- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Workflow for High-pressure Freezing and Freeze Substitution of the Caenorhabditis elegans Embryo for Ultrastructural Analysis by Conventional and Volume Electron Microscopy
(*contributed equally to this work) Published: Vol 11, Iss 7, Apr 5, 2021 DOI: 10.21769/BioProtoc.3981 Views: 6584
Reviewed by: Gunar FabigAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2020
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The free-living nematode Caenorhabditis elegans is a popular model system for studying developmental biology. Here we describe a detailed protocol to high-pressure freeze the C. elegans embryo (either ex vivo after dissection, or within the intact worm) followed by quick freeze substitution. Processed samples are suitable for ultrastructural analysis by conventional electron microscopy (EM) or newer volume EM (vEM) approaches such as Focused Ion Beam Scanning Electron Microscopy (FIB-SEM). The ultrastructure of cellular features such as the nuclear envelope, chromosomes, endoplasmic reticulum and mitochondria are well preserved after these experimental procedures and yield accurate 3D models for visualization and analysis (Chang et al., 2020). This protocol was used in the 3D reconstruction of membranes and chromosomes after pronuclear meeting in the C. elegans zygote (Rahman et al., 2020).
Keywords: High-pressure freezingBackground
C. elegans is a free-living nematode with many properties that make it amenable to scientific study: (1) the worms are ~1 mm long; (2) they are easy to grow, handle and maintain; (3) they proliferate rapidly and (4) they are amenable to genetic manipulations. The reader is encouraged to consult Corsi et al. (2015) for an excellent primer to C. elegans and its use as a model organism in biology. Embryonic cell division events in C. elegans are largely invariant both spatially and temporally, making the organism a robust eukaryotic model system (Oegema and Hyman, 2006). Transient changes in dynamic cellular components like the nuclear envelope and chromosomes during embryonic development can be well described at resolutions afforded by fluorescence microscopy (Cohen-Fix and Askjaer, 2017), but capturing the corresponding ultrastructural changes at higher resolutions, whether in two or three dimensions, is challenging (Altun et al., 2002). The chitinaceous shell of the embryos poses a diffusion barrier to chemicals, precluding conventional aldehyde-based fixation protocols; thus, rapid freezing of samples followed by freeze-substitution and imaging by EM is the preferred way to capture ultrastructural intermediates at nanometer resolutions. As these samples are too large for simple plunge freezing, they have to be high-pressure frozen, ideally in a manner conducive to screening for the correct developmental stage (Muller-Reichert et al., 2003; McDonald et al., 2010). Recently, we published a report describing architectural intermediates in nuclear envelope breakdown during embryonic development (Rahman et al., 2020). We visually followed C. elegans embryos trapped in capillaries until just before high-pressure freezing to ensure that the correct stages were frozen. In an accompanying methods paper, we also reported cryo-fluorescence microscopy of high-pressure frozen whole C. elegans worms followed by correlative FIB-SEM (Chang et al., 2020) to image such structures in the intact worm. In both these advances, central experimental steps included the appropriate high-pressure freezing, freeze-substitution and resin embedding of trapped worms and/or embryos. Here we provide a step-by-step protocol to correctly execute these procedures for downstream vEM analysis, either to replicate our findings, or to answer other questions of interest in C. elegans.
Materials and Reagents
C. elegans maintenance
Tissue culture dish, 35 × 10 mm (Corning, Falcon, catalog number: 353001 )
Worm pick (Genesee Scientific, catalog number: 59-30P16 )
Micropipette tips with barrier (any brand, 1 µl, 20 µl and 1,000 µl)
Serological pipets, 10 ml for OP50 seeding (Corning, Falcon, catalog number: 357551 )
C. elegans Bristol N2 [Caenorhabditis Genetics Center (CGC), https://cgc.umn.edu (Stiernagle, 2006)]
E. coli OP50 strain DA735 [Caenorhabditis Genetics Center (CGC), https://cgc.umn.edu]
Water, molecular biology grade (GE Healthcare, HyClone, catalog number: SH30538.02 )
Agar (RPI, catalog number: A20020-5000 )
Yeast extract (ThermoFisher Scientific, catalog number: BP9727-2 )
Bacto Peptone (BD, Bacto, catalog number: 211677 )
Sodium chloride (Avantor Performance Materials, J.T. Baker, catalog number: 3624-05)
Cholesterol (Sigma, catalog number: 1580-01 )
Ethanol 200 proof (Decon Labs, catalog number: 2716 )
Trizma-Cl (Roche, catalog number: 10812846001 )
Trizma-OH (Roche, catalog number: 10708976001 )
Luria-Bertani broth, bacterial culture medium (KD medical, catalog number: BLC-5020 )
M9 buffer (IPM Scientific USA, catalog number: 11006-517 )
Levamisole hydrochloride (Sigma, catalog number: 196142 )
High-pressure freezing and freeze substitution
Glass microscopy slides (ThermoFisher Scientific, catalog number: 10144633B )
Tubes, 1.5 ml for BSA solution aliquots (ThermoFisher Scientific, catalog number: 05-408-129 )
Micropipette tips with barrier (any brand, 1 µl, 20 µl and 1,000 µl)
Micropipette tips without barrier (Eppendorf, 1 µl, catalog number: 30072.014 )
Cellulose capillary tube (Leica, catalog number: 16706869 )
Needles, 21-gauge (G) x 1½ inch (Covidien Monoject, catalog number: 305167 )
Syringe, disposable (VWR, EMS, catalog number: 72508 for 2.5 ml or 72509 for 5 ml)
Alcohol swabs (BD medical, catalog number: 326895 )
Type A gold-coated copper planchette (Leica, catalog number: 16770152 )
Type B gold-coated copper planchette (Leica, catalog number: 16770153 )
Cryomarker, Black (ThermoFisher Scientific, Nalgene, catalog number: 22-026-700 )
Sample holder Cartridge system, D3 mm half cylinder (Leica, catalog number: 771849 )
Sample holder Cartridge system, D3 mm middle plate (Leica, catalog number: 771813 )
Nalgene cryovials (ThermoFisher Scientific, catalog number: 5000-1012 )
Polypropylene tubes, 50 ml (BD Falcon, catalog number: 352098 )
String or twine (VWR, Twine, catalog number: 30-33113 )
Disposable cellulose acetate filter, 0.22 µm mesh size, diameter 25 mm (Millipore, Cameo syringe filter, catalog number: 1213657 )
Erlenmeyer glass flasks, 250 ml (VWR, Pyrex, catalog number: 4444-250 )
Pasteur pipettes, Borosilicate glass, 5 ml (ThermoFisher Scientific, catalog number: 13-678-20A )
Pasteur pipettes, plastic, 2 ml and 7 ml (Globe Scientific, catalog numbers: 137040 and 134090 )
Serological pipette, Borosilicate glass, 1 ml (VWR, catalog number: 93000-682 )
Plastic cups (for weighing resin) (VWR, Therapak, catalog number: 74850 )
Beem capsules (Ted Pella, catalog number: 69910-01 )
Beem capsule holder (Ted Pella, catalog number: 132-B )
Double Edge Carbon steel blade (Ted Pella, Feather, catalog number: 121-9 )
Kimwipes 05517 (ThermoFisher Scientific, Kimberly-Clark Kimtech Science Precision wipes, catalog number: 06-677-72 )
Styrofoam container or tray (e.g., a shipping container lid, typically 15 × 20 cm and 2 cm deep)
Disposable polypropylene spatula for osmium handling (VWR, catalog number: 80081-194 )
Bovine Serum Albumin (BSA), heat shock fraction (Sigma, catalog number: A3294 )
Nail polish (any color)
Osmium tetroxide (OsO4) granules (EMS, catalog number: 19134 )
Acetone (ThermoFisher Scientific, catalog number: 9011 )
Uranyl acetate (EMS, catalog number: 22400 )
Methanol (Mallinckrodt, catalog number: 3016 )
Poly/Bed 812 embedding kit with DMP-30 (Polysciences, catalog numbers: 0 8792 and 0 8791 )
Dry ice (in-house supply)
Double deionized water (in-house supply)
Dry liquid nitrogen (LN2) (in-house supply)
Dodecenyl succinic anhydride (DDSA) (Polysciences, catalog number: 0 8792 , kit same as 35)
Nadic Methyl anhydride (NMA) (Polysciences, catalog number: 0 8792 , kit same as 35)
Modified Youngren’s, Only Bacto-peptone (MYOB) plates for worm maintenance (see Recipes)
Cholesterol stock solution (see Recipes)
E. coli OP50 stock (see Recipes)
Cellulose capillary attachment (see Recipes)
20% BSA solution (see Recipes)
25 mM Levamisole solution (see Recipes)
Quick Freeze-substitution (QFS) cocktail (see Recipes)
Poly/Bed 812 resin mix (see Recipes)
Equipment
Micropipettes (any brand, for 1 µl, 20 µl and 1,000 µl volumes)
Sharp-point tweezers (EMS, Dumont, catalog numbers: 78320-51T , 72919-0A , and 78340-51S ) (Figure 1a-1c)
Tweezers insulated with PVC (EMS, Dumont, catalog number: 3C-linox-E ) (Figure 1d)
Forceps insulated with PVC, long (Leica, VOMM, catalog number: 22SAESD ) (Figure 1e)
Crimping tool, scalpel #20 (Bard-Parker, catalog number: 371620 ) (Figure 1f)

Figure 1. Tools for sample handling. a-c. Sharp-point tweezers for sample handling (Section A Steps 4-5). d-e. Insulated tweezers for frozen sample recovery and transfer (Section B Steps 4-9, and Section C Steps 6-7). f. Crimping tool: #20 scalpel manually blunted by rubbing the blade gently on a metal surface (McDonald et al., 2010).Table-top centrifuge (Eppendorf, model: 5424 or similar)
Bottom illuminated stereomicroscope with frosted glass stage (Leica, model: SM2745 or similar)
Leica high-pressure freezer, ultra-low temperature equipped with stereomicroscope and funnel to fill LN2 (Leica, model: ICE )
Frozen sample recovery cryobox (stainless steel tray, deep) with frozen sample release station and 3 mm punch, rod, and plug (Leica, Austria; EM ICE high-pressure freezer package)
Mini benchtop orbital shaker (VWR, catalog number: 97109-890 )
Large surface slide warmer (Premiere, catalog number: XH-2002 )
Infrared thermometer (General Tools & Instruments, catalog number: IRT207 )
Analytical balance (Sartorious Corp, model: BCE64-15 )
Stirring plate (Corning, catalog number: PC420D or similar)
Standard desiccator connected to a mechanical pump (Ted Pella, model: VRD4 )
Metal blocks with 12 mm holes (ThermoFisher Scientific, catalog number: 88880152 )
Dry ice buckets, multiple (VWR, Scienceware Magic Touch 2 with lid, catalog number: M16807-2001 )
Bunsen burner
Chemical safety hood
Laboratory timer
Chemical scale (Mettler, model: AE240 )
Temperature-controlled oven (Quincy Lab, model: 20GC )
Standard autoclave
Hairdryer (any brand with hot air fan)
Sample storage, large LN2 dewar (Taylor-Wharton, catalog number: HC34 )
Portable LN2 dewar on a rolling base, 25 L (Worthington, catalog number: LD25 )
Portable LN2 dewar, 4 L (Worthington, catalog number: LD4 )
PPE (lab coat, face shield and cold-resistant gloves) for handling LN2 (any brand)
Facemask, surgical (Halyard health, catalog number: 28806 ; or any brand)
Procedure
High-pressure freezing (HPF) of C. elegans individual embryos
Start a worm culture by placing 20-30 starved L1 larva on a new MYOB agar plate seeded with E. coli OP50 bacteria (see Recipes). Maintain worms by transferring 2-3 adults to a new MYOB plate every fourth morning (Stiernagle, 2006).
72 h prior to the HPF experiment, transfer 5-6 gravid adults to each of several new MYOB plates (at least 3 separate plates to ensure enough young adults).
In the morning of the HPF experiment, take out two aliquots (500 µl each) of 20% w/v BSA solution from 4 °C and spin at 94 x g (~1,000 rpm) for 5 min in a table-top centrifuge to remove bubbles. Keep at room temperature.
Place 3-4 pairs of clean sharp-point tweezers (Figure 1a and 1b) next to the dissecting scope, and 1-2 pairs (Figure 1c) next to Leica ICE high-pressure freezer. Tweezers 1a and 1b are suitable for cellulose tube transfer while tweezer 1c is suitable for planchette transfer. It is critical to have alcohol swabs next to the tweezers (Figure 2a): unless you clean them frequently, the tweezers become sticky with 20% BSA solution, resulting in sample loss during transfer.
Place one micropipette (1 µl) set at 0.8 µl and another micropipette (20 µl) set at 12 µl next to alcohol swab, tweezers, crimping tool (white arrow), and worm pick (yellow arrow) as in Figure 2a. Set up worm dissecting tool (syringe with 21G needles, Figure 2b). Place multiple cellulose tubes attached to pipette tips (Figure 2c, purple arrows and inset). Keep one micropipette (1 µl) dedicated for capillary tube handling (Figure 2c).
Take out 9-10 planchettes (type A and type B separately) in 35 mm Petri dishes (Figure 2c), and mark type A cavities with a black marker to distinguish it from type B later.
Note: Type A planchettes have a single 300 µm deep cavity on one face while the other face is flat; the samples will be placed later in the shallow cavity of a type B planchette (Step A14).
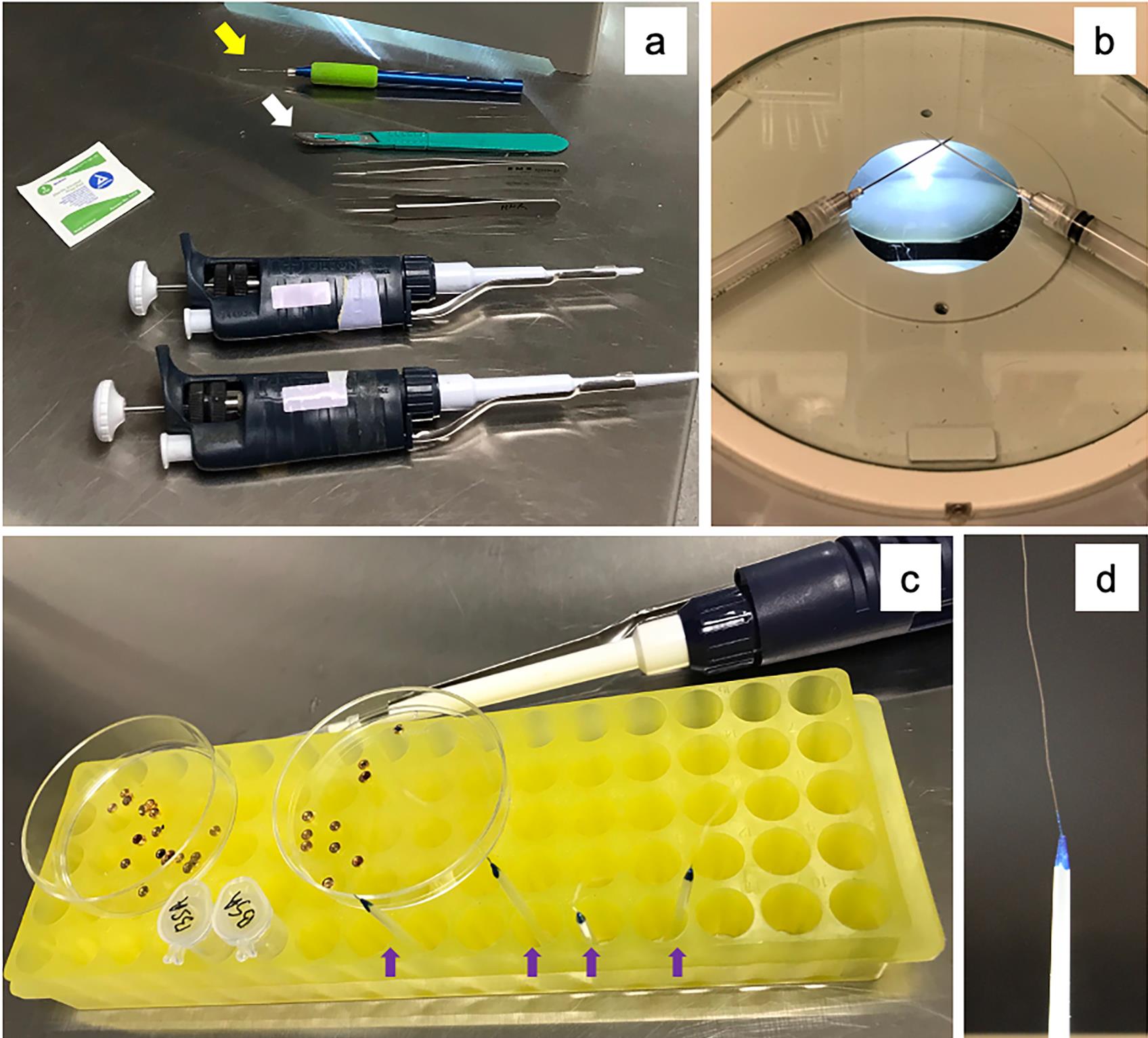
Figure 2. Setup for worm dissection prior to HPF. a. Micropipettes (1 µl and 20 µl), sharp-point tweezers, alcohol swab, crimping tool (white arrow) and worm pick (yellow arrow). b. Worm dissecting tool – a pair of 21G needles on 2.5 ml syringe to be used as scissors to cut worms. c. A dedicated micropipette (1 µl) for handling cellulose capillaries attached to pipette tips (purple arrows and d). Planchettes type A and type B are placed in separate plastic Petri dishes. d. A cellulose capillary attachment (see Recipe 4) for embryo and/or worm collection.Check liquid nitrogen (LN2) transport dewars to make sure they are empty and dry. Fill one large dewar (25 L) and two medium dewars (4 L) with LN2.
Turn on the Leica ICE high-pressure freezer (Figure 3a) and wait to hear the compressor turn on (identified by a hum and mild vibration), which should also be reflected in the monitor status screen. The loading station (Figure 3b) should be clean and dry. Fill the freezer chamber (Figure 3c) with LN2 from the 25 L dewar slowly with multiple brief pauses to avoid triggering the alarm and a false indication that the tank is full. A significant amount of LN2 evaporates in the process of chilling the tank.
Note: You may need help from a second person to pour ~18 liters of LN2 safely.
Assemble HPF sample storage dewar (Figures 3d-3f). Note the trisection pod in the dewar can hold up to three cartridge systems in each of three cups, equating to a maximum of nine HPF “shots” before the sample storage dewar is full. The high-pressure freezer requires ~20 min to equilibrate to LN2 temperature; once equilibrated, fill the HPF sample storage dewar (Figure 3f) with LN2 and insert into Leica ICE freezer chamber (Figure 3g, underneath the loading station in Figure 3a). You must wait until the freezer is ready, otherwise the container will collect and freeze condensation from the air forming ice crystals. Crystalline ice or frost must be avoided in this procedure, as with other cryogenic experiments. So, maintain dry conditions, and work at a quick pace to minimize exposure of the sample and tools to humidity.
Assemble plastic adapters on the two halves of the Leica ICE high-pressure freezer loading station (Figures 3h-3j, and arrows in Figure 3b). Place the planchette holder with a hole in it (Figure 3i) on top of a plastic adapter placed on the bottom steel surface (Figure 3b, yellow arrow). Run a blank HPF cycle by manually closing the red lid (Figure 3b) to ensure the freezer is working properly (Figure 5).

Figure 3. Setting up the Leica EM ICE for HPF. a. Leica EM ICE high-pressure freezer. b. Loading station, enlarged from boxed area in 3a. Arrows show where planchette holders are placed for each HPF run, with a half cylinder on top (white arrow and 3h), and planchette holder with a hole in it (3i) atop the other half cylinder placed on the bottom black steel half (yellow arrow and 3j). c. LN2 filling port behind grey door (long black arrow). d-f. Assembly of HPF sample storage dewar comprising the trisection pod (3d) and segmenting insert (grey cylinders in 3e). Insets in 3e show top view after assembly; blue arrow, “bayonet” or release button and yellow arrowhead, lock/open position. The bayonet is pressed and rotated counter-clockwise or clockwise to open or lock respectively (left inset – bayonet at open position, and right inset – bayonet at close position). g. Collection holder unit inserted into receptacle before automatic withdrawal into the freezer unit, seen as a black outlined panel below the loading station in 3a. The notch with the “in >” label (3e inset, yellow ring) should face the HPF instrument holding unit. h-j. Leica cartridge system contains two identical half cylinders and a planchette holder with a hole in it.Collect 3-4 young adult worms with a worm pick and place in a drop (12 µl) of 20% BSA solution on a clean glass slide. Use two needles as shears (Figure 4a) to cut the worms open (at the middle of the body) to release the embryos (McDonald, 1999; Muller-Reichert et al., 2007; McDonald et al., 2010).
Note: If whole worms are collected, use 20% BSA solution with 25 mM levamisole and incubate 2-3 min to anesthetize the worms. The worms can be taken up into capillaries individually, as below.
Visually scan the embryos to find one at the desired stage of embryonic development, keeping in mind that the steps leading to HPF will take approximately 2 min. Avoid prolonged observation under the dissecting scope as the small volume of 20% BSA solution will quickly dry out due to evaporation.
To collect an embryo of desired stage, place the open end of a cellulose capillary tube, attached to pipette tip (Figure 4b, cartoon) close to the embryo. Due to capillary action, the embryo will get into the capillary tube and there is no need to pipette it in (Figure 4c, cartoon).
Use a crimping tool to press gently on the capillary tube (otherwise it will cut open and embryo will float away) on both sides of the trapped embryo to seal the capillary tube (refer to Figure 4c, cartoon). Use the same tool to separate the section (Figure 4c, inset) with trapped embryo away from dissected worms. Remember to keep the length of this section around or below 2 mm (Figure 4c, inset) so that it will fit in the cavity of the type B planchette (Muller-Reichert et al., 2008). Follow embryonic development of the trapped embryo under a dissecting scope.
Approximately 1 min prior to the desired embryonic developmental stage, add 0.8 µl of 20% BSA solution into the 100 µm cavity of the type B planchette (Figure 4d). The planchette must be filled to the top to avoid air bubbles, but not overfilled to avoid excessive liquid wicking into the loading area. A gentle positive meniscus typically suffices. Transfer the capillary tube section with the embryo (Figure 4c, inset) with sharp-point tweezers into the type B planchette filled with 20% BSA solution (for ease of visualization, capillary tube sections with intact worms are shown in Figure 4d). Place a type A planchette (flat side) on top of the type B planchette (Figure 4g).
Transfer the planchette sandwitch with the embryo secured inside (Figures 4e-g) to the Leica EM ICE loading station (Figure 3d, pre-assembled cartridege system; also see Figures 4h-i). Manually closing the red flap (Figure 4j) will initiate the freezing process, where the embryo secured inside the cartridge system is cooled to LN2 temperatures within tens of milliseconds and under high pressures of approximately 2,000 bar (refer to Figure 5).

Figure 4. Sample preparation for HPF. a-c. Dissection of young adult worms in a drop of 20% BSA solution. Steps are depicted in a cartoon above each image. An embryo of choice is collected into a cellulose capillary and trapped with the crimping tool (c, inset). d. Planchette with whole worms inside cellulose capillaries in 20% BSA solution filled to the top. e-g. A sharp-point tweezer which is used to transfer the capillary piece (c, inset) to the type B planchette cavity, is used to place the type A planchette (flat side) on top of type B planchette. h-j. The planchette sandwich (g, right) is placed securely in the holder (h, orange arrow). Manually closing the red lid (j) on the loading station will plunge the sample into LN2 under high pressure.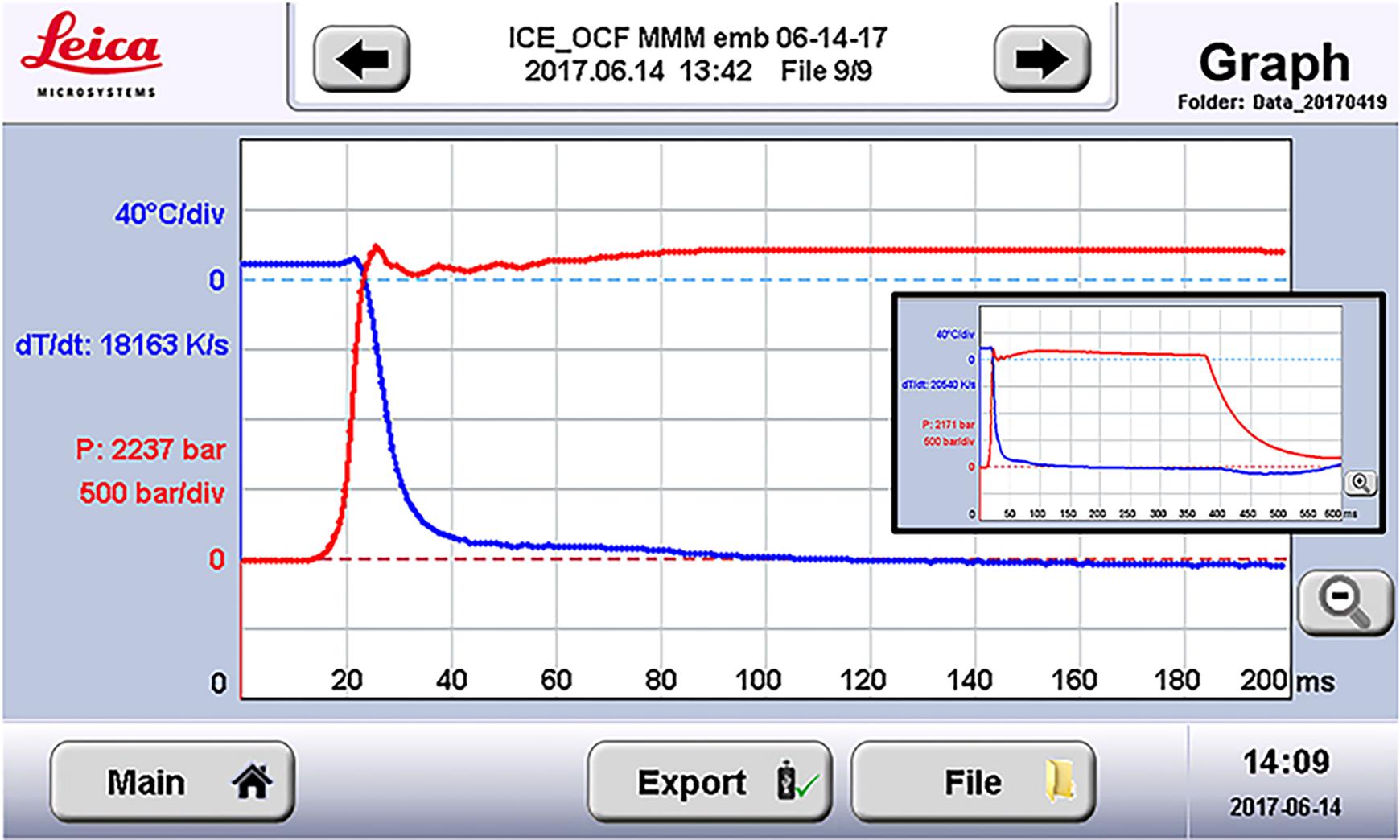
Figure 5. Temperature and pressure plot of a high-pressure freezing cycle. The x axis is time in milliseconds (ms), and the y axis is either pressure in bar, red, or temperature in Kelvin (K), blue. For only illustration purpose, here is a screenshot of an acceptable execution of a single run from the high-pressure freezer, with pressure reading of 2237 bar at time sample cooling started at a rate (dT/dt) of 18163 K/s. Using the zoom out button (right bottom corner) pressure, the temperature status can be obtained for up to 600 ms (see inset). For details refer to Muller-Reichert et al. (2007 and 2008).Repeat the Steps A10 to A15 until you have frozen the necessary number of samples. The maximum capacity of a single sample storage dewar in the Leica ICE high-pressure freezer is nine samples, however a blank HPF run is typically executed at the start of the experiment, leaving space for up to eight samples. Note that it is possible to switch the sample collection to a new and dry holder (Figure 3d) to collect another set of up to nine samples. Other high-pressure freezers do not have a limit of nine samples.
High-pressure frozen samples can be stored under LN2 for years. For long-term storage of samples go to protocol B. For freeze substitution, go to protocol C directly, skipping protocol B.
Recovery of frozen samples from HPF machine
To store frozen samples, prepare a 50 ml polypropylene tube by perforating its side wall with a hot end of a sharp mini screwdriver (place the tip on a Bunsen burner briefly to heat). The hole prevents pressure build up and a potential explosion hazard in case of accidental warming. Attach a long (~3 feet) string securely in the cap (Figure 6) and a tape label so the tube holding frozen samples can be pulled out of a large LN2 tank.

Figure 6. Preparation of storage tube. A 50 ml blue cap tube is prepared as described in Section B Step 1. Inset: An example of label on the tube and tape label to the string.Prepare multiple 1 ml cryovials by perforating its side wall (similar to Step B1). The hole prevents pressure buildup in the tube. Label each cryovial with date, experiment number and sample number using a pencil. Pencil marks are stable in acetone and help prevent mix-up later.
Use a hairdryer to remove moisture completely from the sample unloading chamber and accessory units (Figure 7a). Fill the chamber slowly with LN2, and wait until there are no bubbles, that is, the LN2 surface is calm. Keep the plexiglass cover closed to minimize ice or frost contamination.
Place all tweezers and other accessories on a slide warmer set at 45 °C next to the chamber and keep the hairdryer plugged in as you will need it again. Be quick and alternate the tweezers kept on a slide warmer to prevent depositing ice crystals into sample unloading chamber.
Note: Wearing a surgical facemask reduces condensation and fog in chamber filled with LN2.
Take the sample storage dewar out from Leica ICE high-pressure freezer. Transfer the segmenting insert and attached trisection pod (Figure 7c) to the designated slot (Figure 7a, left side) in the cryobox and release the trisection pod containing the planchette assemblies (Figures 7c-7d) by pressing and twisting the “bayonet” or release button from the locked to open position on the segmenting insert (Figure 7c, blue arrow).
Dip a dry insulated tweezer (Figure 1d) in LN2 away from the samples for about 20 s to chill it to LN2 temperature. Watch for the bubbles and wait until the hissing sound ceases.
Remove plastic adapters one at a time out of the sample holder (Figure 7e) leaving only the holder with frozen planchettes. Quickly transfer the holder with frozen planchettes (keeping submerged in LN2 all the time) to the sample release station (Figures 7e-7f, right side).
With the release handle (Figure 7a, white arrow), punch out the frozen planchettes from the holder into the reservoir (Figure 7g, yellow arrow). Keep the plexiglass cover closed to minimize ice contamination when not transferring/releasing samples.
Place prelabeled cryovials into the cryobox and allow to chill to LN2 temperature for a few minutes. Use dry tweezers (Figure 1d) to transfer frozen planchettes into respective cryovials (keeping submerged in LN2 all the time). Warm up each cryovial cap with your palm, and then quickly close. If the cap is not warm and moisture free, then it will freeze to the tube and it is nearly impossible to reopen the tube without warming up the whole tube, causing damage to the frozen sample.

Figure 7. Sample recovery after high-pressure freezing. All photographs were taken without LN2 for clarity. a. Sample recovery cryobox showing a tray for sample holder (left) and sample release station with release handle (white arrow) and collection reservoir on the right. b-d. Sample holder from ICE high-pressure freezer transferred to sample recovery cryobox. Twisting the bayonet (c, blue arrow) counter clockwise releases the sample collection container unit from the cylinders. e-g. One sample holder at a time is transferred to the release station. Frozen planchettes are collected into the reservoir (g, orange arrow).Final Check: Inspect for any split planchettes, and if found, collect the ones without the black dot. The type-A planchette (flat bottom) used as a cover is marked with a black dot, and can be easily identified under LN2 and discarded, as the frozen embryo is inside the cavity of an unmarked, type B planchette.
Collect all cryovials into a prelabeled 50 ml polypropylene tube (prechilled in LN2 in a portable dewar), close it with a cap with the string with tape label visible outside (see Figure 6), and deposit into a LN2 storage tank. Samples can be stored for years with appropriate care.
Freeze substitution of HPF embryos for electron microscopy
Note: This process takes 3-4 days in total and likely more than 8 h on the first day due to long incubation times. Confirm that you have enough dry ice and LN2 supply. Reagents must be freshly prepared, so plan your day accordingly. Use a slide warmer and hairdryer to keep all instruments moisture free!
Prepare the QFS cocktail as per recipe 7 (Rahman et al., 2020), and aliquot 1 ml each in prelabeled cryovials (label as per sample number, one frozen sample per vial).
Place a metal block with 12 mm holes facing up in dry ice bucket. Fill with LN2 and allow to chill to LN2 temperature. Add LN2 frequently to keep the block submerged. Place a lid on the bucket to prevent condensation. The bucket must be placed in a chemical fume hood, and subsequent steps performed in a fume hood as osmium is toxic and volatile.
Freeze the QFS cocktail by placing the tubes into the metal block submerged in LN2. Do not add too much LN2, otherwise the tubes will float out of the block.
Collect the sample cryovials (HPF embryos or worms) from the LN2 storage tank into a transfer dewar filled with LN2 (Figure 8a, blue arrow).

Figure 8. Sample transfer for QFS. a. Frozen samples are transferred in LN2 dewar (bottom, blue arrow). QFS cocktail submerged in LN2 in a dry ice bucket (top left, yellow arrow). b. A shallow Styrofoam container with frozen QFS cocktail cryovial (top left) and HPF sample cryovial (top right) laid flat submerged in LN2. One tube at a time, cryovials are opened (top right), frozen planchettes are spilled into LN2 on a shallow Styrofoam container, and then transferred into frozen QFS cocktail in cryovials laid flat on the same surface (top left) as in Steps C5 to C7 with tweezers. c. QFS tubes are placed on dry ice sideways (in a prechilled metal block). d. The bucket is filled with more dry ice, covered with a lid, and then placed on an orbital shaker (McDonald and Webb, 2011).Take a clean and dry shallow Styrofoam container or tray. A lid of a shipping container, typically 15 × 20 cm and 2 cm deep, will suffice. Add LN2 to the tray and allow it to cool just prior to the subsequent steps.
Take out one cryovial at a time. Uncap the cryovial, and quickly lay down the vial flat on a shallow Styrofoam container filled with LN2 and spill out the planchette under LN2. Next to it also uncap and lay down a frozen QFS-containing vial flat in the LN2. Keep the cap of this vial warm in your palm or on a slide warmer to avoid condensation otherwise it will freeze and get stuck.
Transfer one planchette into each vial with frozen QFS cocktail with a sharp-point tweezer. Quickly stand up the cryovial (keeping most of the vial submerged in LN2) and screw the warm cap onto the tube. Immediately transfer it to the metal block submerged in LN2 (Step C2). It is possible to place multiple planchettes in a single QFS cocktail tube if samples are easily identifiable, however we warn that the planchettes can jostle against each other and dislodge large samples during QFS.
Frequently check and add LN2 to keep the metal block submerged the whole time. Place the lid on top to restrict condensation (as much as possible) from the moisture in the air.
Repeat Steps C4 to C7 until all sample-containing planchettes are transferred to individually labeled QFS-containing cryovials. We advise not to attempt more than eight tubes at a time.
Carefully decant LN2 and fill the ice bucket halfway with dry ice. Rotate the metal block so cryovials are now lying flat on their side. Add more dry ice to cover the metal block completely and cover the bucket with a lid. Place it on an orbital shaker set at 60 cycles/min for 3 h inside a chemical safety hood (McDonald and Webb, 2011).
Discard dry ice after 3 h. Place the lid back and continue to rotate at 60 cycles/min for another hour.
Remove the lid and continue to shake at 60 cycles/min for another hour (or as needed) until the metal block reaches a temperature of about 4 °C. Frequently check the temperature of the block with an infrared thermometer. This is a critical step! Long exposure at 4 °C or higher may significantly darken the entire volume, making it hard to identify a single embryo inside the capillary.
Replace the QFS cocktail with 100% acetone (stored at 4 °C). First, carefully remove the QFS cocktail with a glass pipette (take care to leave the planchette undisturbed). Use a fresh pipette, and gently refill by releasing the solution (100% acetone) against the side wall of the cryovial. Replace the solution with the following mixture after 1 h incubation in each (use an orbital shaker set at 60 cycles/min).
1:2 resin:acetone
1:1 resin:acetone
2:1 resin:acetone
Discard all pipettes, used and unused reagents, paper towels and kimwipes used in the process in accordance with institutional biohazard disposal procedure.
Transfer the planchettes into 100% resin and leave overnight (14 h to 16 h) at room temperature on an orbital shaker set at 60 cycles/min.
Next morning, replace overnight resin with freshly prepared 100% resin and immediately proceed to the next step.
Use a razor blade to cut off the bottom of a Beem capsule; uncap the capsule and flex the hinge so that the lid lies flat on a surface. Place the planchette sample side up along with a few drops of resin on the inside of the Beem capsule lid. Carefully close the capsule onto the lid, keeping the planchette sample side up. The bottom of the capsule with the cut-out hole should be facing up. Fill the capsule through this opening with 100% resin (see Figures 9a-9i), and bake it to cure in an oven at 75 °C for 60 h to 65 h.
Once cured, use a razor to cut off the plastic beam capsule (see Figures 9j-9m). Now carefully remove all the resin around the metal planchette under a dissecting scope. Important: be sure to completely remove all resin on the sides – the shiny metal of the planchette side should be totally exposed (see Figures 9o-9p). Now quickly immerse the exposed metal base of the planchette into LN2 until it reaches cryogenic temperature, then heat with a hairdryer until all condensation has disappeared and the planchette is warm. Repeat the cold/hot cycle multiple times as required, until the planchette or the metal carrier pops out and falls away due to differential thermal expansion between the metal and resin. This will leave the resin-embedded sample intact (Figure 9r). Do not use force to pry the planchette loose, as this risks fracturing the resin leaving the resin-embedded sample still in the cavity. You can strore resin-embedded samples either with or without planchette attached for years in a dustproof storage container at room temperature before sectioning for imaging (TEM or vEM).
Note: The QFS protocol described here is adequate for FIB-SEM imaging and array tomography in our hands. For conventional TEM imaging, the samples may be sectioned and are typically post-stained to enhance contrast (Hayat, 2000). For other volume EM approaches such as serial block face SEM, users may wish to further enhance metallization of the sample for high-resolution work by adapting room temperature en-bloc staining protocols (Hayat and Giaquinta, 1970; Hua et al., 2015).

Figure 9. Resin embedding and sample processing. a-c. A razor blade is used to cut off the bottom of a Beem capsule. d-e. Flex the hinge so that the lid lies flat on surface. Place the planchette (from QFS Step C15) sample side up and add a few drops of resin with a Pasteur pipette. f-g. Carefully close the capsule on the lid keeping sample side of the planchette up. h-i. Fill the Beem capsule with 100% resin and transfer to an oven (see Step C16) to cure. After curing, the stained sample is at the bottom of the capsule (9j, back view). k-m. Use a razor blade to remove the Beem capsule. n-p. Under a dissecting scope carefully remove the resin using a razor blade to expose the planchette. q. Once the planchette is exposed multiple times, immerse it in LN2 followed by heating with a hairdryer until the metal carrier pops out. r. Exposed sample embedded in 100% resin after removal of planchette or metal carrier.
Notes
Working with liquid nitrogen is potentially dangerous. Appropriate Personal Protective Equipment (PPE) must be used to protect your eyes, face and exposed skin. Also see your institute’s safety procedure.
Osmium, a heavy metal, poses health risk if inhaled. You must wear a surgical mask and prepare the freeze substitution cocktail mix in a chemical hood connected to an exhaust system.
Recipes
Modified Youngren’s, Only Bacto-peptone (MYOB) plates for worm maintenance
Bacto Agar 20 g
Sodium chloride (NaCl) 2 g
Trizma-HCl 0.55 g
Trizma-OH 0.24 g
Bacto Peptone 3.1 g
Deionized water to 1 liter
Autoclave for 20 min (liquid cycle), allow to cool down
Add 1.6 ml cholesterol from stock solution (see below)
Mix thoroughly with a magnetic stirrer prior to pouring into 35 mm tissue culture dishes, about 5 ml per plate (~200 plates per liter)
Once the agar plates have solidified and are dry, apply ~200 µl E. coli OP50 culture (see below) and let stand for two days at room temperature until the bacterial solution has dried and a bacterial lawn is formed (Church et al., 1995)
Cholesterol stock solution
Cholesterol 5 mg
Ethanol (200 proof) 100 ml
Use a small magnetic stir bar to dissolve cholesterol (slow speed for about 2 h)
E. coli OP50 stock
From a frozen stock, streak bacteria on an agar plate (without any antibiotics) and incubate overnight (~14-16 h) at 37 °C. Inoculate a few isolated colonies into 100 ml LB media (without any antibiotics) and incubate for 6-7 h at 37 °C without shaking. Seed MYOB plates with 200 µl culture (Stiernagle, 2006). Dispose of unused bacterial culture appropriately.
Cellulose capillary attachment
See Figure 10
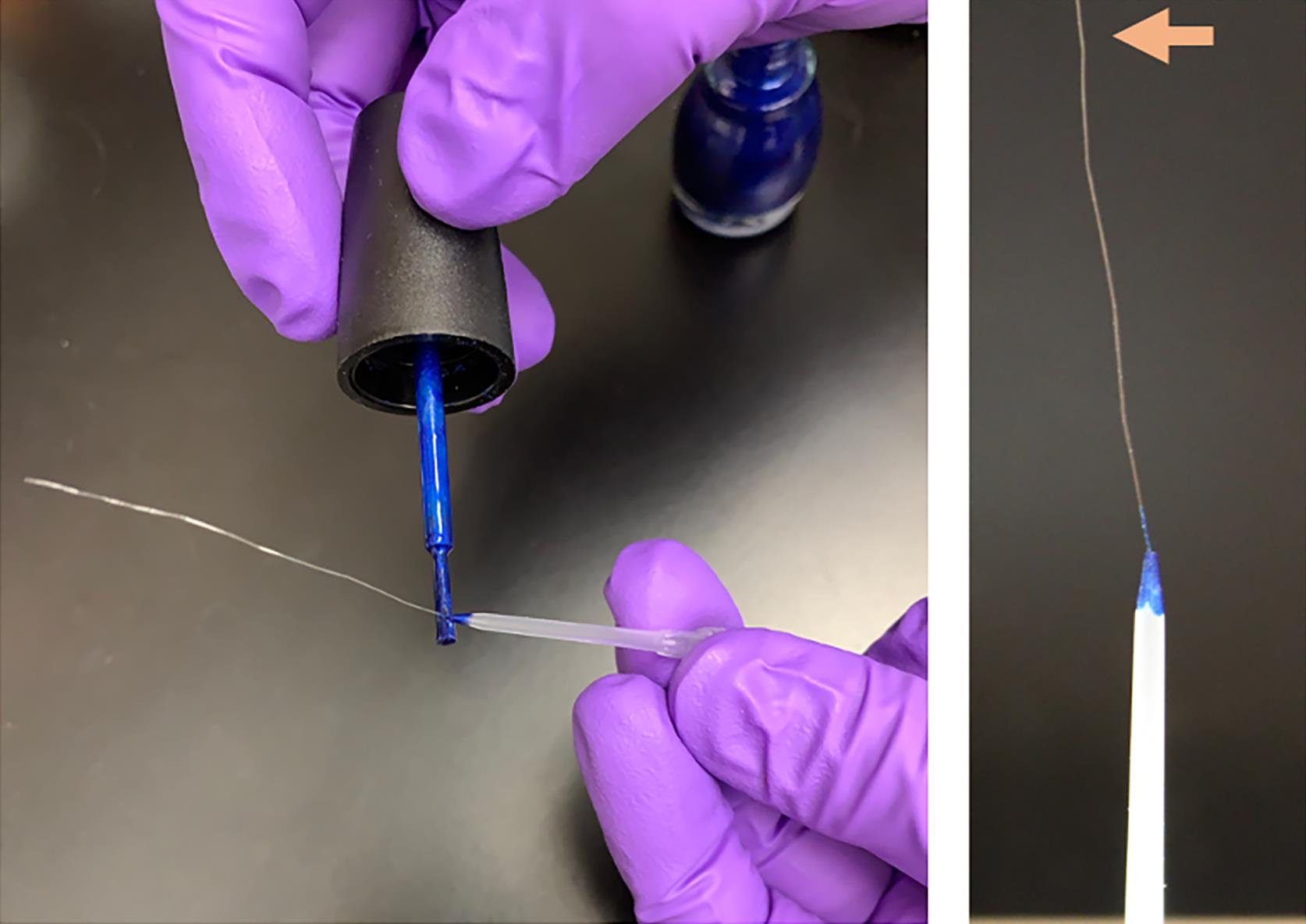
Figure 10. Cellulose capillary attachment for sample collection. Cut cellulose capillary into ~4 cm pieces using sharp scissors. Under a dissecting scope, push or pull the capillary through a 1 ul pipette tip (without a barrier) until the edge of the capillary tube is close to the edge of the tip. Apply nail polish to glue the capillary tube to the tip of the pipette tip and allow it to dry at room temperature for an hour (Muller-Reichert et al., 2003). Ensure the other end of the capillary tube is open; if not, cut to open near the edge with sharp scissors (see arrowhead).20% BSA solution
BSA 10 g
M9 buffer to 50 ml
Prepare the solution in multiple steps, that is, each time add a small amount of BSA into 35 ml of M9 buffer (in a 50 ml blue cap tube)
Mix gently on a table-top orbital shaker at low speed to avoid generating bubbles
Once BSA is completely dissolved, bring volume to 50 ml. Mix gently to homogeneity and store at 4 °C, or -20 °C in small aliquots (long-term preservation) for up to one year
25 mM Levamisole solution
Add 0.051 g levamisole into 10 ml of 20% BSA solution
Mix gently on a table-top orbital shaker at low speed to avoid generating bubbles
Note: The solution can be used for up to two to three months.
Quick Freeze-substitution (QFS) cocktail
Note: Must prepare inside a chemical hood. Use a borosilicate glass pipette to measure acetone and methanol. Please follow institutional chemical safety regulations for handling Osmium and Uranium compounds. These are extremely toxic; safety is paramount.
OsO4 granule 0.1 g
Acetone 13.5 ml
At room temperature stir to mix completely for about 10-15 min. Once no residue is seen, add from a freshly opened vial of:
2% Uranyl acetate (UA) in methanol 0.75 ml
Distilled deionized (DD2) water 0.75 ml
At room temperature mix completely and pass through a 0.22 µm cellulose acetate filter to remove any residual undissolved elements. Aliquot 1 ml into separate cryovials, flash freeze in LN2 and store at -80 °C (Rahman et al., 2020)
Poly/Bed 812 resin mix
Poly/Bed 812 14.6 g
Dodecenyl succinic anhydride (DDSA) 8.4 g
Nadic Methyl anhydride (NMA) 7.0 g
DMP-30 0.42 ml
Mix gently with magnetic stirrer until homogeneity is achieved. Prepare fresh from the kit which contains all the chemicals mentioned above (Rahman et al., 2020)
Acknowledgments
We thank Heather Berensmann for critical reading of the manuscript and help with photography.
M.M. Rahman and O. Cohen-Fix were supported by the National Institute of Diabetes and Digestive and Kidney Disease (intramural grant DK069012). I.Y. Chang, and K. Narayan were supported by the National Cancer Institute (Contract No. 75N91019D00024). This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. 75N91019D00024. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Competing interests
The authors declare no competing financial interests.
Ethics
Hazardous chemicals were disposed according to institutional guidelines.
References
- Altun, Z. F., Herndon, L. A., Wolkow, C. A., Crocker, C., Lints, R. and Hall, D. H. (Eds.) Worm Atlas, 2002-2020.
- Chang, I. Y., Rahman, M. M., Harned, A., Cohen-Fix, O., and Narayan, K. (2020). Cryo-fluorescence microscopy of high-pressure frozen C. elegans enables correlative FIB-SEM imaging of targeted embryonic stages in the intact worm. Methods Cell Biol https://doi.org/10.1016/bs.mcb.2020.09.009.
- Church, D. L., Guan, K. L. and Lambie, E. J. (1995). Three genes of the MAP kinase cascade, mek-2, mpk-1/sur-1 and let-60 ras, are required for meiotic cell cycle progression in Caenorhabditis elegans. Development 121(8): 2525-2535.
- Cohen-Fix, O. and Askjaer, P. (2017). Cell Biology of the Caenorhabditis elegans Nucleus. Genetics 205(1): 25-59.
- Corsi, A. K., Wightman, B. and Chalfie, M. (2015). A Transparent window into biology: A primer on Caenorhabditis elegans.WormBook 1-31. doi:10.1895/wormbook.1.177.1
- Hayat, M. A. (2000). Principles and techniques of electron microscopy: biological applications (4th edition). Cambridge, UK ; New York: Cambridge University Press.
- Hayat, M. A. and Giaquinta, R. (1970). Rapid fixation and embedding for electron microscopy. Tissue Cell 2(2): 191-195.
- Hua, Y., Laserstein, P. and Helmstaedter, M. (2015). Large-volume en-bloc staining for electron microscopy-based connectomics. Nat Commun 6: 7923.
- McDonald, K. (1999). High-Pressure Freezing for Preservation of High Resolution Fine Structure and Antigenicity for Immunolabeling. In: Hajibagheri, N. (Ed.). Methods in Molecular Biology: Electron Microscopy Methods and Protocols (Vol. 117, pp. 77-97). Totowa, NJ: Humana Press Inc.
- McDonald, K. L. and Webb, R. I. (2011). Freeze substitution in 3 hours or less. J Microsc 243(3): 227-233.
- McDonald, K., Schwarz, H., Muller-Reichert, T., Webb, R., Buser, C. and Morphew, M. (2010). "Tips and tricks" for high-pressure freezing of model systems. Methods Cell Biol 96: 671-693.
- Muller-Reichert, T., Hohenberg, H., O'Toole, E. T. and McDonald, K. (2003). Cryoimmobilization and three-dimensional visualization of C. elegans ultrastructure. J Microsc 212(Pt 1): 71-80.
- Muller-Reichert, T., Mantler, J., Srayko, M. and O'Toole, E. (2008). Electron microscopy of the early Caenorhabditis elegans embryo. J Microsc 230(Pt 2): 297-307.
- Muller-Reichert, T., Srayko, M., Hyman, A., O'Toole, E. T. and McDonald, K. (2007). Correlative light and electron microscopy of early Caenorhabditis elegans embryos in mitosis. Methods Cell Biol 79: 101-119.
- Oegema, K. and Hyman, A. A. (2006). Cell division. WormBook 1-40. doi:10.1895/wormbook.1.72.1
- Rahman, M., Chang, I. Y., Harned, A., Maheshwari, R., Amoateng, K., Narayan, K. and Cohen-Fix, O. (2020). C. elegans pronuclei fuse after fertilization through a novel membrane structure. J Cell Biol 219(2). doi:10.1083/jcb.201909137.
- Stiernagle, T. (2006). Maintenance of C. elegans. WormBook 1-11. doi:10.1895/wormbook.1.101.1.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Rahman, M. M., Chang, I. Y., Cohen-Fix, O. and Narayan, K. (2021). A Workflow for High-pressure Freezing and Freeze Substitution of the Caenorhabditis elegans Embryo for Ultrastructural Analysis by Conventional and Volume Electron Microscopy. Bio-protocol 11(7): e3981. DOI: 10.21769/BioProtoc.3981.
- Rahman, M., Chang, I. Y., Harned, A., Maheshwari, R., Amoateng, K., Narayan, K. and Cohen-Fix, O. (2020). C. elegans pronuclei fuse after fertilization through a novel membrane structure. J Cell Biol 219(2). doi:10.1083/jcb.201909137.
Category
Developmental Biology > Cell growth and fate
Biophysics > Microscopy
Cell Biology > Cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.