- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Quantitative Characterization of the Amount and Length of (1,3)-β-D-glucan for Functional and Mechanistic Analysis of Fungal (1,3)-β-D-glucan Synthase
Published: Vol 11, Iss 8, Apr 20, 2021 DOI: 10.21769/BioProtoc.3995 Views: 4808
Reviewed by: Alexandros Alexandratosdeepika jaiswalAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
(1,3)-β-d-Glucan synthase (GS) is an essential enzyme for fungal cell wall biosynthesis that catalyzes the synthesis of (1,3)-β-d-glucan, a major and vital component of the cell wall. GS is a proven target of antifungal antibiotics including FDA-approved echinocandin derivatives; however, the function and mechanism of GS remain largely uncharacterized due to the absence of informative activity assays. Previously, a radioactive assay and reducing end modification have been used to characterize GS activity. The radioactive assay determines only the total amount of glucan formed through glucose incorporation and does not report the length of the polymers produced. The glucan length has been characterized by reducing end modification, but this method is unsuitable for mechanistic studies due to the very high detection limit of millimolar amounts and the labor intensiveness of the technique. Consequently, fundamental aspects of GS catalysis, such as the polymer length specificity, remain ambiguous. We have developed a size exclusion chromatography (SEC)-based method that allows detailed functional and mechanistic characterization of GS. The approach harnesses the pH-dependent solubility of (1,3)-β-d-glucan, where (1,3)-β-d-glucan forms water-soluble random coils under basic pH conditions, and can be analyzed by SEC using pulsed amperometric detection (PAD) and radioactivity counting (RC). This approach allows quantitative characterization of the total amount and length of glucan produced by GS with minimal workup and a d-glucose (Glc) detection limit of ~100 pmol. Consequently, this approach was successfully used for the kinetic characterization of GS, providing the first detailed mechanistic insight into GS catalysis. Due to its sensitivity, the assay is applicable to the characterization of GS from any fungi and can be adapted to study other polysaccharide synthases.
Keywords: (1,3)-β-D-Glucan synthaseBackground
Characterization of polysaccharides is fundamental to our understanding of various biological processes, such as cell wall biosynthesis in bacteria, fungi, and plants, biofilm formation by microbes, and the formation of extracellular matrix in humans. Although the characterization of short and soluble oligosaccharides can be performed using a variety of methods including thin-layer chromatography, high-performance liquid chromatography, and mass spectrometry, significant technical challenges remain with respect to the characterization of long, biologically relevant polymers. As a result, molecular details regarding the size and mechanism of biosynthesis of large polysaccharides remain mostly unknown.
Over the past two decades, many methods have been developed to study charged and water-soluble polysaccharides. For example, hyaluronan has been characterized using paper chromatography (Tlapak-Simmons et al., 2005) or electrophoresis (Krupa et al., 2007), and bacterial peptidoglycan has been characterized by electrophoresis (Barrett et al., 2007); however, few methods are available for the characterization of charge-neutral and water-insoluble polysaccharides. Among such polysaccharides, (1,3)-β-d-glucan is an essential structural component of the fungal cell wall (Munro, 2013), and its biosynthetic enzyme, (1,3)-β-d-glucan synthase (GS), is a proven target of FDA-approved antifungal antibiotics (Douglas, 2001). Thus, characterizing the mechanism of catalysis and inhibition of GS is imperative to understand fungal cell wall biosynthesis and the mechanism of action and resistance against GS-targeted antifungal drugs. However, despite GS activity having been known since the 1980s (Shematek et al., 1980), detailed mechanistic characterization has not been possible due to the absence of appropriate methods to quantitatively evaluate the amount and length of (1,3)-β-d-glucan. Consequently, many fundamental aspects of this enzyme, such as the product length specificity, remain ambiguous.
GS has been characterized using radioactive assays (Shematek et al., 1980) that quantitate the total amount of water-insoluble (1,3)-β-d-glucan by determining the amount of incorporated d-glucose (Glc). While this method determines the overall activity of GS, it does not reveal the length of the polymer products. The length of GS products has been characterized using reducing end modification (Shematek et al., 1980), where the reducing end of (1,3)-β-d-glucan is reduced to sorbitol and the polymer hydrolyzed to monosaccharides. Subsequently, the length is determined based on the ratio between Glc and sorbitol. This analysis suggests that the average length of (1,3)-β-d-glucan produced by GS in a crude membrane preparation is 60–80 mer (Shematek et al., 1980); however, this approach requires a large amount of (1,3)-β-d-glucan, usually in millimolar quantities. Moreover, the method can underestimate the length by cleavage via a peeling reaction or other mechanisms during the workup or purification (Chhetri et al., 2020); therefore, there currently exists no suitable method for the detailed mechanistic characterization of GS.
More recently, size exclusion chromatography (SEC) has been used to characterize the length of water-insoluble polysaccharides; however, the chromatography conditions and detection methods frequently limit its use in detailed mechanistic investigations. For example, fungal cell wall glucan and chitin were analyzed by SEC after carboxymethyl derivatization with monochloroacetic acid using radioactivity as the detection method (Cabib and Duran, 2005; Cabib, 2009). This chemical derivatization solubilizes otherwise water-insoluble glucan and chitin and allows SEC analysis using an aqueous solvent. However, since the derivatization is not quantitative, the absolute length of the polymers was not determined, and only the relative lengths of glucan- and chitin-containing isolated cell walls were determined (Cabib et al., 2012).
SEC has also been applied to characterize bacterial cellulose synthase. In this case, cellulose was solubilized in dimethylacetamide containing 8% LiCl (w/v) and analyzed by gel permeation chromatography coupled to multi-angle light scattering (GPC-MALS) (McManus et al., 2018). This approach avoids the pitfalls of chemical derivatization and is potentially applicable to many other polysaccharides. However, the length was determined only under the steady-state of enzyme catalysis, and elongation of the cellulose polymer was not detectable, likely due to the limited sensitivity of refractive index detection.
Here, we report the protocol used for the determination of the amount and length of (1,3)-β-d-glucan using SEC in aqueous sodium hydroxide with pulsed amperometric detection (PAD) and radioactivity counting (RC). This protocol overcomes the aforementioned limitations: PAD and RC allow the characterization of the length and distribution of glucan polymers at sensitivities appropriate for the mechanistic characterization of GS, and the use of aqueous sodium hydroxide as a solvent allows solubilization of the otherwise water-insoluble (1,3)-β-d-glucan. The detection limit of this approach (~100 pmol) is greater than four orders of magnitude lower than reducing end modification previously reported for (1,3)-β-d-glucan characterization. One important limitation of this assay is that it requires reasonably pure GS. So far, the method does not work with crude membrane fractions due to the presence of proteins that perturb the migration of glucan through the SEC column. Therefore, in this protocol, we describe both the preparation of partially purified GS using the product entrapment method and the SEC assay. The product entrapment yields GS with 20–30% purity on SDS-PAGE and a specific activity of ~1,000 nmol/min/mg. While our characterization suggests that the impurities in this preparation does not affect the apparent function of GS (either glucan length or kinetics) (Chhetri et al., 2020), it is critical to remove proteins from the glucan samples by washing with SDS. This protocol has been used to study the mechanism of GS catalysis and successfully detected (1,3)-β-d-glucan elongation between ~1,000 and ~8,000 mer for the first time in the over 40-year-long history of GS (Chhetri et al., 2020). The facile chain length determination was also coupled to blocked substrate analogs to unambiguously determine the direction of polymerization, one of the key but challenging mechanistic questions in polysaccharide biosynthesis (Chhetri et al., 2020). These applications demonstrate the significance of the SEC-based GS activity assays. Similar approaches could be adapted to study the activities of other polysaccharide synthases such as glycogen and hyaluronan synthases.
Materials and Reagents
50 ml Falcon tubes (VWR, catalog number: 89039-656)
70 ml polycarbonate bottle assembly, 38 × 102 mm (Beckman Coulter, catalog number: 355622)
3.5 ml open-top thick-wall polycarbonate ultracentrifuge tubes, 13 × 51 mm (Beckman Coulter, catalog number: 349622)
Acclaim SEC-1000 column 7 µm 4.6 × 300 mm (Thermo Fisher, catalog number: 079724)
Acclaim SEC-1000 Guard Column 7 µm 4.6 × 33 mm (Thermo Fisher, catalog number: 082739)
Trans-Blot Turbo RTA Transfer Kit LF PVDF (Bio-Rad, catalog number: 1704274)
MultiScreenHTS FC Filter Plate, 1.2/0.65 µm (Millipore Sigma, catalog number: MSFCN6B5)
MultiScreenHTS Vacuum Manifold (Millipore Sigma, catalog number: MSVMHTS00)
6” wood handle cotton swab (VWR, catalog number: 89031-270)
Peptic digest of animal tissue (Meat peptone; Criterion, catalog number: C7482)
Saccharomyces cerevisiae BY4741 (ATCC, catalog number: 201388)
Yeast extract (Criterion, catalog number: C7342)
d-Glucose (VWR, catalog number: BDH9230)
Agar (Acros, catalog number: 443570010)
0.5 mm glass beads (Scientific Industries, catalog number: SI-BG05)
Note: Prior to use, the glass beads should be cleaned with bleach, soaked overnight, and subsequently washed with deionized water until the pH of the water wash is neutral based on pH testing strips, usually after 10 washes. Finally, the beads should be washed twice with isopropanol and dried overnight.
Liquid nitrogen (Airgas, catalog number: NI 240LT22)
Ethylenediaminetetraacetic acid, proteomics grade (EDTA; VWR, catalog number: M101)
Sodium chloride (EMD Millipore, catalog number: SX0420-5)
Phenylmethylsulfonyl fluoride (PMSF; Acros Organics, catalog number: 215740100)
Tris base (Sigma, catalog number: T6066)
β-Mercaptoethanol (VWR, catalog number: M131)
Glycerol (EMD Millipore, catalog number: GX0185-6)
Pierce 660-nm Assay (Thermo Scientific, catalog number: 1861426)
EZQ Protein Quantitation Kit (Thermo Scientific, catalog number: R33200)
3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS; VWR, catalog number: 0465)
Cholesteryl hemisuccinate Tris Salt (CHS; Anatrace, catalog number: CH210)
Dithiothreitol (DTT; VWR, catalog number: 97061-338)
Guanosine 5′-[γ-thio]triphosphate tetralithium salt (GTPγS; Sigma, catalog number: G8634)
4–20% Mini-PROTEAN TGX Stain-Free Protein Gels, 10 well, 50 µl (BioRad, catalog number: 4568094)
UDP-[U-14C]-d-Glucose (UDP-[14C]Glc; American Radioactive Chemicals, catalog number: ARC0154)
UDP-d-Glucose disodium salt (UDP-Glc; Carbosynth, catalog number: MU08960)
Sodium dodecyl sulfate (SDS; Sigma, catalog number: 75746)
Ethanol 190 proof (Koptec, catalog number: V1101)
Trichloroacetic acid (TCA; EMD Millipore, catalog number: TX1045)
50% (w/w) sodium hydroxide solution (NaOH; Fisher Chemical, catalog number: SS254-500)
ASTM Type I water (Ricca, catalog number: 9150-5)
Isopropanol (Fisher Chemical, catalog number: A416P).
Pullulan standard (Showa Denko K.K., catalog number: P-82)
Primary antibodies:
Anti-Fks1p (a gift from J.-P Latgé, Institut Pasteur) (Beauvais et al., 2001)
Anti-Rho1p (a gift from Y. Ohya, University of Tokyo) (Qadota et al., 1996)
Goat anti-rabbit IgG-HRP secondary antibody (Southern Biotech, catalog number: 4030-05)
Radiance HRP substrate for CCD imaging (Azure Biosystems, catalog number: AC2101)
Dionex ED electrochemical detector disposable electrodes, gold on PTFE (Thermo Fisher, catalog number: 066480)
40% sterile glucose solution (see Recipes)
Yeast Peptone Dextrose (YPD) media (see Recipes)
0.5 M EDTA stock, pH 8.0 (see Recipes)
Breaking Buffer (see Recipes)
Membrane Buffer (see Recipes)
KF Mix (see Recipes)
2× Assay Buffer (see Recipes)
Equipment
500 ml baffled flasks (Chemglass, catalog number: CLS-2044-05)
2.8 L baffled flasks (Chemglass, catalog number: CLS-2022)
-80°C freezer (VWR symphony Ultra-Low Temperature Freezer, model: DW-86L638H)
-20°C freezer (VWR, catalog number: 82027-388)
Tweezers (VWR, catalog number: 82027-388)
Magnetic stir bar (VWR, catalog number: 58948-98)
7 ml Dounce tissue grinder with type A and B pestles (Kimble, catalog number: D9063)
Sample pestle with a tube, 1.5 ml (Research Products International, catalog number: 199226)
Bead beater (Biospec, model: BeadBeater)
Type 45-Ti rotor (Beckman Coulter, model: 339160)
Beckman L7-55 Ultra-speed Centrifuge (Beckman, model: L7-55)
TLA 100.3 Fixed-Angle Rotor (Beckman Coulter, model: 349490)
Beckman Optima TL-100 Ultracentrifuge (Beckman, model: Optima TL-100)
Stir plate (Labnet Accuplate Analog Magnetic Stirrer, model: D0310)
BioVortexer (Biospec, model: 1083)
Water bath sonicator (NEY, model: ULTRAsonik 28H)
ICS-5000+ DC Chromatography/Detector System (Thermo Fisher, model: ICS-5000+)
Scintillation counter (Beckman Coulter, model: LS 6500)
Fraction collector (Pharmacia Biotech, model: FRAC-100)
Software
Microsoft Excel (Microsoft, https://www.microsoft.com/en-us/microsoft-365/excel)
Chromeleon 7.2 Thermo Scientific Dionex Chromeleon Chromatography Data System (Thermo Fisher Scientific, https://www.thermofisher.com/order/catalog/product/CHROMELEON7#/CHROMELEON7)
Procedure
Saccharomyces cerevisiae culture
Streak S. cerevisiae from glycerol stocks onto fresh YPD agar plates (2% agar) and incubate the plates at 30°C for 2–3 days. Plates usually last ~2 weeks.
Prepare 2 × 200 ml YPD media in 500 ml baffled flasks and 12 × 1.5 L YPD media in 2.8 L baffled flasks.
Pick a single colony from the plate and inoculate 2 ml YPD media in a culture tube. Incubate cultures with 200 rpm shaking at 30°C for ~8 h. This is usually performed early in the morning of the day before the large-scale culture.
At the end of the day, inoculate 2 × 200 ml YPD media in 500 ml baffled flasks with 1 ml pre-culture started in the morning, and incubate with 200 rpm shaking overnight at 30°C. Cultures should reach OD600 of 10–12.
The next morning, add an aliquot (30 ml) of the overnight pre-culture to each of 12 flasks containing 1.5 L YPD media, resulting in an initial OD600 of 0.2–0.3. Incubate the cultures with 200 rpm shaking at 30°C until the OD600 reaches 0.8–1.2; typically, this takes ~6 h from inoculation.
Harvest cells by centrifugation at 5,000 × g, 8°C for 25 min.
Wash the pellets once with 10 mM EDTA buffer (pH 8.0) (~30 ml per 20 g cell paste).
Determine the wet cell weight and store pellets in a -80°C freezer until use. Typically, 50–60 g wet cell paste is obtained.
GS preparation
Membrane fraction preparation
Thaw the frozen cell pellets on ice.
Resuspend the cell pellets in 150 ml ice-cold breaking buffer.
Transfer the suspension to the bead beater chamber with ~150 ml ice-cold, clean 0.5 mm glass beads. Top up the chamber with buffer to minimize the available air space once the chamber is assembled, and ensure that the chamber is well covered by the ice-water. Lyse cells by bead beating with 5 × 1 min pulses, with a 10-min rest between each pulse.
Centrifuge at 1,500 × g for 15 min at 4°C to clear the lysate; separate the clarified lysate.
Resuspend the insoluble material in breaking buffer (100 ml) and repeat Steps B1c and B1d to ensure breaking > 95% of cells.
Combine the clarified lysates from the two bead beating cycles and transfer to 70-ml ultracentrifuge tubes.
Centrifuge the lysate at 100,000 × g for 1 h at 4°C. A brown pellet should be visible at the bottom of the tubes. This pellet consists mostly of membrane proteins, lipids, and any other molecules such as glycans tightly associated with them.
Resuspend the pellets in ~80 ml membrane buffer using a 7-ml Dounce homogenizer and the loose pestle (labeled by the manufacturer as “A”) for ~10 strokes, or until no more large pellet chunks remain, and then the tight pestle (labeled by the manufacturer as “B”) for an additional ~10 strokes to ensure thorough resuspension of the membrane pellet.
Determine the protein concentration using the 660 nm Assay Kit and BSA standards according to the manufacturer’s protocol. To ensure appropriate reading, the resuspended membrane fraction is typically diluted 50–200-fold.
Adjust the final volume of the resuspended membrane fractions with membrane buffer to a final concentration of ~5 mg/ml protein. Typically, an 18 L culture yields ~100 ml resuspended membrane fraction.
Flash-freeze 20 ml aliquots in 50 ml Falcon tubes in liquid nitrogen, and store in a -80°C freezer until ready to proceed to the next step (Figure 2, lane 1).
Note: We call this resuspension a “membrane fraction,” which can be stored with minimal loss of activity if freeze–thawing is minimized.
Detergent solubilization
Thaw aliquots of the membrane fraction on ice and transfer to an appropriately sized, chilled Erlenmeyer flask containing a stir bar and place in an ice-water bath. Due to the ability to store the purified GS, we suggest using the entire prepared membrane fraction to obtain a 2.5-ml batch of purified GS.
While stirring the thawed membrane fractions, add GTPγS (27.5 μM final concentration), DTT (6.88 mM), and NaCl (192 mM).
Add a 10% (w/v) CHAPS/2% (w/v) CHS solution dropwise to achieve final concentrations of 0.688% CHAPS and 0.138% CHS.
Allow the suspension to stir for 30 min in an ice-water bath using a stir bar and stir plate. It is important to mix the solution gently to prevent inactivation of GS; thus, set the stirring such that the surface of the solution forms a small vortex without any bubbles. This step can also be performed using a rotator.
Transfer the solution to 70-ml centrifuge tubes and balance.
Centrifuge at 100,000 × g for 30 min at 4°C.
Retain the supernatant as the “detergent-solubilized fraction” (Figure 2, lane 2). If this solution is not immediately used for the product entrapment purification, it can be flash-frozen and stored at -80°C with minimal loss of activity.
Product entrapment purification (see Figure 1 for a flow chart of the procedure)
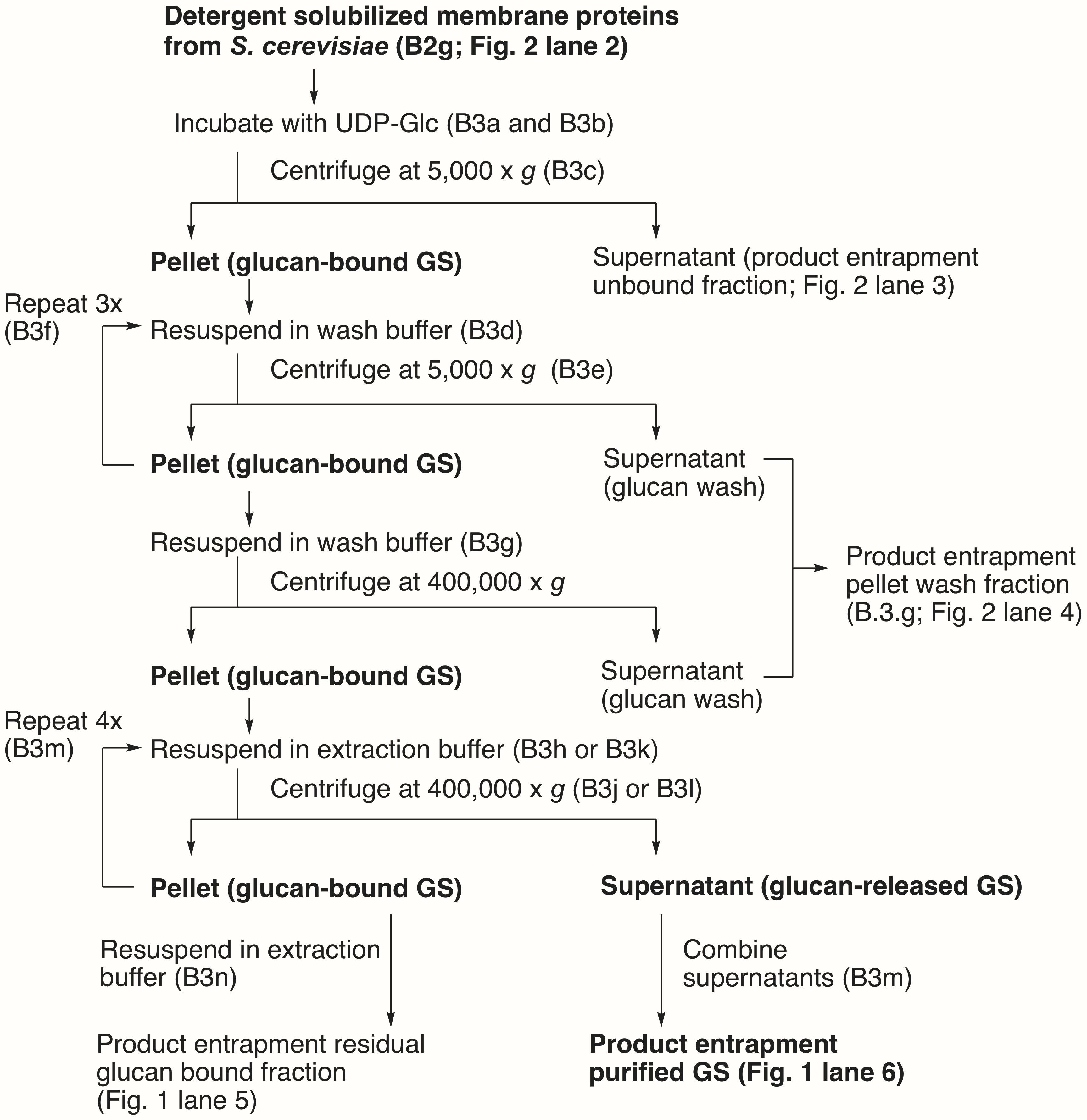
Figure 1. Flow chart of the product entrapment purification of GS. Steps in the protocol and the corresponding SDS-PAGE lanes in Figure 2 are described in parentheses. Key fractions containing GS are bolded.Add a 0.125× volume of KF mix and UDP-Glc (4 mM final concentration) to the detergent-solubilized fraction. Incubate at 30°C for 20–30 min. Periodically invert the tube to ensure sufficient mixing.
Chill the reaction on ice for 15 min. Insoluble glucan should become visible in the solution.
Collect the insoluble glucan and bound proteins by centrifugation at 5,000 × g for 5 min at 4°C (see Figure 3 for an image of the pellet). Save an aliquot of the supernatant (product entrapment unbound fraction; Figure 2, lane 3) for protein concentration determination (Step B3o) and SDS-PAGE analysis (Step B3p).
Wash the resulting glucan/protein pellet by resuspension in 1.5 ml wash buffer and homogenization using the 7-ml homogenizer and the tight pestle “B”.
Collect the glucan/protein pellet by centrifugation at 5,000 × g for 5 min at 4°C. Save the supernatant for later characterization.
Repeat Steps B3d and B3e three more times.
Resuspend the pellet in 1.5 ml wash buffer as described in Step B3d, transfer the resuspension to a 3.5-ml ultracentrifuge tube, and collect the pellet by ultracentrifugation at 400,000 × g, 4°C for 10 min. Combine the supernatant from Steps B3e and B3g and store as the “product entrapment pellet wash fraction” (Figure 2, lane 4) for later characterization.
Release GS from the insoluble glucan by resuspending the pellet in 0.5 ml extraction buffer, followed by homogenization for ~10 s using the hand-held electronic homogenizer (BioVortexer).
Incubate the resuspension overnight at 4°C.
Centrifuge the resuspension at 400,000 × g, 4 °C for 10 min and separate the supernatant and pellet.
Resuspend the pellet from Step B3j in 0.5 ml extraction buffer, followed by homogenization for ~10 s using the hand-held electronic homogenizer.
Incubate the suspension for 10 min on ice, centrifuge the resuspension at 400,000 × g (100,000 rpm using the TLA100.3 rotor), 4°C for 10 min, and separate the supernatant and pellet.
Repeat Steps B3k and B3l three more times.
Combine the supernatants from Steps B3j and B3l to obtain ~2.5 ml “product entrapment purified GS” (Figure 2, lane 6).
Resuspend the pellet in 0.5 ml extraction buffer as described in Step B3k, and adjust the volume to 1.0 ml with extraction buffer to obtain the “product entrapment residual glucan bound fraction” (Figure 2, lane 5).
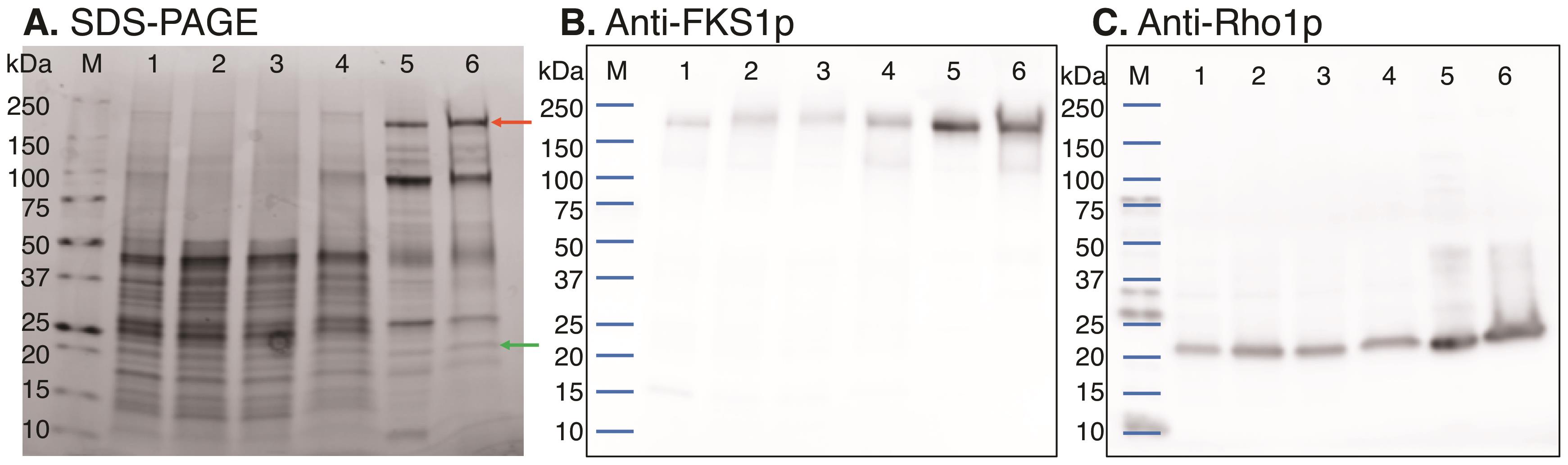
Figure 2. Characterization of GS purification. SDS-PAGE (A) and Western blots using the anti-Fks1p (B) or anti-Rho1p (C) antibody. Each lane was loaded with 5.3 μg protein. Lane: 1, membrane fraction (see Procedure section B1k); 2, detergent-solubilized fraction (see Procedure section B2g); 3, product entrapment unbound fraction (see Procedure section B3c); 4, product entrapment pellet wash (see Procedure section B3g); 5, product entrapment residual glucan bound fraction (see Procedure section B3n); 6, product entrapment purified GS (see Procedure section B3m). The red arrow indicates the band corresponding to Fks1p, and the green arrow indicates the band corresponding to Rho1p. Reprinted with permission from Chhetri et al. (2020).
Figure 3. Image of a glucan pellet formed after product entrapment Step B3c. The pellet was prepared from 30–40 ml product entrapment reaction (Step B3a) in a 50-ml centrifuge tube.Determine the protein concentration at each step of the purification using the EZQ Assay Kit and BSA as the standard. Typically, this protocol yields 0.73 ± 0.12 mg GS from 18 L yeast culture. Knowledge of the protein concentration is critical to performing the GS activity assay and determining the specific activity of GS.
Analyze 1–10 µg protein from each purification step using SDS-PAGE with a 4–20% stain-free gel (Bio-Rad). Determine the purity of GS according to densitometry of the Fks1p band relative to the other bands (see Figure 2A). We typically obtain GS at 20–30% purity. Western blotting can also be used to ensure the copurification of Fks1p and Rho1p, two known components of GS.
Flash-freeze and store aliquots of the purified GS at -80°C.
Typical enzyme activity assays
Prepare stock and working solutions of UDP-[14C]Glc
To prepare a stock solution of radioactive UDP-Glc, dilute 500 µl UDP-[14C]Glc in 70% EtOH to a total volume of 3.5 ml with diH2O by washing the vial with at least 3 × 500 µl volumes of diH2O. Divide the solution into 4 tubes and freeze at -20°C. Once frozen, lyophilize the tubes overnight to dryness. Prepare a 50 mM solution of non-radioactive UDP-Glc and add 3 µmol (60 µl) to each tube, resulting in UDP-Glc solutions of ~10,000 cpm/nmol. Typically, we prepare a stock solution with high radioactivity and dilute based on the purpose of the experiment.
Dilute 1 µl resulting solutions 1,000× with water.
Determine the concentration of the diluted UDP-[14C]Glc prepared in Step C1b using the absorbance at 261 nm and the molar extinction coefficient (ϵ) of uridine at 261 nm (10.1 mM/cm).
To determine the radioactivity of the diluted UDP-[14C]Glc used in Step C1b, transfer 0, 2, 4, 6, 8, 10, 20, 40, 60, 80, 100, and 200 µl diluted sample to scintillation vials, add 3 ml scintillation fluid, and count the radioactivity on a liquid scintillation counter (LSC).
Plot the amount (nmol) of UDP-[14C]Glc according to the radioactivity (cpm) of each sample. The specific radioactivity can be calculated by the slope of a linear fit (cpm/nmol).
To prepare a working solution of UDP-[14C]Glc, dilute the stock solution with non-radioactive UDP-Glc and determine the specific radioactivity by following the Steps C1b–C1e. In our experience, UDP-[14C]Glc with ~800 cpm/nmol specific radioactivity allows detection of glucan formation from 5 µl reaction solution with as short as a 30-s incubation.
Thaw aliquots of stored GS on ice. We typically perform activity assays with a 30–50 µg/ml final concentration of protein in a 30-µl scale.
Prepare a 2× assay buffer as described in the Recipe section.
Incubate the 2× assay buffer and GS in a 30°C water bath for 15 min.
Prepare filter plates for quenching and filtering the insoluble glucan. Add 200 µl 10% w/v TCA to each filter plate well. Each well will be used to quench each timepoint.
Initiate reactions by adding 1 volume of GS to 1 volume of 2× assay buffer. Mix the solution by pipetting up and down as well as closing the caps of the tubes and gently flicking and briefly vortexing before returning to the water bath. Note that this is important for reproducible results as the GS is stored in 30% glycerol, which requires thorough mixing to yield a homogeneous solution.
Incubate the reaction at 30°C.
At each time point, transfer a 5–20 µl aliquot of the reaction to the filter plate wells containing 200 µl 10% w/v TCA (prepared in Step C5). Mix the solution by pipetting up and down.
After quenching all the time points, apply a vacuum using the vacuum manifold to filter the quenched solution. Wash the filter with 3 × 200 µl 10% TCA, followed by 2 × 200 µl EtOH to remove unincorporated UDP-[14C]Glc.
The filters can be allowed to dry overnight or can be analyzed immediately. Remove the plastic support from the bottom of the plate and position the well over an empty scintillation vial. Using a cotton swab, push the filter through the PVDF membrane into the scintillation vial. Break the swab handle so that the vial cap can be closed. If the PVDF membrane remains attached to the plate, transfer to the scintillation vial using a pair of tweezers.
Add 3 ml scintillation fluid, tightly close the vial, and vortex and shake to ensure that the filter is submerged.
Determine the radioactivity of each vial. We set the scintillation counter to a preset time of 3 min, or a % error of 5.0, whichever was shorter.
Quantitative detection of GS product by SEC-PAD analysis
Thaw the frozen GS solution on ice.
Prepare a 2× assay buffer.
Preincubate both the GS and the 2× assay buffer for 15 min in a 30°C water bath.
Prepare the filter plates for quenching and filtering the insoluble glucan. Add 100 µl or an appropriate volume of 10% w/v TCA to each well of the plate. See Step D7.
Initiate the reaction by mixing equal volumes of GS and 2× assay buffer, and mix well by pipetting, flicking, and brief vortexing before returning to the water bath. Note that this is important for reproducible results as the GS is stored in 30% glycerol, which can result in poor mixing and irreproducible results. See Step C6.
Incubate the reaction at 30°C.
At defined time points, remove two aliquots and quench separately.
Quench the first aliquot by mixing 5 µl reaction mixture with 100 µl 10% TCA in one of the filter plate wells. This quenched solution will be used to determine the total amount of glucan.
Quench the second aliquot of reaction mixture (25–200 µl) with 5–10 equivalents of 2% (w/v) SDS solution in a separate well. This quenched solution will be used for length determination by SEC. Do not use acid quenching for length determination since acid-denatured membrane proteins are difficult to remove and interfere with the SEC analysis.
Note: As the filter plate wells only fit ~250 µl volume, for larger aliquots, reactions need to be quenched in larger tubes and transferred to a single well for workup.
Wash the well(s) from Step D7a with 3 × 200 µl 10% TCA, followed by 3 × 200 µl EtOH.
Wash the well(s) from Step D7b with 5 × 200 µl 2% SDS, 5 × 200 µl H2O, and 5 × 200 µl EtOH. The wash with SDS is critical for removing proteins that interact with glucan since they interfere with the SEC analysis.
Dry the filters in the wells overnight under a vacuum.
Transfer the filters from Step D8 to liquid scintillation vials for LSC to determine the total amount of glucose incorporated in the reaction at each timepoint. See Step C10.
Transfer the filters form Step D9 to a 1.5-ml screw top vial and add 200 µl 1 M NaOH.
Note: Use freshly diluted NaOH from a 50% w/w solution.
Briefly centrifuge to ensure that the filter is well submerged in the solution, and then sonicate in an ice-water bath for 10 min.
Note: Longer sonication or sonication at higher temperatures can result in more significant degradation of the glucan polymers and affect the final length calculation and elution profile of the glucan product.
Briefly centrifuge the mixture again and transfer 100 µl to a freash 1.5-ml vial.
Using a wide-bore pipet tip (or regular pipet tip with the extremity cut off to increase the diameter), break apart the filter and resuspend in the solution.
Briefly centrifuge the mixture, remove an additional ~80 µl from the tube, and combine with the solution in Step D14.
Centrifuge the combined solution from Step D16 at 16,000 × g for 10 min at 4°C to remove any particulates.
Transfer 150 µl supernatant to an HPLC vial insert for SEC analysis.
Transfer a 5-µl aliquot of the supernatant to a scintillation vial and analyze by LSC to determine the glucan recovery yield. We typically recover 60–90% of the radioactivity.
Analyze 25 µl each sample by SEC-PAD-RC using the ICS-5000+ DC Chromatography/Detector System equipped with an Acclaim SEC-1000 column at 30°C. Chromatography is performed by isocratic elution with 10 mM NaOH at a flow rate of 0.3 ml/min and monitored by PAD (Gold, Carbo, Quad waveform).
Using a fraction collector, fractionate the elution every 0.5 min during 5–15 min of the chromatography, transfer 120 µl fraction to scintillation vials, and analyze by LSC to determine the radioactive glucose in each fraction.
To determine a standard curve for calibration of the SEC, inject pullulan standards P-82 (Showa Denko K.K.) and analyze under the same conditions as described in Step D20.
Export the chromatography data from the Chameleon software into Excel to calculate the properties of the samples, as detailed in section Data analysis B.
Data analysis
SEC standard curve
Determine the retention time of the pullulan standards at the peak maximum.
Using the peak maximum molecular weight (Mp) for the pullulan standards provided by the manufacturer, plot the log (Mp) of the standard vs. retention time of the pullulan standards to generate a linear standard curve (see Figure 4).
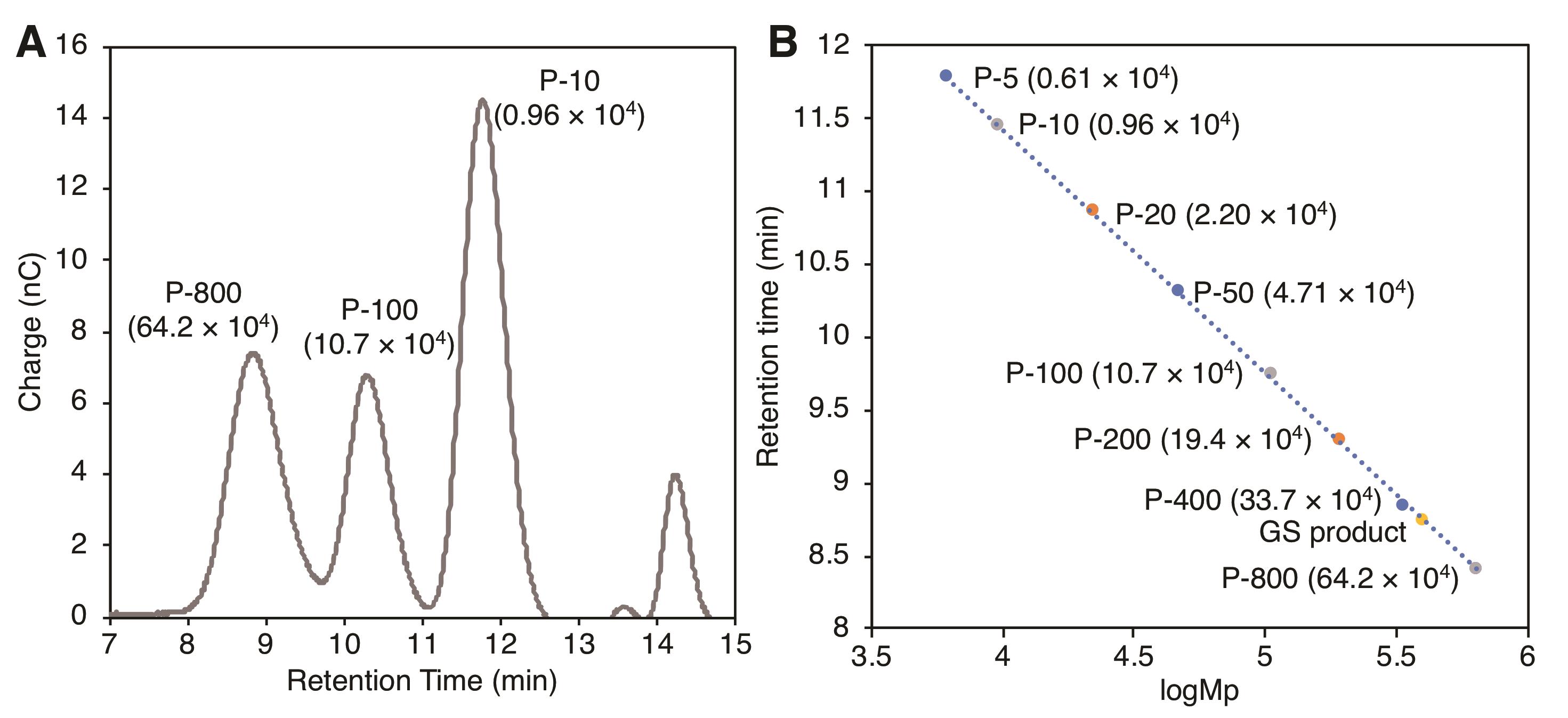
Figure 4. Calibration curve of SEC-PAD using commercial pullulan as a standard. A. Representative chromatograms of the commercial pullulan standards P-800, P-100, and P-10. Numbers in parentheses represent the Mp of the standards. B. A calibration curve with pullulan standards compared with the GS product. Modified with permission from Chhetri et al. (2020).
Analysis of SEC-PAD data
Export the SEC-PAD data for the GS assays in text format and plot the chromatogram (Figure 5A).
Calculate the average retention time (ti, Figure 5B) between every two consecutive data points in the SEC-PAD chromatogram. For our experiments, this resulted in 2,400 slices (PAD data collected every 1/120th s).
Using the calibration curve determined in Step A2, calculate the molecular weight (Mi) at the average retention time (ti) for each slice (Figure 5B).
Calculate the average PAD signal (Pi) for each slice by taking the average of the two PAD data points in each slice (Figure 5B).
Determine the area of each slice (Ai) using the following equation: Ai = Mi × Pi.
To determine the total area of the peak of interest (A, Figure 5C), calculate the sum of Ai for all the slices in the peak: A = ∑Ai.
To determine the weight fraction (Wi) of each slice i of the peak of interest, divide Ai for each slice by the total area of the peak (A).
To determine the weight average molecular weight (Mw), calculate the sum of Mi × Wi for all the slices in the peak: Mw = ∑(Mi × Wi).
To determine the number average molecular weight (Mn), divide 1 by the sum of Wi/Mi for all the slices in the peak: Mn = 1/∑(Wi/Mi).
Calculate the polydispersity index (PDI) for the sample: PDI = Mw/Mn.
The Mp, Mw, and Mn can be converted to the peak maximum degree of polymerization (Xp), weight average degree of polymerization (Xw), and number average degree of polymerization (Xn) by dividing each by the mass of polymerized glucose (C6H10O5, 162.14).
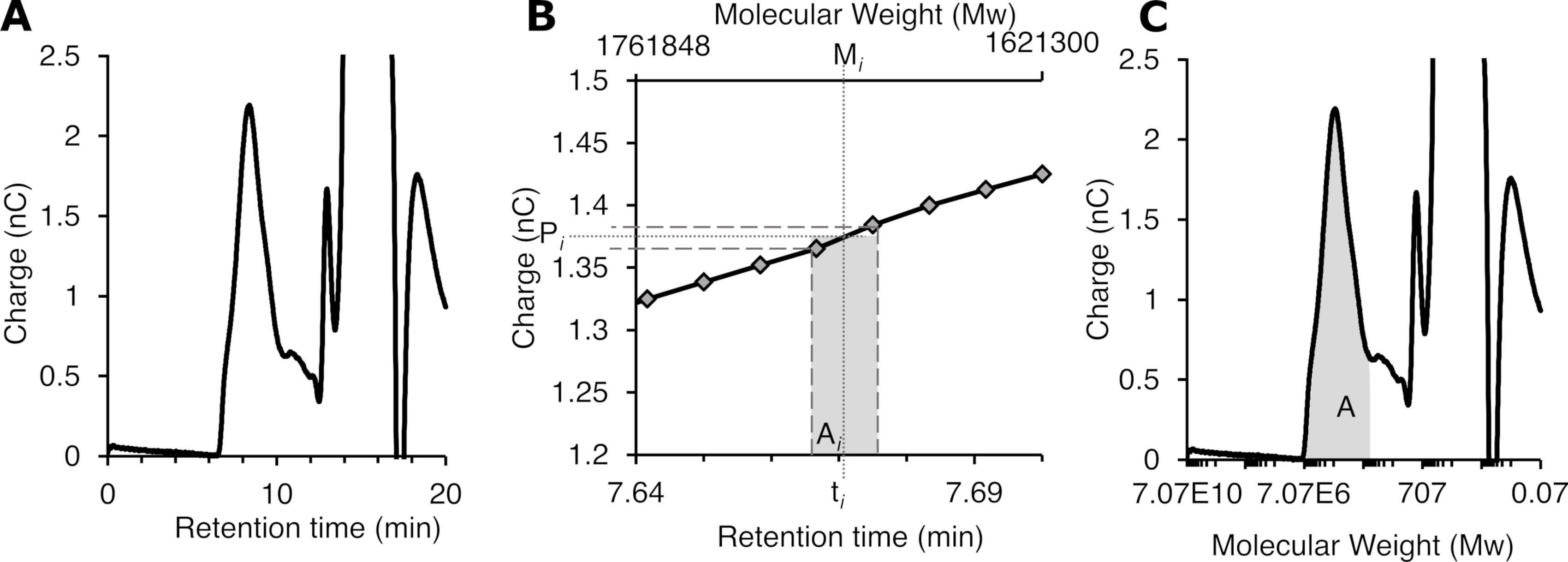
Figure 5. Determination of glucan molecular weight parameters from an SEC-PAD chromatogram. A. Chromatogram plotted with a retention time as the x-axis. B. Magnified view of the chromatogram in A with molecular weight as the x-axis. Representative slice i is highlighted. (C) Chromatogram plotted with molecular weight as the x-axis. The peak area used for the determination of the polymer length parameters is highlighted.
Analysis of radioactivity data
Enter the LSC readout for the fractions in cpm in MS Excel. Adjust for the delay in the retention times of RC relative to that of PAD caused by the tubing between the PAD and the fraction collector.
Using the specific radioactivity of UDP-[14C]Glc, convert the cpm into nmol to determine the amount of Glc incorporated into (1,3)-β-d-glucan. Remember to adjust for the difference in the quenched volumes, dilutions, and yield recovery during the workup for each data point.
Preparation of the overlay of SEC-PAD and radioactivity chromatograms
Overlay of the PAD and RC chromatograms helps visual assessment of the data. For the overlay, we established a quantitative approach as described below.
Plot the PAD and RC chromatograms as described in Sections B and C (Figure 6A).
Calculate the difference (Di) between the PAD value and radioactivity at each data point of the RC chromatogram.
Calculate the sum of the square of Di, ∑(Di)2 for the peak of interest.
Determine the scaling factor for the PAD chromatogram that minimizes ∑(Di)2. For example, a scaling factor of 5 was determined for Figure 6A since 5× magnification of the PAD chromatogram minimized ∑(Di)2.
Using the scaling factor, adjust the y-axis scale in the PAD chromatogram (Figure 6B).
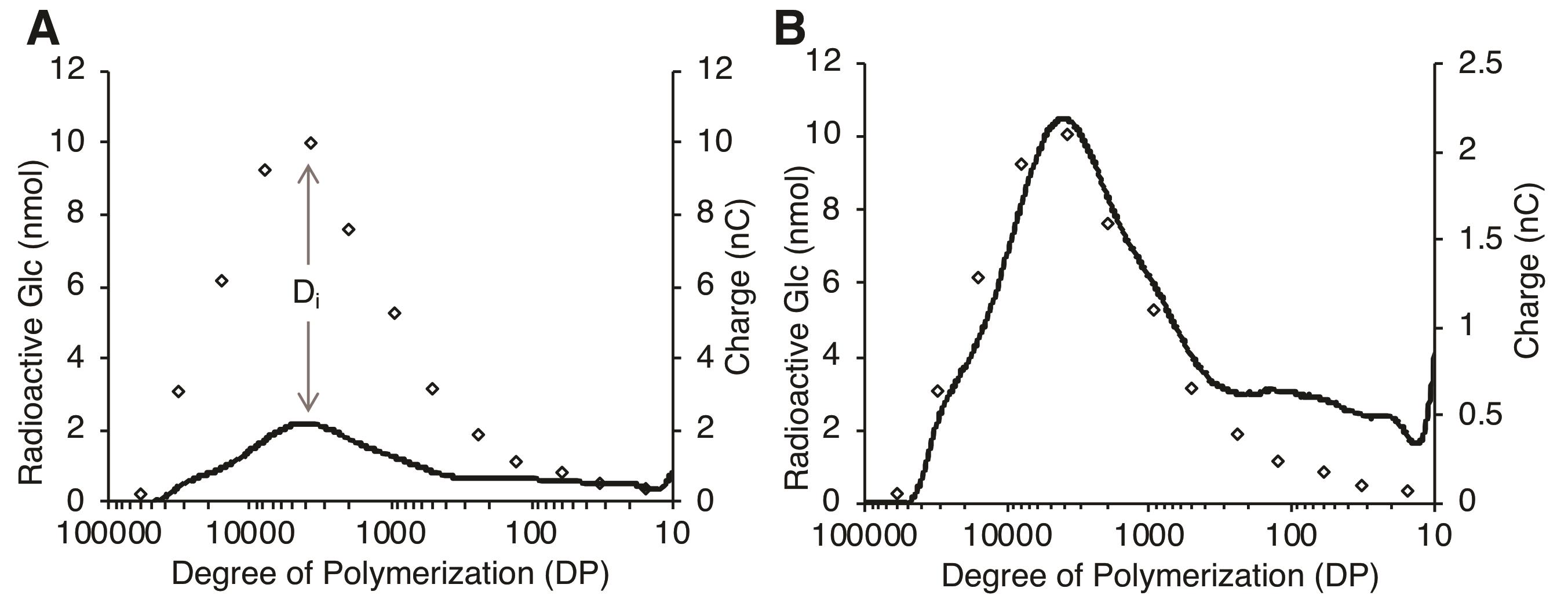
Figure 6. Preparation of the overlay of SEC-PAD and RC chromatograms using least-squares function approximation. A. Original unmodified PAD (solid line) and RC (diamonds) chromatograms. Di represents the difference between the PAD and RC values at data point i. B. Scale-adjusted overlay of the PAD (solid line) and RC (diamonds) chromatograms. The y-axis of the PAD chromatogram was adjusted by minimizing the ∑(Di)2 in (A).
Notes
When preparing the 2× assay buffer, remember to account for the 2-fold dilution of the UDP-Glc upon mixing with GS.
Store the 50% (w/w) NaOH solution with minimal exposure to CO2 to minimize contamination with sodium carbonate and bicarbonate that interfere with PAD analysis.
Make the SEC-PAD-RC eluents with degassed ASTM Type I water to minimize ion contaminants that can affect the background signal of the PAD response.
Recipes
40% sterile glucose solution
400 g (40% w/v) d-glucose
Adjust to 1 L with diH2O
Filter-sterilize
Yeast Peptone Dextrose (YPD) media
20 g peptic digest of animal tissue (meat peptone)
10 g yeast extract
For solid media, add 20 g agar
Adjust to 950 ml with diH2O
Autoclave
Add 50 ml (2% w/v) 40% sterile glucose solution (filter sterilized)
0.5 M EDTA stock, pH 8.0
186.12 g EDTA disodium dihydrate salt
Adjust to 800 ml with diH2O
Cool solution to 4°C in an ice-water bath
Adjust pH to 8.0 with NaOH
Adjust volume to 1,000 ml with diH2O
Breaking Buffer
29.22 g (0.5 M) sodium chloride
20 ml (10 mM) 0.5 M EDTA stock, pH 8.0
Adjust volume to 1,000 ml with diH2O
Add 10 ml (1 mM) 100× PMSF stock solution in 2-propanol (storable at -20°C) immediately prior to use
Membrane Buffer
50 mM Tris, pH 8.0
10 mM EDTA, pH 8.0
333.3 ml (33% v/v) glycerol
Adjust volume to 1,000 ml with diH2O
Add 70.92 μl (1 mM) β-mercaptoethanol and cool to 4°C prior to use
KF Mix
200 mM Tris stock, pH 8.0
4 mM EDTA stock, pH 8.0
871.5 mg (0.3 M) KF
Adjust volume to 50 ml with diH2O
10% CHAPS, 2% CHS stock
5 g (10% w/v final) CHAPS
Dissolve in 40 ml diH2O with sonication
Add 1 g (2% w/v final) CHS Tris salt
Note: CHS Tris salt was found to be more soluble than CHS.
Sonicate ~1 h then rock overnight at RT
Adjust volume to 50 ml with diH2O
Store at -20°C
Extraction Buffer
48 ml membrane buffer
2 ml 10% CHAPS (0.4% final) and 2% CHS stock (0.08% final)
1 mM DTT
Add a stock solution of GTPγS at 4 μM final concentration
Wash Buffer
50 ml extraction buffer
5 mM UDP-Glc
2× Assay Buffer
10 mM (or any appropriate concentration) UDP-[U-14C]Glc (with specific activity of ∼800 cpm/nmol, or adjusted as necessary for experiments)
150 mM Tris, pH 7.5
1.5 mM EDTA
1.5% (w/v) BSA
50 mM KF
0.04 mM GTPγS
10% TCA
100 g TCA
Add diH2O up to 1 L
2% SDS
2 g (2% w/v) SDS
Add diH2O up to 100 ml
Acknowledgments
This work was funded in part by Duke University Medical Center and the National Institute of General Medical Sciences Grant R01 GM115729 (to K.Y.). A.L. was supported by the Tri-Institutional Molecular Mycology and Pathogenesis Training Program from the National Institute of Allergy and Infectious Diseases (T32AI052080). The authors thank Dr. Yoshikazu Ohya at the University of Tokyo for the anti-Rho1p antibody and Dr. Jean-Paul Latge at the Institut Pasteur for the anti-Fks1p antibody.
This protocol is a detailed version of the protocol used in a recent publication by the authors (Chhetri et al., 2020).
Competing interests
The authors declare no conflicts of interest.
References
- Barrett, D., Wang, T. S., Yuan, Y., Zhang, Y., Kahne, D. and Walker, S. (2007). Analysis of glycan polymers produced by peptidoglycan glycosyltransferases. J Biol Chem 282(44): 31964-31971.
- Beauvais, A., Bruneau, J. M., Mol, P. C., Buitrago, M. J., Legrand, R. and Latge, J. P. (2001). Glucan synthase complex of Aspergillus fumigatus. J Bacteriol 183(7): 2273-2279.
- Cabib, E. (2009). Two novel techniques for determination of polysaccharide cross-links show that Crh1p and Crh2p attach chitin to both beta(1-6)- and beta(1-3)glucan in the Saccharomyces cerevisiae cell wall. Eukaryot Cell 8(11): 1626-1636.
- Cabib, E. and Duran, A. (2005). Synthase III-dependent chitin is bound to different acceptors depending on location on the cell wall of budding yeast. J Biol Chem 280(10): 9170-9179.
- Cabib, E., Blanco, N. and Arroyo, J. (2012). Presence of a large beta(1-3)glucan linked to chitin at the Saccharomyces cerevisiae mother-bud neck suggests involvement in localized growth control. Eukaryot Cell 11(4): 388-400.
- Chhetri, A., Loksztejn, A., Nguyen, H., Pianalto, K. M., Kim, M. J., Hong, J., Alspaugh, J. A. and Yokoyama, K. (2020). Length Specificity and Polymerization Mechanism of (1,3)-beta-d-Glucan Synthase in Fungal Cell Wall Biosynthesis. Biochemistry 59(5): 682-693.
- Douglas, C. M. (2001). Fungal beta(1,3)-D-glucan synthesis. Med Mycol 39 (Suppl 1) 55-66.
- Krupa, J. C., Shaya, D., Chi, L., Linhardt, R. J., Cygler, M., Withers, S. G. and Mort, J. S. (2007). Quantitative continuous assay for hyaluronan synthase. Anal Biochem 361(2): 218-225.
- McManus, J. B., Yang, H., Wilson, L., Kubicki, J. D. and Tien, M. (2018). Initiation, Elongation, and Termination of Bacterial Cellulose Synthesis. ACS Omega 3(3): 2690-2698.
- Munro, C. A. (2013). Chitin and glucan, the yin and yang of the fungal cell wall, implications for antifungal drug discovery and therapy. Adv Appl Microbiol 83: 145-172.
- Qadota, H., Python, C. P., Inoue, S. B., Arisawa, M., Anraku, Y., Zheng, Y., Watanabe, T., Levin, D. E. and Ohya, Y. (1996). Identification of yeast Rho1p GTPase as a regulatory subunit of 1,3-beta-glucan synthase. Science 272(5259): 279-281.
- Shematek, E. M., Braatz, J. A. and Cabib, E. (1980). Biosynthesis of the yeast cell wall. I. Preparation and properties of beta-(1 leads to 3)glucan synthetase. J Biol Chem 255(3): 888-894.
- Tlapak-Simmons, V. L., Baron, C. A., Gotschall, R., Haque, D., Canfield, W. M. and Weigel, P. H. (2005). Hyaluronan biosynthesis by class I streptococcal hyaluronan synthases occurs at the reducing end. J Biol Chem 280(13): 13012-13018.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Chhetri, A., Loksztejn, A. and Yokoyama, K. (2021). Quantitative Characterization of the Amount and Length of (1,3)-β-D-glucan for Functional and Mechanistic Analysis of Fungal (1,3)-β-D-glucan Synthase. Bio-protocol 11(8): e3995. DOI: 10.21769/BioProtoc.3995.
Category
Biochemistry > Carbohydrate > Polysaccharide
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.