- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Single Cell Analysis and Sorting of Aspergillus fumigatus by Flow Cytometry
Published: Vol 11, Iss 8, Apr 20, 2021 DOI: 10.21769/BioProtoc.3993 Views: 5313
Reviewed by: Emilia KrypotouPreeti SharmaSimab Kanwal
Original research article
The authors used this protocol in:

May 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Experimental results in fungal biology research are usually obtained as average measurements across whole populations of cells, whilst ignoring what is happening at the single cell level. Microscopy has allowed us to study single-cell behavior, but it has low throughput and cannot be used to select individual cells for downstream experiments. Here we present a method that allows for the analysis and selection of single fungal cells in high throughput by flow cytometry and fluorescence activated cell sorting (FACS), respectively. This protocol can be adapted for every fungal species that produces cells of up to 70 microns in diameter. After initial setting of the flow cytometry gates, which takes a single day, accurate single cell analysis and sorting can be performed. This method yields a throughput of thousands of cells per second. Selected cells can be subjected to downstream experiments to study single-cell behavior.
Keywords: Flow cytometryBackground
Fungal biology research has often been dependent on average measurements of total populations of cells, thereby missing what is happening at the individual cell level. This is specifically the case for filamentous fungi, since hyphae grow by apical extension and branch subapically, and may fuse with neighboring hyphae. This behavior results in an interconnected, entangled mass of hyphae, the colony, which makes it challenging to study single-cell behavior. Therefore, most research on single cells has focused on non-overlapping hyphae that are located in the colony margin using microscopy (Vinck et al., 2005 and 2011; Bleichrodt et al., 2012 and 2015). Since hyphae are attached to the colony, it is challenging to segment them using image analysis. Here we developed a flow cytometry protocol to analyze and select single cells with high throughput. This protocol is suited for e.g. analyzing germination of spores; selection of fluorescent transformants; minimum inhibitory concentration (MIC) testing; obtaining cells for single cell omics approaches; identifying heterogeneous subpopulations of cells; cell aggregation assays, which can be relevant for studying the initial stages of pellet formation in fermenters; and analysis and selection of any fluorescently labeled cellular target, whether stained or genetically encoded, that is smaller than 70 microns (Bleichrodt and Read, 2019).
Materials and Reagents
Falcon 5 ml flow cytometry tubes (STEMCELL Technologies, catalog number: 38057)
Corning cell culture flasks, 25 cm2, vented cap (Merck, catalog number: CLS430639-200EA)
Inoculation loops (VWR, catalog number: SIMPL200-2)
Sterile cotton swabs (VWR, catalog number: HERE1030619)
Easystrainer 70 µm filter, for 50 ml tubes (Greiner Bio-One, catalog number: 542070)
Corning 50 ml centrifuge tubes (Merck, catalog number: CLS430828-100EA)
1.5 ml Eppendorf tubes (VWR International, catalog number: 0030125150)
Microscopy slides and coverslips
96-well plates, optically clear bottom µ-Plate, 96 Well Black (Ibidi, catalog number: 89626)
Pipette filter tips
10 ml pipettes (VWR, catalog number: BURK7550-0010)
4-peak beads (Spherotech, Lake Forest, IL, catalog number: RCP-35-5). Store at 4 °C for up to 1 year
Accudrop beads (Beckton Dickinson Biosciences, catalog number: 345249). Store at 4 °C for up to 1 year
Calcium-/magnesium-free phosphate buffered saline (PBS) tablets (ThermoFisher Scientific, Gibco, catalog number: 18912014)
Ethanol (Fisher Scientific, catalog number: 12468750) diluted to 70% v/v with Milli-Q water. Store at room temperature (RT) for up to 1 year
Sodium hypochlorite (Fisher Scientific, catalog number: 11448842). Store at room temperature for up to 1 year
Potato dextrose agar (Merck Millipore, catalog number: 1.10130.0500)
Fluorescent Brightener 28 disodium salt solution (Calcofluor white solution), 25% (Merck, catalog number: 910090-20ML, CAS Number: 4193-55-9). Store at 4 °C for up to 1 year
Sheath fluid (see Recipes)
50% glucose solution (see Recipes)
Minimal medium (MM) (see Recipes)
Phosphate-buffered saline (see Recipes)
Saline Tween solution (ST) (see Recipes)
Calcofluor white working solution (see Recipes)
Vishniac solution (see Recipes)
Equipment
Hemocytometer (VWR, catalog number: BRND718905)
Set of micropipettes
BD Influx cell sorter (Becton Dickinson) with 100-µm nozzle tip
Sonicating water bath (Grant Instruments Ltd, model XUBA3)
Autoclave (Prestige Electrolysis + Spa Supply, model: MED 2100, catalog number: SAP2100)
Temperature-controlled incubators (Binder, Series BD Classic Line, model: BD 400)
Light microscope (ZEISS, model: Axiolab 5)
Fluorescence microscope (ZEISS, Axio Observer Research Inverted Microscope)
Pipet controller (VWR, model: Pipet boy, catalog number: 612-0928)
Tabletop centrifuge (Fisher Scientific, model: EppendorfTM Benchtop Microcentrifuge, catalog number: 10216522)
Software
Sortware version 1.2.0.142 (Becton Dickinson)
FlowJo version 10.7 (Becton Dickinson) (https://www.flowjo.com/)
Rstudio version 1.3 (optional) (https://rstudio.com/products/rstudio/)
Procedure
Note: All steps must be performed in a Class II Biosafety cabinet.
Growing cells for flow cytometry analysis
Pipet 10 ml of autoclaved 4% potato dextrose agar in Milli-Q water (PDA) into 25-cm2 vented cell culture flasks and allow to solidify.
To obtain dormant spores (Figure 1A), take spores from frozen glycerol stock with a sterile loop and evenly spread spores on the agar medium.
Incubate for 3 days at the optimal growth temperature (37 °C for A. fumigatus), depending on the species.
Harvest spores by pipetting 10 ml ST on the culture, close the cap and mix vigorously by shaking the culture from side to side.
If the spores do not separate easily from the colony, scrape them off with a sterile loop or cotton swab while the agar medium is covered with ST.
Place a 70-µm filter on a 50-ml tube and pour the total culture through the filter.
Count the spores in the filtrate (stock) using a hemocytometer after making a 1/100 dilution in ST, and dilute from stock to 5 × 106 spores/ml using ST (total 1 ml in a 1.5-ml Eppendorf tube). Keep on ice until fluorescent labeling is performed. Never keep spore solutions in the refrigerator for longer than 1 day, as the spores tend to aggregate, which hampers single-cell analysis.
To obtain isotropically grown spores (Figure 1B), inoculate 20 ml of MM with 5 × 106 spores/ml from fresh spore solution in a 25-cm2 vented cell culture flask.
Suspend the spores by pipetting the solution up and down.
Incubate for 6 h at 37 °C as a standing culture, in the case of A. fumigatus. Using a standing cultures limits cell aggregation.
To obtain germlings (Figure 1C), inoculate 20 ml of MM with 5 × 106 spores/ml from fresh spore solution in a 25-cm2 culture flask.
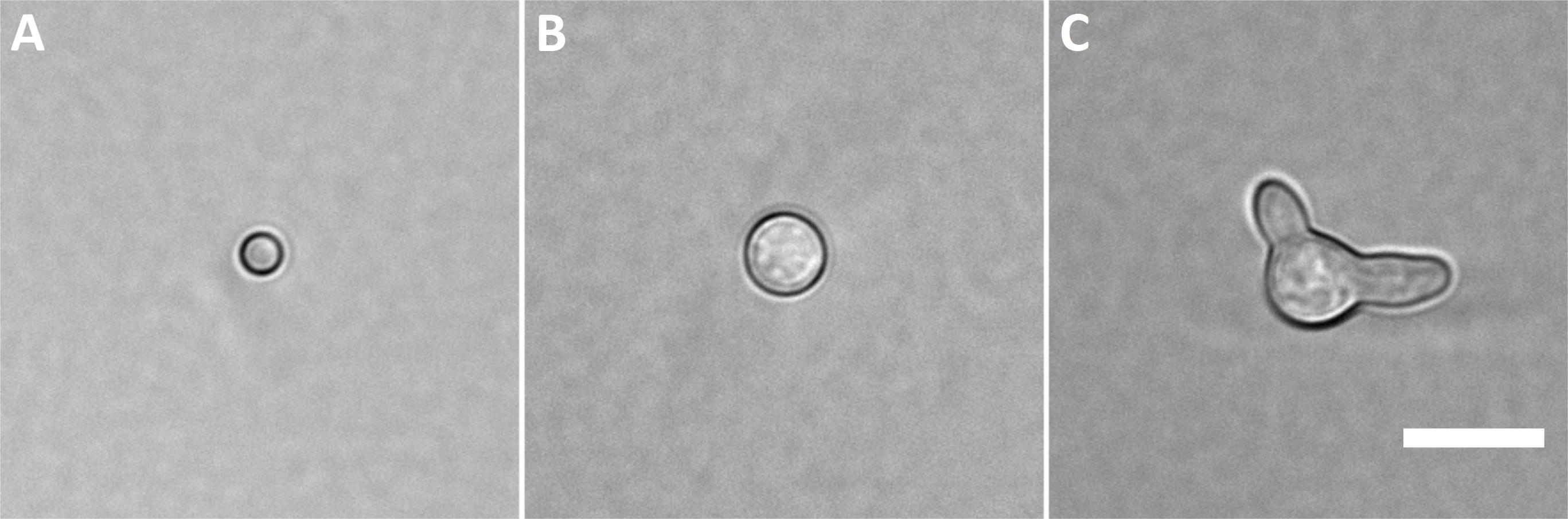
Figure 1. Cell types that can be analyzed using flow cytometry and selected by FACS. A. Dormant spore; B. Isotropically grown spore; C. Germling. Scale bar represents 10 µm.Suspend the spores by pipetting the solution up and down.
Incubate for 16 h at 30 °C as a standing culture, in the case of A. fumigatus.
The next day, harvest isotropically grown spores and germlings from the standing cultures.
Vigorously mix the cultures by shaking them from side to side.
Place a 70-µm filter on a 50-ml tube and pour the total culture through the filter. Keep on ice until fluorescent labeling.
Fluorescent labeling of fungal cells
Prepare dye solutions in ST or PBS at working concentrations. For example, the fungal cell wall can be labeled with Calcofluor white. The specific working concentration (25 nM–1 µM) must be determined experimentally and can be checked using a fluorescence microscope. Alternatively, organelles such as mitochondria can be fluorescently labeled to assess their activity. Another option is to use genetically encoded fluorophores (Bleichrodt and Read, 2019). As an example, we here describe fluorescent labeling of the cell wall using Calcofluor white, which binds to chitin.
Pipet 1 ml, from the harvested culture produced as described above (Step A7 or A14) into a 1.5-ml Eppendorf tube.
Spin the sample down for 10 min at 6,000 × g at RT to pellet the cells.
Remove the supernatant with a pipet tip.
Add 1 ml Calcofluor white working solution (250 nM) to the pellet and resuspend by vortexing.
Incubate for 10 min at RT and spin down to pellet cells for 10 min at 6,000 × g at RT.
Remove the supernatant with a pipet tip.
Wash the cells by adding 1 ml ST and vortexing.
Spin the sample down for 10 min at 6,000 × g at RT to pellet the cells.
Remove the supernatant with a pipet tip.
Add 1 ml ST and vortex.
Keep the cells on ice until flow cytometry analysis.
Initializing the flow cytometer
Check that the waste tank is empty and sufficient sheath fluid is available.
Prior to starting the cell sorter, exchange the ethanol shutdown tank for a recently filled sheath tank containing calcium-free and magnesium-free PBS (sheath fluid).
Empty the waste container and add 50 ml of undiluted sodium hypochlorite solution to the tank.
Switch on the PC and cell sorter, along with all power supplies for the lasers.
Start the flow cytometry software.
Sonicate the 100-µm ceramic nozzle tip in a sonicating water bath for 5 min while the existing ethanol solution in the system is flushed and replaced with sheath fluid.
Following sonication, insert the nozzle into the flow cell assembly and run the stream.
Allow the stream to run and settle for a minimum of 45 min prior to alignment and calibration (Procedure D and E).
Alignment and calibration
Use 4-peak rainbow calibration particle (Spherotech) fluorescent beads to align the lasers.
Prepare 4-peak rainbow calibration particles by adding 1 drop of beads (~60 µl) to 2 ml of Milli-Q water. The remaining beads can be stored at 4 °C and are stable for up to 1 month.
On the BD Influx, manually align the laser to the stream along 3 axes using the micrometers. Correct alignment is achieved when 4 populations are clearly resolved and the spread, measured with the coefficient of variance (CV) of the brightest peak, is <6% as indicated by the software.
Record the laser power by drawing a gate for each of the 4 fluorescent populations of the 4-peak rainbow calibration particles.
Stop the run.
Save the gates in the flow cytometry software.
Before starting a new experiment, calibrate the laser power by running the 4-peak rainbow calibration particles and align their events in the 4 respective loaded gates. This can be done by adjusting the voltages of the lasers individually in the software. This procedure ensures that fluorescent labeling between individual experiments can be compared absolutely.
Following alignment, set the drop delay using Accudrop beads (Beckton Dickinson).
Add one drop of Accudrop beads (~60 µl) to 1 ml of Milli-Q water.
Run the beads at a flow rate that enables 2,000–3,000 events per second to be visualized on the software.
Set a sort gate within the software, which ensures that all beads will be apportioned to the sort stream.
Adjust the drop delay to ensure >95% of Accudrop beads are present in the sort stream. The drop delay is the time period (in microseconds) required for a particle of interest (e.g., a cell or a spore) to travel from the point of identification by the software to the end of the intact stream. To ensure this timing is set correctly, we utilize 6-μm diameter Accudrop fluorescent beads and adjust the drop delay time until >95% of the beads are present in the side (sort) stream and no longer in the waste stream.
Note: The stability of the stream and the relative position of the breakoff point are monitored visually throughout the sorting process.
Set up index sorting
Index sorting is a single-cell or particle technique where the relative light scatter and fluorescence of each individually sorted particle can be traced back to the original data. Index sorting is performed within the Sortware software by selecting the single-cell mode of sorting and checking the appropriate index sorting box.
Prior to sorting, align the sort stream to the capture plate by adjusting the deflection plate voltages and ensure the test sort droplets are central to each well. This procedure will ensure that the single droplets, which contain individual spores, are directed to the correct well along the entire 96-well plate. Each sorted particle is assigned its own unique X and Y coordinates on the plate, and this information is recorded within the data set for recall later during analysis.
Sorting single cells to determine gating strategy (only perform this step once for each new fungus and cell type)
Pipet 1 ml of the sample into a 5-ml flow cytometry tube and load the sample in the flow cytometer.
Draw a new flow cytometry data plot.
Run the sample and adjust the axis of the data plot to SSC (side scatter) and FSC (forward scatter) (Figure 2A).
Dormant spores, isotropically grown spores, and germlings are usually located in the mother gates shown in Figures 2A, 2C and 2E, respectively. Draw the gates accordingly.
Select the data from the mother gate (Figures 2A, 2C or 2E) and plot this in a new data plot using trigger pulse width (TPW) and FSC on the axes. On flow cytometers that have no TPW setting, plot FSC-Width against FSC-Area. For circular cells, such as dormant and isotropically grown spores, the few cells located outside the main diagonal population of cells are aggregates.
To select single cells, draw a daughter gate on the main diagonal population of cells (Figures 2B and 2D).
For germlings (Figure 2F), draw a daughter gate just above the main diagonal population of cells, but taking only the left side of the population from the FSC signal, and only up to the TPW containing the majority of cells (green area).
To test these gates for your fungal species, sort cells on a slide of each daughter gate, apply a cover slip, and observe the cells under a light microscope to determine whether you can predominantly see single cells of the desired cell type.
If this is not the case, some optimization is required.
Place a 96-well plate with an optically clear bottom suited for microscopy in the sorting chamber of the flow cytometer.
Now sort 1 cell per well from the daughter gate using index sorting.
Find each cell in the well using microscopy and score which wells exclusively contain single cells of the desired type.
In the flow cytometry software, select all the wells that contained a single cell and plot this data.
Now plot the events in the initial gate in a new window and draw a daughter gate on this data, so that all of the events fall in the new gate.
Steps F10-F14 should be repeated for each cell type individually (e.g., dormant spores, isotropically grown spores and germlings; Figure 1).
The specific gates can be stored in the flow cytometry software and loaded before each new session.
Calibrating the laser power before each run (Procedure D) is essential to ensure accurate gating.
For measuring and/or sorting additional samples
Back flush the previous sample in the sample tube and load a new sample.
Use the experimentally determined gates from step F to analyze or sort single cells.
For analysis, plot all events of the daughter gates (see daughter gates Figures 2B, 2D and 2F) in a new plot and select appropriate axes (e.g., SSC versus FSC for unlabeled cells or fluorescence intensity (excitation/emission) corresponding to the fluorescent label of the cells).
Select the appropriate stopping (daughter gates) and storing gates (all events) in the software and acquire data. This procedure ensures that the required number of single cells of a specific cell type is measured, and also stores all events from the total population of cells.
For sorting, place 5-ml sorting tubes or a multiwell plate in the specific sample holder. It is best to pre-fill the tubes/wells with some ST or MM, depending on the planned downstream experiment.
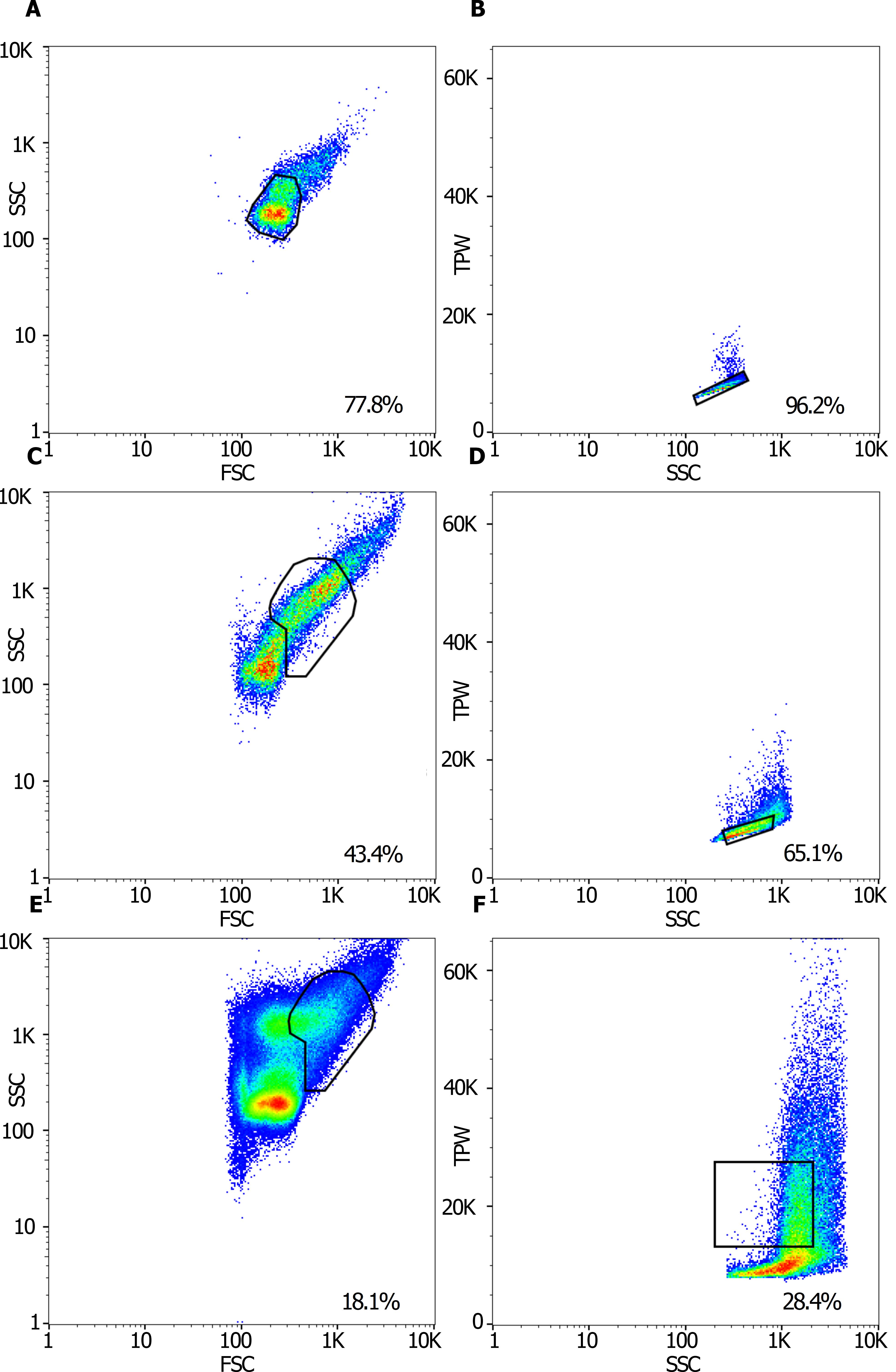
Figure 2. Gating strategy for analyzing and sorting 3 cell types: dormant spores, isotropically grown spores and germlings. Total sample of (A) dormant spores, (C) spores incubated for 6 h at 37 °C, and (E) spores incubated for 16 h at 30 °C showing gates (black shapes) as determined by sorting cells on slides. The rainbow color gradient indicates the event rate, with blue indicating the lowest rate and red indicating the highest rate. The axes show the side scatter (SSC) and forward scatter (FSC) of the signals. The daughter gates (B, D, F) of the gates shown in (A, C, E), as determined by index sorting, that contain predominantly single cells of (B) dormant spores, (D) isotropically grown spores and (F) germlings are shown. The axes show the trigger pulse width (TPW) and side scatter (SSC) of the signals. The percentages of cells from the total population (A, C, E) or the mother population (B, D, F) that fall in the specific gates are indicated in the lower right corner of each panel.
Cleaning procedure
Following each analysis or sorting procedure, it is important to establish and run an effective decontamination protocol. We tested various protocols for spore survival, and the most effective protocol was implemented in these sorting experiments.
Backflush the sample line with system sheath fluid to ensure that a minimal number of spores remain in the sample lines.
Using a 5-ml tube, run 4 ml of 70% ethanol through the sample lines for 5 min.
Stop the run and allow the lines to bathe in ethanol for 15 min.
Remove the 5-ml tube of ethanol and exchange it for a tube of 10% v/v sodium hypochlorite and repeat the procedure, i.e., run the solution for 5 min, followed by bathing for 15 min.
Finally, using a 5-ml tube, run 3 ml of Milli-Q water through the system for 20 min to flush out the bleach.
Collect 1 ml of stream fluid, add it to liquid growth medium, and incubate for 3 days. No spore outgrowth should be visible.
Shut down
Shut down the cell sorter following the decontamination procedure.
Stop the stream and remove the tube of water.
Exchange the sheath tank for a tank of 70% ethanol and start the stream.
Run ethanol through all parts of the system for 15 min.
Switch off the stream and leave the cell sorter in ethanol.
Backflush the nozzle with Milli-Q water and leave the nozzle submerged.
Switch off all the lasers and shut down the system.
Remove the waste tank. Do not empty the waste tank until the next day to ensure that no organisms can grow in the waste.
Data analysis
Exported flow cytometry data files can be opened and visualized in FlowJo and exported as csv files. For optional statistical analysis of cell subpopulations and heterogeneity thereof we refer the reader to Bleichrodt et al. (2020) (see Materials and Methods section and Text S1). The code for statistical analysis in R can be obtained from https://github.com/rbleichrodt/Conidial-heterogeneity and run in Rstudio.
Notes
In principle, this protocol can be used with any flow cytometer that allows index sorting.
For A. fumigatus, we generally obtain sorting efficiencies of ~98%, 91% and 80% for dormant and isotropically grown spores and germlings (Figure 1), respectively.
Decontamination procedures should be tested for each individual species. The decontamination procedure in this protocol serves only as a guideline for A. fumigatus.
Recipes
Note: All recipes should be made up using Milli-Q water unless stated otherwise.
Sheath fluid
Calcium-free/magnesium-free PBS tablets dissolved in Milli-Q water and autoclaved
Store at RT for up to 2 weeks
50% glucose solution
500 g/L d-glucose. Sterilize by filtration
Store at RT for up to 1 year
MM, pH 6.0
6 g/L NaNO3, 1.5 g/L KH2PO4, 0.5 g/L KCl, 0.5 g/L MgSO4·7H2O and 0.2 ml/L Vishniac solution (Pontecorvo et al., 1953; Vishniac and Santer, 1957)
Sterilize by autoclaving
Allow to cool and add 20 ml/L 50% glucose solution, to obtain a final concentration of 1% glucose
Store at RT for up to 1 year
Vishniac solution, pH 4.0
10 g/L ethylenediaminetetraacetic acid (EDTA)
4.4 g/L ZnSO4·7H2O
1.0 g/L MnCl2·4H2O
0.32 g/L CoCl2·6H2O
0.32 g/L CuSO4·5H2O
0.22 g/L (NH4)6Mo7O24·4H2O
1.47 g/L CaCl2·2H2O
1.0 g/L FeSO4·7H2O
Sterilize by filtration and store at 4 °C for up to 1 year
Saline Tween solution (ST)
0.9% (w/v) NaCl
0.005% (v/v) Tween 80
Sterilize by autoclaving and store at RT for up to 1 year
PBS, pH 7.4
NaCl 8 g/L
KCl 200 mg/L
Na2HPO4 1.44 g/L
KH2PO4 245 mg/L
Sterilize by autoclaving and store at RT for up to 1 year
Calcofluor white (Fluorescent Brightener 28) working solution
Dilute from 25% (w/v) stock to a final concentration of 250 nM in ST
Sterilize by filtration and keep at 4 °C for up to 1 month
Acknowledgments
This project received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement FUNFIT No. 659003. This work was derived from Bleichrodt et al. (2020). Some figures were combined and panel text was modified, and the main body of the text was modified. Creative Commons Attribution 4.0 International license https://creativecommons.org/licenses/by/4.0/.
Competing interests
The authors declare no competing interests.
References
- Bleichrodt, R. J., van Veluw, G. J., Recter, B., Maruyama, J., Kitamoto, K. and Wösten, H. A. B. (2012). Hyphal heterogeneity in Aspergillus oryzae is the result of dynamic closure of septa by Woronin bodies.Mol Microbiol 86(6): 1334-1344.
- Bleichrodt, R. J., Vinck, A., Read, N. D. and Wösten, H. A. B. (2015). Selective transport between heterogeneous hyphal compartments via the plasma membrane lining septal walls of Aspergillus niger. Fungal Genet Biol 82: 193-200.
- Bleichrodt, R. J. and Read, N. D. (2019). Flow cytometry and FACS applied to filamentous fungi. Fungal Biol Rev 33(1): 1-15.
- Bleichrodt, R. J., Foster, P., Howell, G., Latgé, J. P. and Read, N. D. (2020). Cell wall composition heterogeneity between single cells in Aspergillus fumigatus leads to heterogeneous behavior during antifungal treatment and phagocytosis.mBio 11(3): e03015-19.
- Pontecorvo, G., Roper, J. A., Hemmons, L. M., Macdonald, K. D. and Bufton, A. W. (1953). The genetics of Aspergillus nidulans. Adv Genet 5: 141-238.
- Vinck, A., Terlou, M., Pestman, W. R., Martens, E. P., Ram, A. F., van den Hondel, C. A. M. J. J. and Wösten, H. A. B. (2005). Hyphal differentiation in the exploring mycelium of Aspergillus niger. Mol Microbiol 58(3): 693-699.
- Vinck, A., de Bekker, C., Ossin, A., Ohm, R. A., de Vries, R. P. and Wösten, H. A. B. (2011). Heterogenic expression of genes encoding secreted proteins at the periphery of Aspergillus niger colonies. Environ Microbiol 13(1): 216-225.
- Vishniac, W. and Santer, M. (1957). The thiobacilli. Bacteriol Rev 21(3): 195-213.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Howell, G. J. and Bleichrodt, R. (2021). Single Cell Analysis and Sorting of Aspergillus fumigatus by Flow Cytometry. Bio-protocol 11(8): e3993. DOI: 10.21769/BioProtoc.3993.
Category
Microbiology > Microbial cell biology > Cell-based analysis
Cell Biology > Single cell analysis > Flow cytometry
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.