence is shown in Figure 6. Firstly, a Hahn echo (HE) is created by a π/2 and a π-pulse applied at νprobe and affecting spin A. Next, a pump pulse πpump at the frequency νpump inverts spin B. Finally, a refocusing π-pulse at νprobe is applied, refocusing the Hahn echo from spin A. In the PELDOR experiment, the position of the pump pulse is incremented within the interval τ2 and the integrated intensity of the refocused echo (RE) is measured as a function of the delay T. This yields the so-called PELDOR time trace. If intramolecular dipolar coupling between spins A and B occurs, the coupling frequency will be encoded in the oscillations of the time trace. The overall exponential decay refers to the intermolecular dipolar interactions.
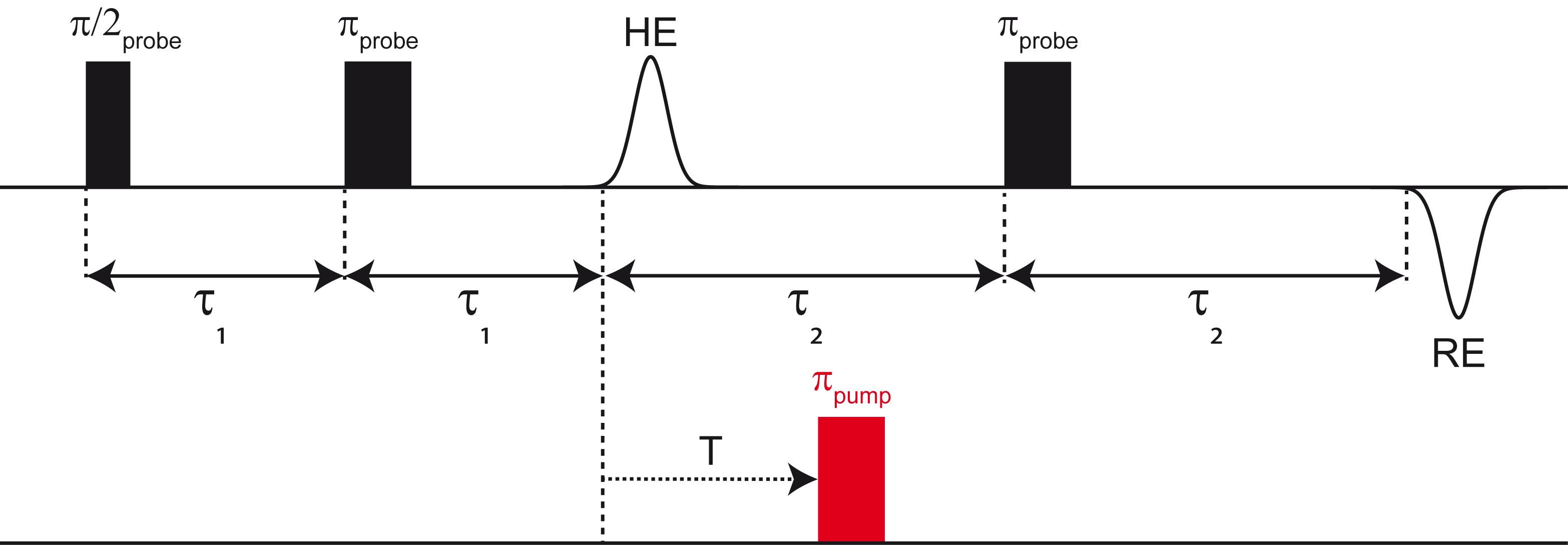
Figure 6. Schematic representation of the PELDOR sequence. Microwave pulses at the probe frequency are shown in black, the pump pulse is displayed in red. “HE” abbreviates the Hahn echo, “RE” the refocused echo. In the PELDOR experiment, the position of the pump pulse is incremented within the first interval τ2 and the integrated intensity of the RE is measured as a function of T.The set-up of the PELDOR experiment consists of various steps, and as PELDOR requires phase cycling and nuclear modulation averaging, it is commonly run from a PulseSPEL program. The Bruker EPR spectrometers are by default equipped with a PulseSPEL program for PELDOR; however, many research groups have developed their own programs featuring custom variable definitions and conventions. Herein, we describe how to set up the PELDOR experiment with our version of the PulseSPEL program, which is available in the Appendix.
Set the variables for the Hahn echo and optimize it at the pump frequency, which is applied in the center of the resonator dip (33.7 GHz).
Load the PELDOR program and the variable definition file into PulseSPEL.
Set the following parameters in PulseSPEL (Table 2):
Table 2. Parameters for setting up the PELDOR experiment. The given parameters are starting values that will be optimized during the course of experimental setup
Parameter Description Value p0 Length of π/2-pulse Use the values which you identified as optimal in Step B3d. p1 Length of π-pulse p2 Length of pump pulse 16 ns as a first guess. d0 Acquisition trigger offset ~360 ns d1 Interpulse delay τ1 Time of the first maximum in the Two-Pulse ESEEM trace, herein 232 ns. d2 Interpulse delay τ2 Set it to a length with enough signal intensity, ~3,000-4,000 ns in this case. SRT Shot-Repetition-Time 3,000 * srtu (with srtu = 1.02 µs). h Number of Shots per Point 10 m Number of steps for nuclear modulation averaging 8 d30 Time increment for the trace 8 ns d31 Time increment for nuclear modulation averaging 16 ns d3 Instrument-related dead time delay Typically ~100 ns Note that the PELDOR experiment is tolerant toward variation of these parameters within certain ranges. However, setting completely improper parameters will result in a poor SNR, a low modulation depth, and data distortion.
Click “Compile,” “Show Program,” and “Validate” in this order.
Select the “2P ESE Setup” experiment. Press the “Run” button in the main window, click “Start” in SpecJet and in the Pulse Tables, and lower the “Attenuation” to 0 dB.
Open the “MPFU control”-tab. Drag the “< +x > Amplitude” slider bar to a level of ~60-70% to maximize the Hahn echo amplitude in SpecJet.
Adjust the global MW phase using the “Signal Phase” slider bar in the FT bridge window. The MW phase should be adjusted such that the signal appears in the real channel of SpecJet and the imaginary channel should be zero on average.
Apply the pump pulse to invert the echo and determine the optimal pump pulse length.
Click “Stop” in the Pulse Tables and in SpecJet.
Select the “3P ELDOR Setup” experiment, set the “Current ELDOR Frequency” to the current spectrometer frequency (~33.7 GHz), and press the “Run”-button in the main window. Write down the “Current ELDOR Frequency” for later use.
Read off the time at which the maximum of the echo occurs in the viewport (Figure 7A, e.g., 72 ns), add it to d0, and set the new value of d0 (e.g., 432 ns) in PulseSPEL. Press “Run” in the main window; the echo should now be maximal at the zero-time on the abscissa in the view port (Figure 7B).
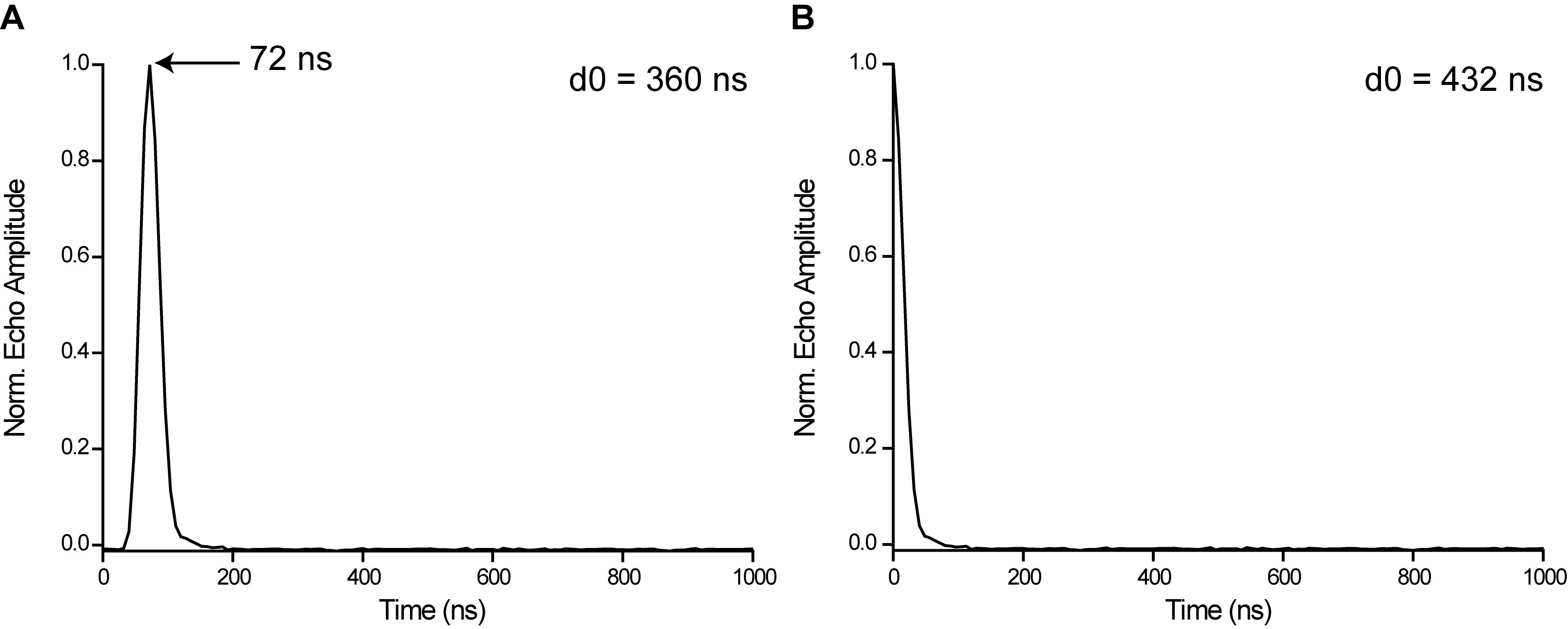
Figure 7. Adjustment of the echo position on the time axis by changing the PulseSPEL variable d0. A. d0 = 360 ns, the echo maximum is located at 72 ns. B. d0 = 432 ns, the echo maximum coincides with the origin of the abscissa.Start SpecJet. Decrease the “ELDOR Attenuation” to 0 dB, which should invert the Hahn echo (Note 9a-d).
Click “Stop” in SpecJet and in the Pulse Tables window. Select the “3P ELDOR Nutation” experiment and press “Run” in the main window; this experiment increments the pump pulse length and measures the amplitude of the Hahn echo. Read off the pump pulse length that leads to the global minimum of the nutation trace and set this value as the parameter p2. Typically, and depending on the sample, optimal pump pulse lengths between 12 ns and 18 ns are obtained (Note 9e).
Set the “Attenuation” to 60 dB, the “ELDOR Attenuation” to 30 dB, and switch the TWT amplifier into the Standby mode. Set the MW Bridge into the “CW”-mode.
Change the MW frequency and optimize the Hahn echo at the probe frequency.
Open the “MW Bridge Tuning Dialog Box” and change the spectrometer frequency by the desired offset (e.g., -80 MHz with respect to the pump frequency) using the “Frequency Slider.”
Perform the safety check as described in the instrument manual and switch the TWT into the Operate mode.
Select the “2P ESE Setup” experiment, the phase cycle option “< + x > none”, and press “Run” in the main window.
Click “Start” in the Pulse Tables window, “Run” in SpecJet, and decrease the “Attenuation” to 0 dB in steps of 10 dB.
Drag the “< + x > Amplitude” slider bar in the MPFU control window to maximize the echo amplitude. Adjust the “< + x > Phase” such that the signal has a negative sign in the real channel. The imaginary channel should be zero on average.
Select the “< - x > none” phase cycle option and again click “Run” in the main window. Adjust the “< - x > Amplitude” slider bar to maximize the Hahn echo. Drag the “< - x > Phase” slider such that the signal has a positive sign in the real channel. The imaginary part of the signal should be zero on average.
Make sure that the absolute echo amplitudes obtained with the phase cycle options “< + x > none” and “< - x > none” are equal.
Choose the length of the dipolar evolution window τ2.
The length of the interval τ2, i.e., the length of the PELDOR time trace, is governed by two aspects, namely transverse electron spin relaxation and the inter-spin distance to be resolved. Transverse electron spin relaxation dictates the maximum of the dipolar evolution window, this can be read off the Hahn echo decay curve recorded in Step B5. The minimal length of τ2 is given by the need to resolve at least 1.5 dipolar oscillations encoding the most probable distance in the sample for reliable data analysis.
If you have prior knowledge of the expected inter-spin distance (e.g., from in silico spin labeling), compute the expected oscillation period and set τ2 accordingly to resolve at least 1.5 oscillations in the time trace.
If there is no information available on the expected distance, choose a value for τ2 according to the Hahn echo decay curve. Set τ2 to a value at which sufficient signal intensity is still left in the Two-Pulse ESEEM-trace. If you later realize that a longer dipolar evolution time window is needed to resolve 1.5 oscillations, abort the PELDOR run, increase τ2, and re-start the measurement. Note that artefacts may occur at the end of the time trace, which have to be cut off later (see section on Data analysis). Take this into account and hence increase τ2 sufficiently.
Set the chosen value for τ2 in PulseSPEL and activate it by clicking “Compile,” “Show Program,” and “Validate.” Depending on the length of the time trace, consider increasing or decreasing the time increment d30. Find a trade-off between a sufficiently high resolution of the dipolar oscillations and acceptable measurement times (Note 10).
Optimize the refocused Hahn echo.
Set the “ELDOR Attenuation” to 0 dB.
Select the “4P DEER Setup” program with the phase cycle option “2-step”. Click “Run” in the main window, “Start” in the Pulse Tables window, and “Run” in SpecJet.
Set the “No. of Averages” in SpecJet to 1.
Increase the “Video Gain” amplification such that the echo fills the whole display of SpecJet without clipping at the top or bottom edge.
In SpecJet, set the “No. of Averages” to a higher value (100-1,000) such that the echo can be recognized clearly. Depending on the number of transient averages to accumulate, this may take a while.
Press “Run” in the main window to transfer the echo from SpecJet into the viewport. Read off the time position at which the echo is maximal.
Set the integration gate width (parameter pg in PulseSPEL) to the length of the longest pulse in the PELDOR sequence, most likely the π-pulse at the probe frequency.
Adjust the acquisition trigger offset d0 such that the echo maximum is located at the center of the integration gate. Example: Assume that the refocused echo, recorded with d0 = 360 ns, peaks at 72 ns (Figure 8A). The integration gate length should be 24 ns starting at the zero-point of the time axis (grey rectangle in Figure 8B). Thus, in order to transfer the echo maximum to the position of 12 ns, set the acquisition delay d0 = 360 ns + (72 - 12) ns = 420 ns.
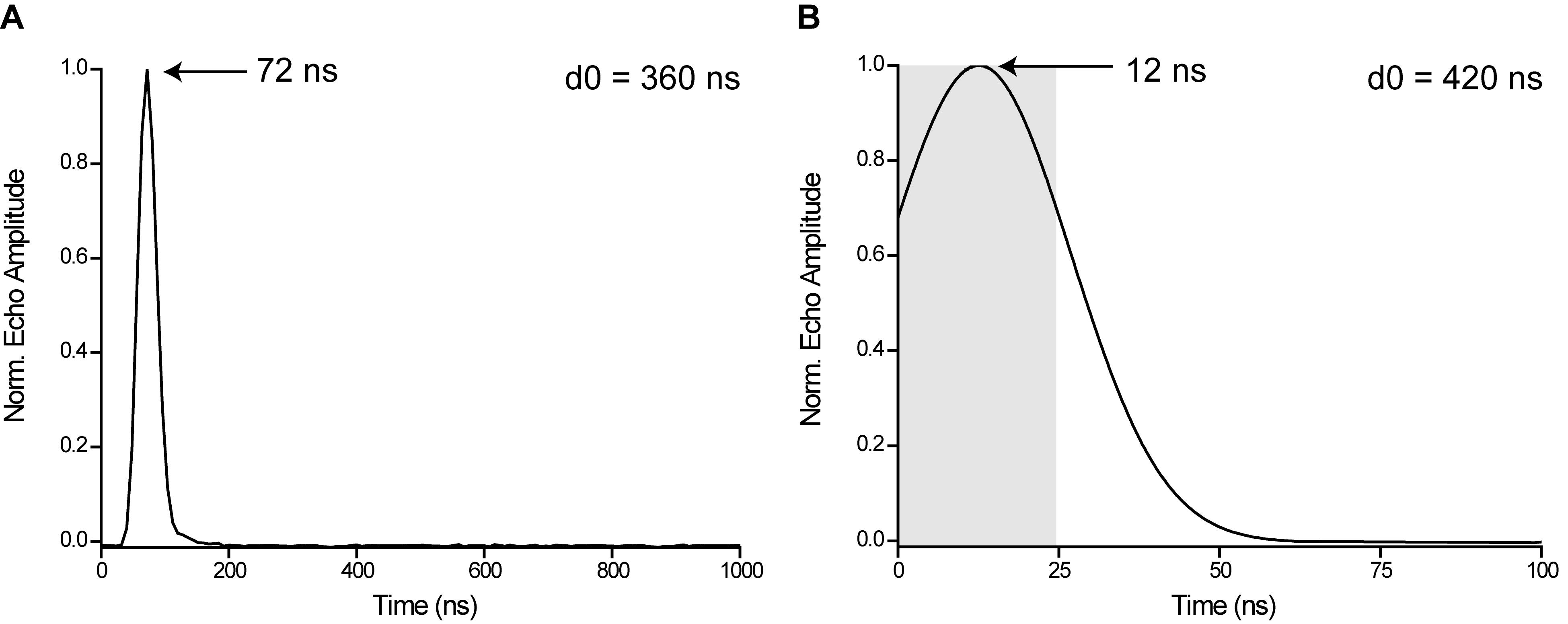
Figure 8. Adjustment of the echo position by changing the PulseSPEL variable d0. A. d0 = 360 ns, the echo maximum is located at 72 ns. B. d0 = 420 ns, the echo peaks at 12 ns. The grey rectangle indicates the integration gate width of 24 ns for the PELDOR experiment.Click “Run” in the main window and check proper positioning of the echo.
Run the PELDOR experiment.
Calculate the number of points to be recorded on the PELDOR time trace and set it as the parameter dim5 in the PulseSPEL program. It is given by:
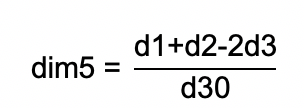
with the interpulse delays d1 and d2 of the PELDOR sequence (τ1 and τ2 in Figure 6), the dead-time delay of the spectrometer d3, and the time increment d30.
Select the “4P DEER” experiment and the 2-step phase cycle from the dropdown menu and click “Run” in the main window. Click the “Re/Im”-button in the main window toolbar to display the imaginary channel and make sure that it is flat and around the zero level. Change the MW phase accordingly, if there is an appreciable amount of signal in the imaginary channel.
Set a sufficiently high number of scans (50-1,500) to achieve a good SNR. The SNR can be defined as the modulation depth divided by the standard deviation of the noise and should amount to at least 20 or better 100 to permit reliable data analysis. Determination of the SNR can be achieved using e.g., the program SnrCalculator. Depending on the spin concentration, the length of the time trace, and the chosen frequency offset, acquisition times between 4 h and 48 h are usual (Note 10).
As the measurement has finished, save the data on disk in the standard Bruker BES3T-format (.DTA / .DSC-files).
A summary of all key steps described above is shown in Figure 9.
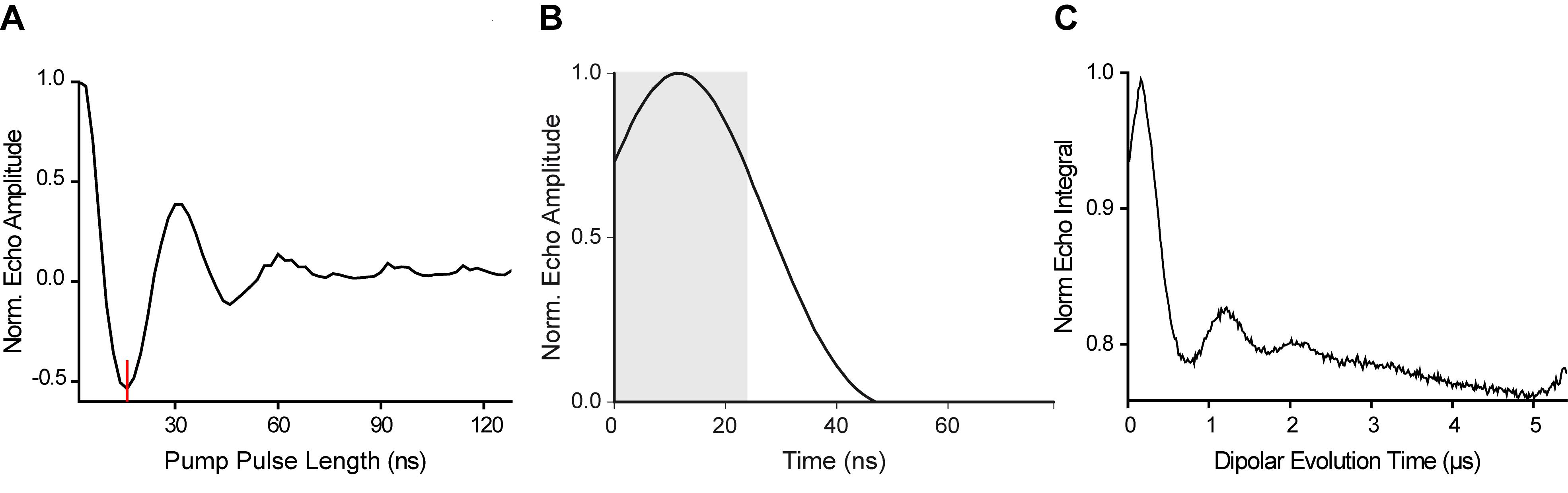
Figure 9. Synopsis of the key steps for setting up a PELDOR experiment. A. Transient nutation experiment. The optimal pump pulse length that leads to a maximal inversion of the echo is given at the global minimum of the trace and marked by a red line. B. Refocused Hahn echo. The integration gate has a length of 24 ns and is indicated by the grey shaded area. C. Raw PELDOR time trace as obtained from the spectrometer prior to data processing.
Switch the TWT into the Standby mode. Set the “Attenuation” to 60 dB and the “ELDOR Attenuation” to 30 dB. Switch the microwave bridge into the “CW”-mode and then to “Standby”.
If the RNA strand and the spin label are rigid, as is the case here and in Wuebben et al., 2020, orientation selection can occur, i.e., not all orientations of the inter-spin vector with respect to the external magnetic field are sampled in one pair of pump and probe frequency. Orientation selection manifests itself in the PELDOR time traces by the dipolar oscillations becoming dependent on the microwave frequency offset Δν. Moreover, the Fourier-transform of the time traces no longer displays the whole Pake pattern.
Regarding data acquisition, two approaches exist to cope with orientation selection. Firstly, the PELDOR experiment can be performed at the frequency offset with minimal orientation selection; in our hands, this is achieved by applying the pump pulse at the maximum of the nitroxide spectrum and the probe pulses at an offset of -80 MHz with respect to νpump.
Secondly, PELDOR measurements can be performed at different combinations of νpump and νprobe. This approach also offers the possibility to take the orientational selectivity explicitly into account during data analysis (see section on Data analysis; Wuebben et al., 2019).
If you opt for the latter approach, repeat this protocol from Step B6c, now using another offset between νpump and νprobe (Figure 10). It is recommended to perform orientation-selective measurements on a particular sample directly one after another and not to remove the sample in between. Thus, the tuning and the sample positioning within the resonator will be equal for all offsets and no changes in spectrometer sensitivity will affect the data quality.
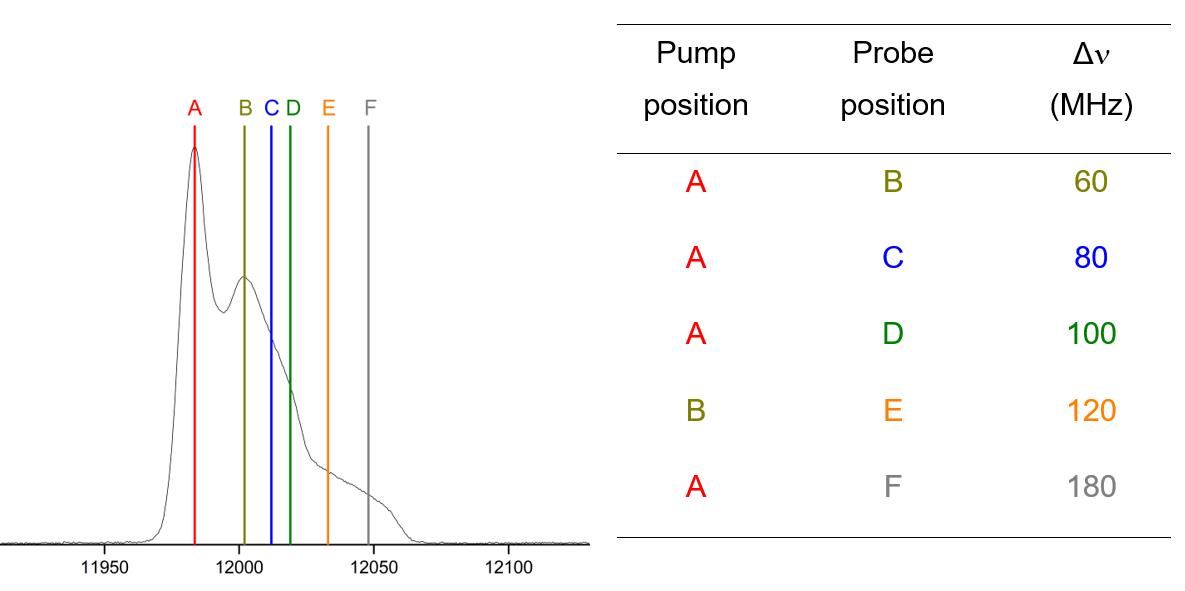
Figure 10. Echo-detected field-swept EPR spectrum with the positions of the pump pulse and the probe pulses indicated for different frequency offsets ΔνIf all measurements on a particular sample have been performed, switch off the membrane pump, wait for the needle of the gasflow controller to drop to zero, and remove the sample rod.
At this point, one can either perform PELDOR measurements on another sample, or completely switch off the spectrometer as described in the instrument manual.
Note: It is good practice to perform the PELDOR experiment on two to three independently prepared samples to ensure data reliability and reproducibility.
Data analysis
If a single PELDOR time trace has been recorded at the frequency offset showing the least orientational selectivity (Δν = 80 MHz, in our hands), perform data analysis starting at point 3.
If PELDOR data have been collected at different combinations of pump and probe positions (Figure 11A), pre-process the time traces in the following way: Divide the raw data recorded at the different offsets by the respective number of scans. Afterwards, sum up all traces and save the result for further processing (Figure 11B). In this way, orientation selection is minimized. This step can be performed either with the data processing tools in the Xepr-software or, more conveniently if many traces are to be processed, a home-written Matlab-script.
Alternatively, and to obtain additional angular information on the orientation of the spin label with respect to the external magnetic field, the program PeldorFit can be used (Abdullin et al., 2015).
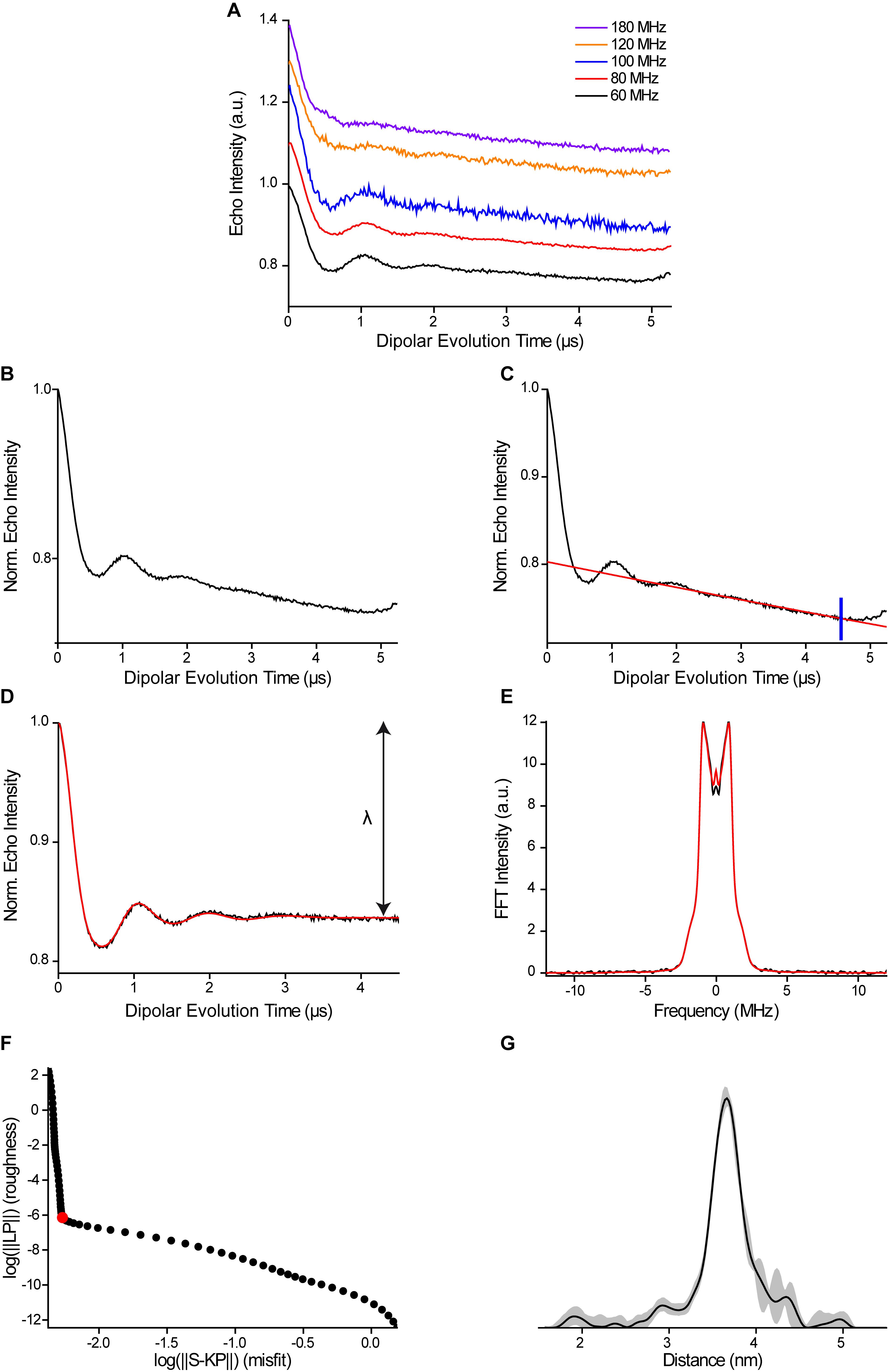
Figure 11. Analysis of orientation-selective PELDOR data. A. Time traces obtained at different offsets between the pump and observer frequency. B. Time trace obtained by summation of the traces shown in (A). C. Time trace from (B) with a background fit indicated as a red line. The cut-off to remove the artefact at the end of the trace is shown in blue. D. Background-corrected time trace with a fit obtained by Tikhonov regularization in red. The arrow indicates the modulation depth λ. E. Pake pattern obtained by Fourier transformation of (D). F. L-curve with the regularization parameter used for computing the distance distribution highlighted in red. G. Distance distribution with the background validation shown as grey shading.Start Matlab. In Matlab, click the “Set Path” button and import the DeerAnalysis folder including its subfolders into Matlab. Click “Save,” “Close,” and then start DeerAnalysis via the command line by entering “DeerAnalysis.” For more detailed instructions, consult the manual and the original publication on DeerAnalysis (Jeschke et al., 2006).
In DeerAnalysis, click the “Load” button, which is located on the right of the window. Select the file containing the PELDOR time trace without orientation selection (either .dat in ASCII-format or .DTA/.DSC in BES3T-format). If you use the BES3T-format, make sure that the .DTA and .DSC-files are both located in the same folder.
Adjust the parameters for raw data processing using the interfaces in the top left corner of the graphical user interface (GUI). Set the zero-time to the maximum of the time trace by clicking the “+” and “-” -buttons or by directly entering a value. Perform a phase correction such that the imaginary part of the dataset is flat and approximately zero. As a starting point, click the “!”-button associated with the phase-correction option.
Set the cut-off parameter to remove artefacts at the end of the time trace (Figure 11C). These artefacts, mostly related to overlap of the pump pulse and the refocusing pulse, manifest themselves by a sudden rise in signal at the end of the trace.
Apply a background fit to remove the intermolecular part of the dipolar coupling. Select the 3D-homogeneous background model in the center panel of the GUI. This background model is based on the function
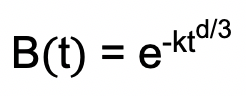 with the density of spins, k, and the dimensionality of the homogeneous distribution, d (Jeschke et al., 2006). Click the “!”-button to auto-optimize the time value for the background start. Inspect the quality of the background correction in the time and frequency domain: In the time domain, the trace should be flat at long dipolar evolution times (Figure 11D). In the frequency domain, the Fourier-transform should have the shape of a Pake pattern and should neither show a deep hole nor a sharp spike at the zero-frequency (Figure 11E). If you identify any of these deficits, change the value for the background start to improve background correction. If you are still unsuccessful, consider changing the background model, e.g., by altering the dimensionality or by selecting a polynomial background.
with the density of spins, k, and the dimensionality of the homogeneous distribution, d (Jeschke et al., 2006). Click the “!”-button to auto-optimize the time value for the background start. Inspect the quality of the background correction in the time and frequency domain: In the time domain, the trace should be flat at long dipolar evolution times (Figure 11D). In the frequency domain, the Fourier-transform should have the shape of a Pake pattern and should neither show a deep hole nor a sharp spike at the zero-frequency (Figure 11E). If you identify any of these deficits, change the value for the background start to improve background correction. If you are still unsuccessful, consider changing the background model, e.g., by altering the dimensionality or by selecting a polynomial background.After background correction, select the “Tikhonov” option in the “Distance analysis”-panel on the right in the GUI and tick the “L-curve” checkbox. Click the “Compute”-button and wait for the L-curve to appear in the GUI (Figure 11F). The L-curve provides a method for solving the ill-posed problem of translating the PELDOR time trace into a distance distribution and permits choosing the regularization parameter for Tikhonov regularization. In the L-curve plot, the abscissa represents the deviation of the fitted time trace from the experimental data and the ordinate indicates the roughness of the resulting distance distribution, both shown on a logarithmic scale. Various criteria can be applied to determine the optimal regularization parameter, e.g., the L-curve corner criterion (Lc), the Generalized Cross Validation criterion (GCV), and the Akaike Information Criterion (AIC). Usually, selecting a regularization parameter close to the corner of the L-curve is a good choice. Uncheck the “L-curve” tick-box above the plot to display the distance distribution.
In order to test for the influence of background removal on the distance distribution (Figure 11G), call the validation routine of DeerAnalysis by clicking the “Validation”-button. Adjust the validation range for the selected parameters and press the “Compute”-button. As the computation has finished, the ensemble of background fits, background-corrected time traces with their respective fits, and distance distributions will be shown. The background-corrected time trace that has the best fit at the selected regularization parameter can be displayed by clicking the “!”-button in the “Parameter set selection” in the center of the GUI. Choose one of the provided parameter sets and click the “Close”-button to quit the validation window. The selected dataset is then transferred into the main window with the distance distribution shown as a bold black line. The uncertainty analysis as obtained from the validation routine is shown as a grey shaded area.
Save the results by clicking the “Save” button. This generates ASCII-files containing the raw data with the background fit (“*_bckg.dat”), the background-corrected trace and its fit (“*_fit.dat”), the dipolar spectrum (“*_spc.dat”), the distance distribution (“*_distr.dat”), and the L-curve (“*_Lcurve.dat”). A summary of data processing including all parameters specified above will be written to the file (“*_res.txt”).
Apart from Tikhonov regularization, DeerAnalysis provides further methods for transforming the dipolar trace into a distance distribution. A rather fast but less elaborate approach is Approximate Pake Transformation (APT), which is used to obtain a first guess of the distance distribution upon loading the time trace. Distance distributions obtained from APT should be regarded as preliminary results only. The DeerNet (Worswick et al., 2018) feature analyzes the time trace using trained neural networks and performs background-correction and translation from the time domain into the distance domain in one step. An advantage of DeerNet is the lack of user interference, i.e., no manual background correction and no choice of a regularization parameter is required. This increases reproducibility of data analysis and prevents data bias, especially in terms of background uncertainty. In addition to these generic analysis methods, parametrized models assuming, e.g., a Gaussian-shaped distance distribution or a random coil, can be used. In this context, DeerAnalysis also permits constructing and implementing user-defined models. For other programs for data analysis, see Note 11.
Plot the results in your preferred data visualization program (e.g., Origin, SciDAVIS, Matlab, Excel, etc.).
Apart from the inter-spin distance, the time trace provides further information on the sample, which should be taken into account for data interpretation: The time trace can be analyzed with respect to the modulation depth (number of coupled spins in a molecule), the background decay (sample concentration and homogeneity), and the signal-to-noise ratio (quality of sample/measurement). The distance distribution can be analyzed in terms of the most probable and mean distances, the width, the modality, and the shape of the distribution. Any features of the distribution to be interpreted must not vanish in the validation. Especially if the shape of the distribution and, e.g., shoulders are to be interpreted, the PELDOR data should be of high quality, should be reproducible, and additional evidence obtained from independent techniques should be taken into account.
Transform your PELDOR-derived distance distribution into a coarse-grained structural model.From the PELDOR measurement, the distance between the spins of the two unpaired electrons is obtained. In order to transform this inter-spin distance into structural information, in silico spin labeling programs such as, e.g., mtsslWizard, MMM, or the GFN/FF-based CREST-MD, can be used. Based on the structure of the biomolecule, which can either be taken from databases or modeled in silico, these programs generate rotamer ensembles of the spin label and further compute an inter-spin distance distribution. Comparing this in silico distribution with the PELDOR-derived one, permits, e.g., refining an existing structural model, monitoring conformational changes, and docking subunits or parts of biomolecular complexes (Duss et al., 2014; Peter et al., 2019). In this regard, it is advisable to record PELDOR data on several constructs of a biomolecule using different labeling positions. Alternatively, the distance between an anchor point, e.g., a spatially fixed metal ion, and various labeling sites can be measured, thus permitting location of the metal ion within the fold of the biomolecule by trilateration (Abdullin et al., 2014). Moreover, the geometry of multimeric biomolecular complexes, and thus the assembly of the subunits, has been reconstructed from distance measurements (Hagelueken et al., 2009; Pilotas et al., 2012; Valera et al., 2016).
Especially for challenging systems, integrated structural biology has turned into a powerful tool: The combination of e.g., FRET, cryo-EM, NMR, X-ray, SAXS, and EPR allows a comprehensive view of the structure. These methods are complementary to each other, each providing pieces of information that can be assembled into a more detailed structural model. Figure 12 summarizes the approach of integrated structural biology and highlights the contribution of EPR spectroscopy.
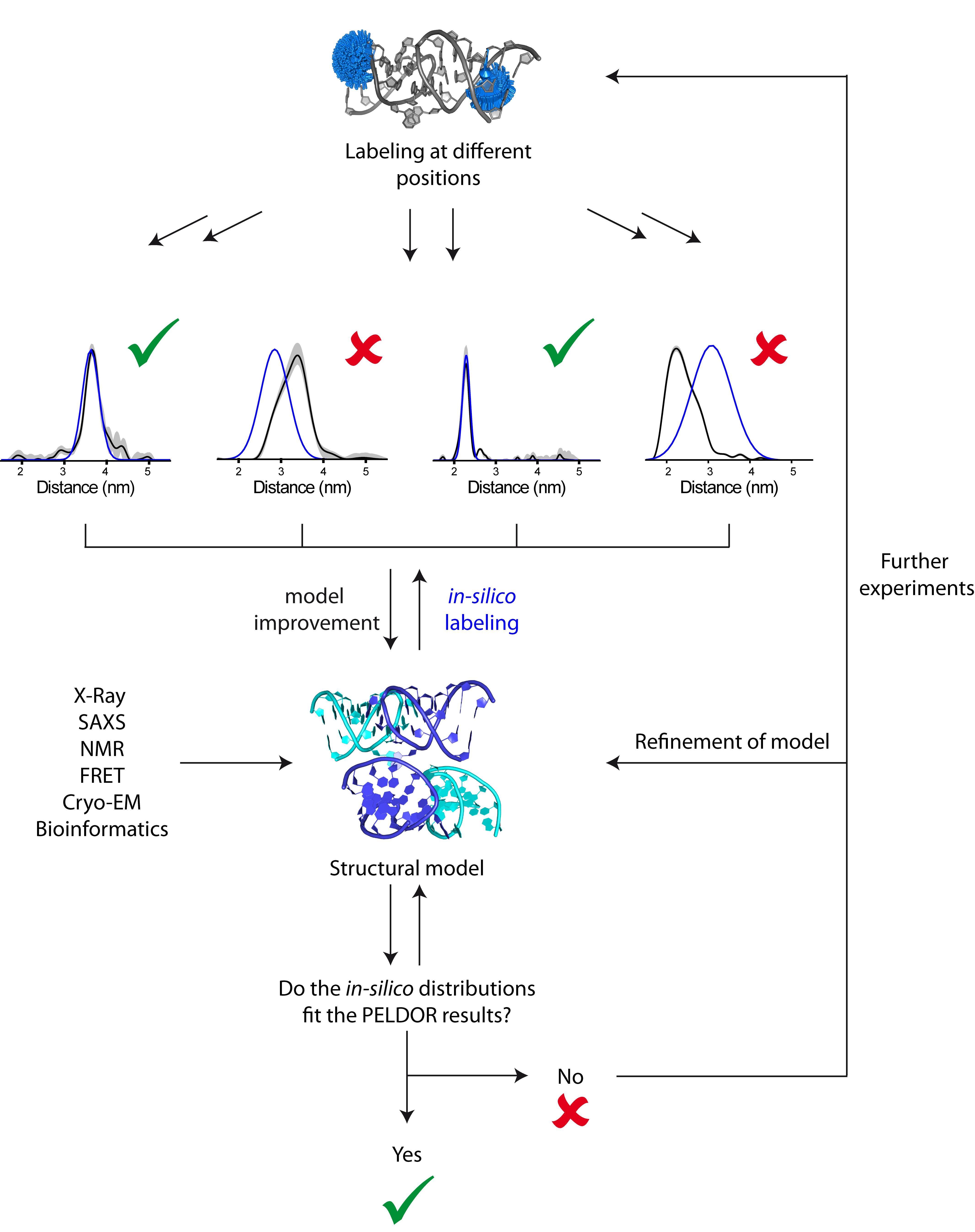
Figure 12. Integrative structural biology. Information from different biophysical methods is combined to a structural model of the biomolecule. Distance distributions derived in-silico from this model are compared with the experimental (PELDOR) ones. They can either match or deviate from each other: if the distributions match, the model is confirmed; if they deviate, further experiments are needed as well as refinement of the model.
Notes
Usually, two labeling reactions can be performed in parallel (in separate Eppendorf tubes), each containing 2.5 nmol RNA. The reaction can be scaled-up to 5 nmol RNA in one Eppendorf tube and in principle, more than 5 nmol RNA (in one Eppendorf tube) should also work. However, when scaling up, you may need to adjust the reaction conditions, e.g., reaction temperature and time. This is also important for each new RNA oligonucleotide sequence you start working with.
It is very important that the complexation of Cu+ with THPTA takes place immediately after dissolving CuI in DMSO, when the solution still has a yellow color. Cu+ is very prone to oxidation by atmospheric oxygen (Hein and Fokin, 2010; Chandrasekaran, 2016) and the formation of Cu2+ in solution can be monitored by the solution’s color: It changes from light to dark yellow when CuI is completely dissolved and turns dark green and finally brown within 5 to 10 min. At this stage, the solution should not be used.
The reaction time needs to be determined individually for each new RNA oligonucleotide. For this, it is recommended to perform a labeling reaction and take samples of 1 or 2 µl every 5 to 10 min. After desalting these samples via an Amicon Ultra centrifugal filter device, you can analyze the reaction progress via HPLC, as described in the procedure Step A5.
EG-d6 acts as a glass-forming agent, i.e., the frozen sample is homogenous. The glass state and the deuterated matrix prolong the phase-memory time of the electron spins, permitting longer dipolar evolution time windows in the PELDOR experiment. Note that some biomolecules can bind or interact with the cryoprotectant (Vagenende et al., 2009; Schmidt et al., 2020). Both the cryoprotectant itself and the ratio of the cryoprotectant and the biomolecule have to be optimized for each individual case. As an alternative to EG-d6, deuterated glycerol (glycerol-d8) or trehalose can be used.
If the temperature does not stabilize at 50K or does not drop at all, address the following points in the given order:
Check the fill level of the helium tank.
Check that all tubing has been connected tightly to the right ports of the membrane pump, the gasflow controller, and the transfer line. Make sure that the helium recovery port is open.
Make sure that the transfer line is permeable and not blocked e.g., with ice.
If the temperature is unstable, make sure that the gasflow amounts to ~1 L/h; much higher or lower flow rates can lead to temperature instabilities. Moreover, check the vacuum within the transfer line insulation and, if necessary, evacuate the transfer line.
Use a small Dewar vessel to cool the EPR tube. In this way, the lower part of the tube can be immersed in the liquid nitrogen, whereas the upper part of the sample rod is outside the coolant. This minimizes the transfer of liquid nitrogen into the cryostat and can prevent difficulties when overcoupling the resonator and removing the EPR tube after the measurement.
If the tuning is unstable, check the following points:
Is the microwave frequency stable? MW frequency instabilities can occur if the electric components were not given enough time to warm up. Moreover, make sure that the cooling water entering the microwave bridge is not too warm (<15°C).
Are the sample and the cryostat in thermal equilibrium and is the temperature stable? After inserting the EPR tube, wait at least 20 min before starting the measurement.
If the resonator tuning dip starts moving without any changes being made to the spectrometer, nitrogen may have condensed in the cryostat. In this case, remove the sample, warm up the cryostat to ~120K for at least 20 min, and re-start from Step B1l. If the problem cannot be solved, warm up the cryostat to room temperature and blow a stream of nitrogen gas through the resonator overnight.
If no echo is seen, try the following steps:
Double-check the parameters such as MW frequency, Center Field, and pulse lengths etc.
Make sure that SpecJet displays enough points to detect the echo at all.
If the Hahn echo amplitude does not cross a maximum when stepwise reducing the attenuation to 0 dB, consider the following aspects:
Check the over-coupling of the resonator. Make sure that the “real” resonator tuning dip is in the center of the MW Bridge Tuning Dialog Box. Especially, make sure to center the “real” dip in the window and not the “fake” dip.
The MW power may not be sufficient to achieve π/2 and π-pulses at the given pulse lengths. Increase the pulse lengths (e.g., to π/2 = 16 ns and π = 32 ns) and again try to optimize the Hahn echo.
If no inversion of the echo can be observed or if the transient nutation experiment reports an optimal pump pulse length greater than 18 ns, address the following points:
Make sure that the “Current ELDOR Frequency” has been properly set, i.e., to the current spectrometer frequency.
Check that the pump pulse length (p2 in the PulseSPEL program) is between 12 ns and 18 ns.
Make sure that the “ELDOR Attenuation” has been set to 0 dB.
Check the tuning and make sure that the tuning dip is centered in the “MW Bridge Tuning Dialog Box” i.e., that the MW frequency coincides with the resonance frequency of the cavity.
Unexpectedly long pump pulse lengths determined from the transient nutation experiment may indicate condensation of liquid nitrogen in the cryostat. See Notes 6 and 7c.
The approximate duration of a PELDOR measurement is given by:
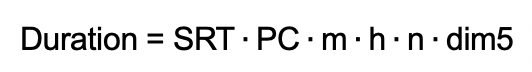
with the shot-repetition-time SRT, the number of phase cycling steps PC (usually 2), the number of nuclear modulation averaging steps m (usually 8), the number of shots per point h, the number of scans to accumulate n, and the number of points to record on the trace dim5.
Apart from the commonly used DeerAnalysis software for translating the PELDOR time trace into a distance distribution, further programs for this task have been published. A selection of these is given below:
DeerLab (Fábregas Ibáñez et al., 2020)
LongDistances (Altenbach, C.; https://sites.google.com/site/altenbach/labview-programs/epr-programs/long-distances)
GLADD/DD (Brandon et al., 2012)
DIPFIT (Steinhoff et al., 1997)
Recipes
DEPC-treated water
Add 0.1-0.2 ml DEPC to 100 ml Milli-Q water, incubate overnight in the fume hood
Due to CO2-formation, the bottle should not be tightly closed
Finally, autoclave the solution to remove the remaining DEPC
Acknowledgments
Funding via the University of Bonn (TRA-2) and the DFG (Reinhart Koselleck grant, Projektnummer 420322655) is gratefully acknowledged.
The protocol is based on the following original research papers:
Wuebben, C., Vicino, M. F., Mueller, M. and Schiemann, O. (2020). Do the P1 and P2 hairpins of the Guanidine-II riboswitch interact?Nucleic Acids Res. 48(18): 10518-10526.
Wuebben, C., Blume, S., Abdullin, D., Brajtenbach, D., Haege, F., Kath-Schorr, S. and Schiemann, O. (2019). Site-Directed Spin Labeling of RNA with a Gem-Diethylisoindoline Spin Label: PELDOR, Relaxation, and Reduction Stability.Molecules 24(24): 4882.
Kerzhner, M., Matsuoka, H., Wuebben, C., Famulok, M. and Schiemann, O. (2018). High-Yield Spin Labeling of Long RNAs for Electron Paramagnetic Resonance Spectroscopy.Biochemistry 57(20): 2923-2931.
Competing interests
No competing interests to declare.
References
- Abdullin, D., Florin, N., Hagelueken, G. and Schiemann, O. (2014). EPR-Based Approach for the Localization of Paramagnetic Metal Ions in Biomolecules. Angew Chem Int Ed 54: 1827-1831.
- Abdullin, D., Hagelueken, G., Hunter, R. I., Smith, G. M. and Schiemann, O. (2015). Geometric model-based fitting algorithm for orientation-selective PELDOR data. Mol Phys 113(6): 644-560.
- Beaucage, S. L. and Radhakrishman, P. I. (1992). Advances in the Synthesis of Oligonucleotides by the Phosphoramidite Approach.Tetrahedron 48: 2223-2311.
- Brandon, S., Beth, A. H. and Hustedt, E. J. (2012). The global analysis of DEER data. J Magn Reson 218: 93-104.
- Chandrasekaran, S. (Ed.) (2016). Click Reactions in Organic Synthesis. ISBN: 978-3-527-33916-7. John Wiley & Sons, Inc, Weinheim, Germany.
- Dastvan, R., Mishra, S., Peskova, Y., Nakamoto, R. K. and Mchaourab, H. S. (2019). Mechanism of allosteric modulation of P-glycoprotein by transport substrates and inhibitors. Science 364(6441): 689-692.
- Duss, O., Michel, E., Yulikov, M., Schubert, M., Jeschke, G. and Allain, F. H.-T. (2014). Structural basis of the non-coding RNA RsmZ acting as a protein sponge. Nature 509(7502): 588-592.
- Fábregas Ibáñez, L., Jeschke, G. and Stoll, S. (2020). DeerLab: a comprehensive software package for analyzing dipolar electron paramagnetic resonance spectroscopy data. Magn Reson 1(2): 209-224.
- Fleck, N., Heubach, C. A., Hett, T., Haege, F. R., Bawol, P. P., Baltruschat, H. and Schiemann, O. (2020). SLIM: A Short-Linked, Highly Redox-Stable Trityl Label for High-Sensitivity In-Cell EPR Distance Measurements. Angew Chem Int Ed 59(24): 9767-9772.
- Geffroy, L., Mangeol, P., Bizebard, T. and Bockelmann, U. (2018). RNA Unzipping and Force Measurements with a Dual Optical Trap. In: Peterman, E. (Ed.) Single Molecule Analysis. Methods in Molecular Biology, vol. 1665, pp. 25-41, Humana Press, New York.
- Goldfarb, D. and Stoll, S. (Eds.) (2018). EPR spectroscopy: Fundamentals and methods. EMagRes Books. ISBN: 978-1-119-16298-8. John Wiley & Sons Ltd, Chichester, West Sussex.
- Hagelueken, G., Ingledew, W. J., Huang, H., Petrovic-Stojanovska, B., Whitfield, C., El Mkami, H., Schiemann, O. and Naismith, J. H. (2009). PELDOR Spectroscopy Distance Fingerprinting of the Octameric Outer-Membrane Protein Wza from Escherichia coli. Angew Chem Int Ed 48: 2904-2906.
- Hagelueken, G., Abdullin, D. and Schiemann, O. (2015). mtsslSuite: Probing Biomolecular Conformation by Spin-Labeling Studies. Methods Enzymol 563: 595-622.
- Hein, J. E. and Fokin, V. V. (2010). Copper-catalyzed azide-alkyne cycloaddition (CuAAC) and beyond: new reactivity of copper(I) acetylides. Chem Soc Rev 39(4): 1302-1315.
- Jeschke, G. (2012). DEER distance measurements on proteins. Annu Rev Phys Chem 63: 419-446.
- Jeschke, G., Chechik, V., Ionita, P., Godt, A., Zimmermann, H., Banham, J., Timmel, C. R., Hilger, D. and Jung, H. (2006). DeerAnalysis2006 – a comprehensive software package for analyzing pulsed ELDOR data. Appl Magn Reson 30 (3-4): 473-498.
- Kerzhner, M., Abdullin, D., Więcek, J., Matsuoka, H., Hagelueken, G., Schiemann, O. and Famulok, M. (2016). Post-synthetic Spin-Labeling of RNA through Click Chemistry for PELDOR Measurements. Chem Eur J22: 12113-12121.
- Kerzhner, M., Matsuoka, H., Wuebben, C., Famulok, M. and Schiemann, O. (2018). High-Yield Spin Labeling of Long RNAs for Electron Paramagnetic Resonance Spectroscopy. Biochemistry 57(20): 2923-2931.
- Kuzhelev, A. A., Krumkacheva, O. A., Shevelev, G. Y., Yulikov, M., Fedin, M. V. and Bagryanskaya, E. G. (2018). Room-temperature distance measurements using RIDME and the orthogonal spin labels trityl/nitroxide.Phys Chem Chem Phys 20(15): 10224-10230.
- Liang, D., Wu, K., Tei, R., Bumpus, T. W., Ye, J. and Baskin, J. M. (2019). A real-time, click chemistry imaging approach reveals stimulus-specific subcellular locations of phospholipase D activity. PNAS 116: 15453-15462.
- Lottspeich, F. and Engels J. W. (Eds.) (2018). Bioanalytics: Analytical Methods and Concepts in Biochemistry and Molecular Biology. ISBN: 978-3-527-33919-8. Wiley-VCH, Weinheim, Germany.
- Malygin, A. A., Krumkacheva, O. A., Graifer, D. M., Timofeev, I. O., Ochkasova, A. S., Meschaninova, M. I., Venyaminova, A. G., Fedin, M. V., Bowman, M., Karpova, G. G. and Bagryanskaya, E. G. (2019). Exploring the interactions of short RNAs with the human 40S ribosomal subunit near the mRNA entry site by EPR spectroscopy.Nucleic Acids Res 47(22): 11850-11860.
- Nikić, I., Kang, J. H., Girona, G. E., Aramburu, I. V. and Lemke, E. A. (2015). Labeling proteins on live mammalian cells using click chemistry. Nat Protoc 10: 780-791.
- Peter, M. F., Tuukkanen, A. T., Heubach, C. A., Selsam, A., Duthie, F. G., Svergun, D. I., Schiemann, O. and Hagelueken, G. (2019). Studying Conformational Changes of the Yersinia Type-III-Secretion Effector YopO in Solution by Integrative Structural Biology. Structure 27 (9): 1416-1426.
- Pilotas, C., Ward, R., Branigan, E., Rasmussen, A., Hagelueken, G., Huang, H., Black, S. S., Booth, I. R., Schiemann, O. and Naismith, J. H. (2012). Conformational state of the MscS mechanosensitive channel in solution revealed by pulsed electron-electron double resonance (PELDOR) spectroscopy. PNAS 109 (40): E2675-E2682.
- Polyhach, Y., Bordignon, E. and Jeschke, G. (2011). Rotamer libraries of spin labelled cysteines for protein studies.Phys Chem Chem Phys13(6): 2356-2366.
- El-Sagheer, A. H. and Brown, T. (2010). Click chemistry with DNA. Chem Soc Rev 39: 1388-1405.
- Salas, D., Gocheva, V. and Nöllmann, M. (2015). Constructing a Magnetic Tweezers to Monitor RNA Translocation at the Single-Molecule Level. In: Boudvillain, M. (Ed.). RNA Remodeling Proteins. Methods in Molecular Biology, vol. 1259, pp. 257-273, Humana Press, New York.
- Schiemann, O. and Prisner, T. F. (2007). Applications of electron paramagnetic resonance to distance measurements in biomolecules. Quart Rev Biophys 40: 1-53.
- Schmidt, T., Wälti, M. A., Baber, J. L., Hustedt, E. J. and Clore, G. M. (2016). Long Distance Measurements up to 160 Å in the GroEL Tetradecamer Using Q-Band DEER EPR Spectroscopy. Angew Chem Int Ed 55: 15905-15909.
- Schmidt, T., Jeon, J., Okuno, Y., Chiliveri, S. C. and Clore, G. M. (2020). Submillisecond Freezing Permits Cryoprotectant-Free EPR Double Electron-Electron Resonance Spectroscopy. ChemPhysChem 21: 1224-1229.
- Schweiger, A. and Jeschke, G. (Eds.). (2001). Principles of pulse electron paramagnetic resonance. ISBN: 9780198506348. Oxford University Press, Oxford.
- Shelke, S. A. and Sigurdsson, S. T. (2014). Site-Directed Nitroxide Spin Labeling of Biopolymers. Struct Bond 152: 121-162.
- Spicher, S., Abdullin, D., Grimme, S. and Schiemann, O. (2020). Modeling of spin-spin distance distributions for nitroxide labeled biomacromolecules. Phys Chem Chem Phys 22: 24282-24290.
- Steinhoff, H. J., Radzwill, N., Thevis, W., Lenz, V., Brandenburg, D., Antson, A., Dodson, D. and Wollmer, A. (1997). Determination of interspin distances between spin labels attached to insulin: comparison of electron paramagnetic resonance data with the X-ray structure. Biophys J 73(6): 3287-3298.
- Theillet, F.-X., Binolfi, A., Bekei, B., Martorana, A., Rose, H. M., Stuiver, M., Verzini, S., Lorenz, D., van Rossum, M., Goldfarb, D. and Selenko, P. (2016). Structural disorder of monomeric A-synuclein persists in mammalian cells. Nature 534: 45-50.
- Tsvetkov, Y. D., Bowman and M., Grishin, Y. (Eds.). (2019). Pulsed Electron-Electron Double Resonance.Nanoscale Distance Measurements in the Biological, Materials and Chemical Sciences. Springer, Berlin, Germany.
- Vagenende, V., Yap, M. G. S. and Trout, B. L. (2009). Mechanisms of Protein Stabilization and Prevention of Protein Aggregation by Glycerol. Biochemistry 48(46): 11084-11096.
- Valera, S., Ackermann, K., Pilotas, C., Huang, H., Naismith, J. H. and Bode, B. E. (2016). Accurate Extraction of Nanometer Distances in Multimers by Pulse EPR. Chem Eur J 22: 4700-4703.
- Ward, R. J. and Schiemann, O. (2014). EPR- based Distance Measurements in Oligonucleotides. Struct Bond 152: 249-282.
- Weber, R. T. (2005). ELEXSYS E580 user’s manual. Bruker BioSpin Corporation, Billerica, USA.
- Widder, P., Berner, F., Summerer, D. and Drescher, M. (2019). Double Nitroxide Labeling by Copper-Catalyzed Azide-Alkyne Cycloadditions with Noncanonical Amino Acids for Electron Paramagnetic Resonance Spectroscopy. ACS Chem Biol 14 (5): 839-844.
- Worswick, S. G., Spencer, J. A., Jeschke, G. and Kuprov, I. (2018). Deep neural network processing of DEER data. Sci Adv 4: eaat5218.
- Wuebben, C., Blume, S., Abdullin, D., Brajtenbach, D., Haege, F., Kath-Schorr, S. and Schiemann, O. (2019). Site-Directed Spin Labeling of RNA with a Gem-Diethylisoindoline Spin Label: PELDOR, Relaxation, and Reduction Stability. Molecules 24(24): 4882.
- Wuebben, C., Vicino, M. F., Mueller, M. and Schiemann, O. (2020). Do the P1 and P2 hairpins of the Guanidine-II riboswitch interact? Nucleic Acids Res 48(18): 10518-10526.
- Yang, Z., Liu, Y., Borbat, P., Zweier, J. L., Freed, J. H. and Hubbell, W. L. (2012). Pulsed ESR dipolar spectroscopy for distance measurements in immobilized spin labeled proteins in liquid solution. J Am Chem Soc 134(24): 9950-9952.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Vicino, M. F., Hett, T. and Schiemann, O. (2021). Spin Labeling of RNA Using “Click” Chemistry for Coarse-grained Structure Determination via Pulsed Electron-electron Double Resonance Spectroscopy. Bio-protocol 11(9): e4004. DOI: 10.21769/BioProtoc.4004.
Category
Biophysics > EPR spectroscopy
Biochemistry > RNA > RNA structure
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.