- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Brain-localized and Intravenous Microinjections in the Larval Zebrafish to Assess Innate Immune Response
Published: Vol 11, Iss 7, Apr 5, 2021 DOI: 10.21769/BioProtoc.3978 Views: 6677
Reviewed by: Chiara AmbrogioMarek WagnerJohn W Peterson
Original research article
The authors used this protocol in:

Jul 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Creating a robust and controlled infection model is imperative for studying the innate immune response. Leveraging the particular strengths of the zebrafish model system, such as optical transparency, ex utero development, and large clutch size, allows for the development of methods that yield consistent and reproducible results. We created a robust model for activation of innate immunity by microinjecting bacterial particles or live bacteria into larval zebrafish, unlike previous studies which largely restricted such manipulations to embryonic stages of zebrafish. The ability to introduce stimuli locally or systemically at larval stages provides significant advantages to examine host response in more mature tissues as well as the possibility to interrogate adaptive immunity at older larval stages. This protocol describes two distinct modes of microinjection to introduce lipopolysaccharide (LPS) or bacteria into the living larval zebrafish: one localized to the brain, and another into the bloodstream via the caudal vein plexus.
Graphic abstract:

Schematic shows the two distinct modes of larval zebrafish microinjection, either in the brain parenchyma or in the blood stream intravenously. Reagents introduced into the zebrafish to assess immune response are depicted in the “injection components” as described in the protocol.
Background
The complex interactions during an infection require the use of in vivo animal models to fully understand the dynamic interplay between the pathogen and its host. Studying this phenomenon requires a controlled and reliable method of pathogen delivery. Zebrafish has been used as a model for studying the immune response to a variety of pathogens (Menudier et al., 1996; Davis et al., 2002; Neely et al., 2002; Prouty et al., 2003; van der Sar et al., 2003; O'Toole et al., 2004; Phelan et al., 2005; Phelps and Neely, 2005; Pressley et al., 2005) for its evolutionarily conserved innate immune system (Herbomel et al., 1999; Traver et al., 2003; Trede et al., 2004), optical transparency, large embryo clutch sizes, genetic tractability and in vivo imaging capabilities (Kimmel, 1989; Kimmel et al., 1988 and 1995; Sullivan and Kim, 2008; Kanther and Rawls, 2010). Previous infection protocols have immersed fish in pathogens by directly adding it to fish water (Davis et al., 2002; O'Toole et al., 2004; Prouty et al., 2003) or injected bacteria into the axial vein or hindbrain ventricle in the early embryo at 28 h post- fertilization (hpf) (van der Sar et al., 2003), or injected LPS at later stages in the larval zebrafish at 3-6 days post-fertilization (dpf) into the yolk to induce a lethal systemic immune response (Yang et al., 2014). Extending from previously published methods, we developed a protocol for microinjection of LPS or bacteria in the larval zebrafish, either directly into the brain parenchyma or into blood circulation to cause a robust innate immune response starting in the 4 dpf larvae. Since adaptive immunity does not begin until about 4 weeks after fertilization in zebrafish (Davis et al., 2002), we can leverage the early larval stages of zebrafish to study specific innate immune functions independent of adaptive immunity. Co-injection of immune activators with a fluorescently labeled dextran, or direct injection of fluorescently tagged immune activators allows for a quick visual verification of a successful injection as well as subsequent labeling of the macrophage response based on phagocytosis of the reporter. This protocol describes two different modes of microinjection with distinct target sites: first, the caudal vein plexus for systemic distribution throughout blood flow, and second, the brain tectum that briefly localizes the injected substance in the brain but is subsequently drained out into circulation (Yang et al., 2020). Although we describe our protocol for 3-5 dpf larvae, these methods are applicable to later larval stages at least up to 10 dpf (Yang et al., 2020).
Materials and Reagents
Polystyrene Petri dish (VWR, catalog number: 25384-342 )
7.5 ml transfer pipettes (VWR, catalog number: 414004-005 )
Thin wall glass capillaries 4 with filament, OD 1.5 mm (World Precision Instruments, catalog number: TW150F-4 )
Escherichia coli (any strain)
Lipopolysaccharides from Escherichia coli O111:B4 (Sigma-Aldrich, catalog number: L3024 ) (store at -20°C)
Lipopolysaccharides from Escherichia coli Serotype 055:B5, Alexa FluorTM 594 Conjugate (Thermo Fisher Scientific, Invitrogen, catalog number: L23353 ) (store at -80 °C and protect from light)
Dextran, Alexa FluorTM 568 (Thermo Fisher Scientific, Invitrogen, catalog number: D-22912 ) (store at -20 °C and protect from light)
Phosphate-buffered saline (PBS), pH 7.4
1.5% low melt agarose (Fisher Scientific, IBI Scientific, catalog number: 50-550-455 ) (see Recipes)
Tricaine (3-amino benzoic acid ethyl ester) (Sigma-Aldrich, catalog number: A-5040 ) (store at room temperature)
Note: Store at -20 °C when made into 25× Tricaine solution (see Recipes).
PTU (N-Phenylthiourea) (Sigma-Aldrich, catalog number: P7629 ) (store at room temperature)
Note: Store at -20 °C when made into 50× PTU solution (see Recipes).
Equipment
Programmable Horizontal Pipette Puller (World Precision Instruments, catalog number: PUL-1000 )
Pneumatic PicoPump (World Precision Instruments, catalog number: SYS-PV820 )
Fluorescent stereomicroscope fully apochromatic corrected with 16:5:1 zoom optics (Leica, model number: M165 MC )
Incubator (Benchmark Scientific, model number: H2200-H )
Stage Micrometer, 1 × 0.01 mm (AmScope, catalog number: MR095 )
Magnetic stand (World Precision Instruments, catalog number: M1 )
Manual micromanipulator (World Precision Instruments, catalog number: M3301 )
Steel base plate, 10 lbs (World Precision Instruments, catalog number: 5052 )
PicoNozzle Kit v2 (World Precision Instruments, catalog number: 5430-ALL )
Dumont #55 Forceps (Fine Scientific Tools, catalog number: 11295-51 )
Procedure
Zebrafish embryo and larval husbandry
Maintain zebrafish in Petri dishes with autoclaved fish water supplemented with 0.003% PTU and incubate at 28.5 °C.
For general questions, the composition of fish water and regular zebrafish husbandry are well described elsewhere including Avdesh et al. (2012).
Replace water with fresh water containing 0.003% PTU daily and monitor health.
Begin experiment when embryos reach your desired larval stage (such as 4 dpf).
Injection mixture preparations
E. coli supplemented with fluorescent dextran: Prepare 3 ml overnight culture derived from a single colony. Centrifuge culture at 3,000 rpm for 1 min, remove supernatant and re-suspend in 500 μl of 1× PBS (pH 7.4). This should be approximately 1.6 × 106 cfu/μl. Add 1 μl of 5 ng/nl fluorescent dextran. Flick the tube with your finger and spin down before use.
LPS supplemented with fluorescent dextran: Mix 9 μl of LPS at 5 ng/nl from Escherichia coli O111:B4 and 1 μl of a 1:10 dilution of 5 ng/nl fluorescently labeled dextran to make a final dilution of 1:100 supplementation with fluorescent dextran. Flick the tube with your finger to mix and spin down before use.
LPS directly conjugated to fluorescent molecules (e.g., Alexa 594) – use directly at 5 ng/nl.
Control vehicle injection using ultra-pure or autoclaved water supplemented with fluorescent dextran – mix 9 μl of water with 1 μl of a 1:10 dilution of 5 ng/nl fluorescently labeled dextran. Flick the tube with your finger to mix and spin down before use.
Needle and microinjector set-up
Microinjection needles are pulled from glass capillaries using a Programmable Horizontal Pipette Puller or similar equipment (Table 1).
Table 1. 4-step protocol for glass capillary tube pullingStep Heat Force Distance Delay 1 690 260 7.3 0 2 500 240 0.5 4 3 500 230 0.5 10 4 380 240 0.5 20 Load ~3 μl of injection solution into needle.
Note: Unless a very fine pipette tip is used, the solution will remain at the mouth of the needle. To force the solution to the tip of the needle, hold needle firmly between middle finger and thumb and flick wrist in a downward motion (see Video 1). Repeat until liquid has reached the narrow tip of the needle.
 Video 1. Loading of injection mixture into a microinjection needle
Video 1. Loading of injection mixture into a microinjection needleTransfer needle into the micromanipulator and set out of the way.
Mounting zebrafish for microinjections (see Video 2)
Video 2. Stepwise demonstration of mounting larvae for microinjectionUse a plastic transfer pipette to transport the larvae to the center of a clean Petri dish lid and remove as much water as possible.
Note: Petri dish lids are used because the sides of the lid have a low profile which allows more latitude to position the needle to the desired target. Several larvae can be mounted at once; for brain injections we can mount upwards of 20-30 zebrafish larvae at once.
Heat 1.5% low-melt agarose (solid form) in microwave to melt it to a liquid form.
Note: Approximately 20 s to less than 1 min is needed to melt 100 ml of agarose. Low melt agarose heats up very fast so you will want to stand nearby to monitor the heating to prevent boil over.
Use pipette to collect a small amount of low-melt agarose.
Immediately after microwaving, the agarose will be very hot. If you see steam/condensation within the pipette then the agarose is too hot and will burn the larvae. Periodically monitor the temperature of the agarose by carefully touching the outside of the pipette where agarose is located. The agarose should be warm to touch, not hot, and remain fluid.
The low-melt agarose will remain fluid at 37 °C and set rapidly below 25 °C. Aim to use the agarose at a temperature between 30 °C and 40 °C. Keeping the agarose on a heat block at 60 °C will preserve the solution in a fluid state for a longer period of time as you work on mounting the larvae.
Cooling the agarose can be expedited by pipetting the hot agarose onto a clean surface (such as another Petri dish) up and down a few times (see Video 2).
Form a circle around the larvae to be injected with the agarose and go in smaller concentric circular motions until the agarose touches the larvae. Mix the agarose with the larvae by using the pipette to gently swirl them together or by pipetting them up and down 1-2 times (see Video 2).
Note: For ease of injection, the goal is to embed the larvae in a thin layer of agarose which allows the fine glass needle to easily penetrate through the gel. Using too much agarose will make orienting the needle more difficult and cause unwanted bending of your glass needle.
Use forceps to orient the larvae.
This is the most time sensitive step because larvae must be correctly positioned before the agarose re-solidifies (1-2 min). Use fine forceps to quickly orient each larva, depending on desired injection site, but without concern for the exact orientation or body alignment. Be cautious to not injure the larvae by poking them, instead use the agarose around them to nudge and move them into position (see Video 2).
For brain and intravenous injections, position the larvae on their dorsal or ventral-lateral sides, respectively.
Wait for agarose to cool down and solidify before starting injections.
Add a small amount of fish system water on top of the agarose to keep larvae in water and healthy during the operation.
Setting up the pneumatic microinjector and calibrating your needle for injection
Review the manufacturer’s recommendations and instructions for the micro-needle puller and the microinjector before operating.
Turn on PicoPump and open air valve. The output pressure should be set to 20 psi. Check that you can feel air eject out from the needle holder (PicoNozzle Kit v2) before putting the needle in place.
Insert your pulled glass needle. Prepare extra needles in case they break or when you need to re-calibrate.
Looking through the microscope orient the tip of the needle into the middle of the field of view by adjusting the manual micromanipulator set on the magnetic stand on a steel base plate that is holding your needle.
Use fine forceps to gently break off small controlled amounts of the glass needle. The objective is to break the minimum amount of the needle to allow the smallest amount of liquid to eject out (see Figure 1).

Figure 1. Calibration of injection needle using a micrometer. To ensure a consist injection volume, use a micrometer to measure the diameter of the injection bubble volume. An injection bubble diameter of 100 μm is equivalent to 0.5 nl.Press the PicoPump foot pedal to see if a noticeable amount of liquid is visible on the tip of the needle. If liquid is not present, continue to carefully clip small pieces off the needle until you can see a tiny amount of liquid.
Use a micrometer to calibrate the size of your needle opening (see Figure 1).
Adjust the opening size of your glass needle by chipping off the end incrementally under the stereoscope while you test how much volume is ejected after each kick by estimating the spherical drop size at the needle tip in air right above a micrometer.
Aim for the spherical drop to have a diameter of about 100 μm on the micrometer to give a 0.5 nl volume per kick on the PicoPump foot pedal (see Figure 1).
Note: To prevent complications from the injection site minimize the size of the needle. We suggest using a smaller needle and administering a 1 nl injection with 2 kicks.
The volume (V) of your droplet from your needle can be determined by the standard formula for a sphere volume (
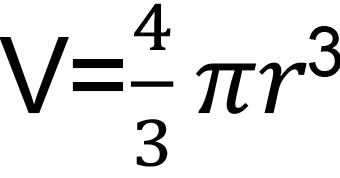 ), where r is the radius of your droplet.
), where r is the radius of your droplet.
Brain microinjection
Conduct the microinjections under a fluorescent stereomicroscope in order to monitor and validate each successful injection by fluorescence. The mounted larvae in low-melt agarose will need to be oriented with brain side up for needle access (see Figure 2).
Place needle directly above the brain tectum (see Figure 2).
Use the micromanipulator to slowly puncture the skin with the needle.
Note: Force of the puncture can drive the needle further into the brain than desired. Pull the needle backwards until the needle is only superficially piercing the brain.
Press the PicoPump foot pedal twice to eject 1 nl.
Screen for successful injection by visualizing fluorescence in the brain tectum at the site of injection (see Figure 2B).
Note: It is normal for the injection liquid to spread into the hindbrain ventricle. Remove any larvae that are not injected correctly by using forceps to grab the larvae out of the agarose.
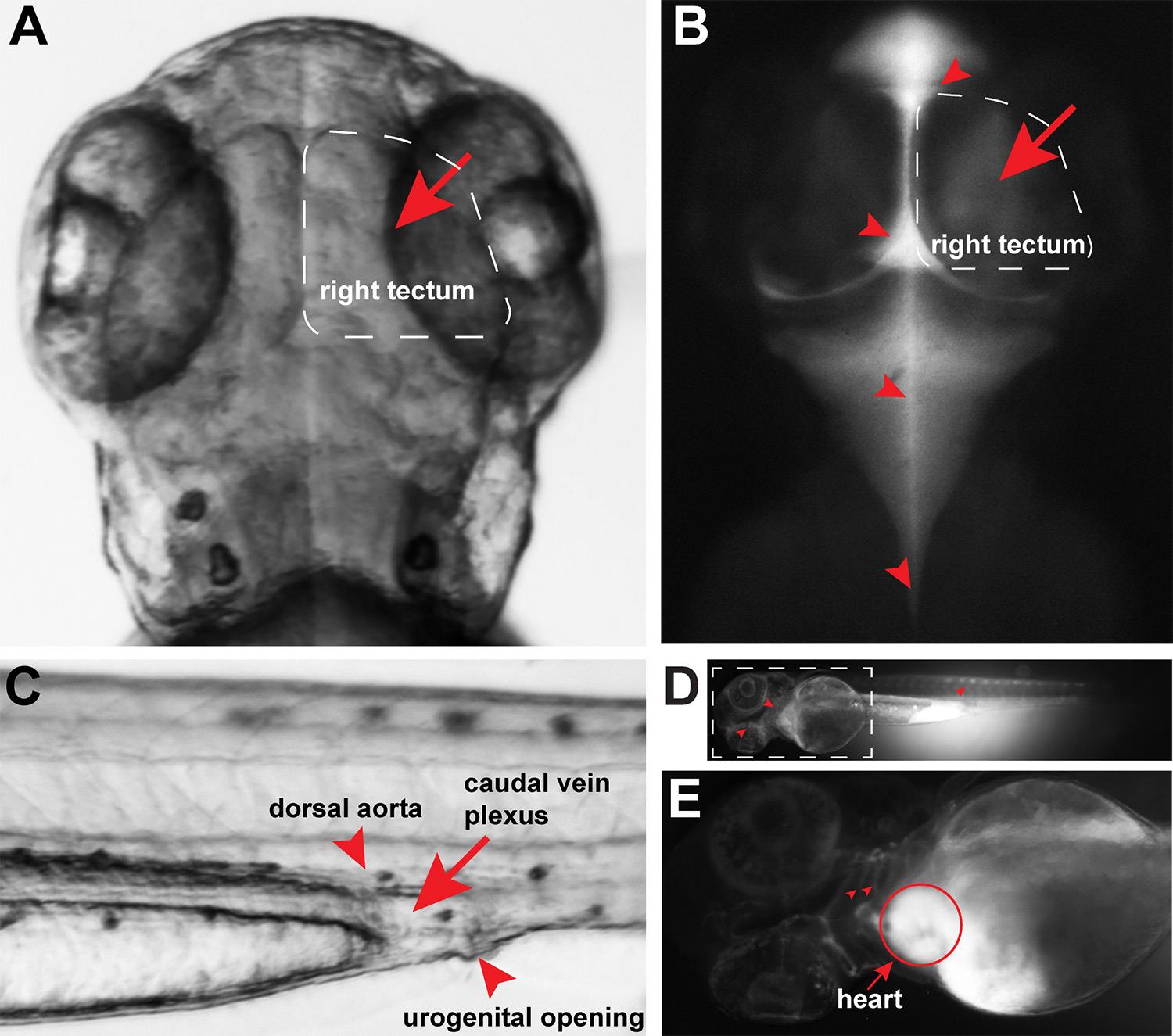
Figure 2. Brain and intravenous microinjection sites. A. Red arrow points to brain injection site in the right tectum and white box outlines the right tectum region of the larval brain at 3 dpf. B. 30 s after fluorescent dextran injection into the brain tectum (red arrow), the fluorescent tracer can be readily observed to disperse into the brain ventricles and into the spinal canal (red arrowheads). C. Red arrow points to the intravenous injection site at the caudal vein plexus located beneath the dorsal aorta (top red arrowhead) and near the urogenital opening (bottom red arrowhead). D. Full body image 30 s after intravenous injection shows fluorescent dextran signal throughout the entire body vasculature. Red arrowheads point to vasculature in the head and trunk made visible by fluorescent dextran. White dotted box, region shown in E. E. Immediately following intravenous injection, the heart (red circle) is filled with fluorescent dextran signal. Red arrowheads point to fluorescent signal in the gill vasculature.
Intravenous microinjection
Conduct the microinjection under a fluorescent stereoscope in order to monitor and validate each successful injection by fluorescence (see Figure 2).
Your mounted larvae in low-melt agarose will need to be oriented with ventral-lateral side up to access the yolk extension side of the tail to inject into the caudal vein plexus (see Figure 2C).
Note: It is most effective to be able to visualize and aim needle into the thin strip of tissue at the caudal vein plexus, which is the most ventral side of the body wall immediately posterior to the end of the yolk extension where continuous circulation can be readily observed in the caudal vein (see Figure 2C).
Place needle directly above the caudal vein plexus close to the urogenital opening (see Figure 2C).
Use the micromanipulator to slowly puncture the skin with the needle.
Note: The force of the puncture can drive the needle pass the vein and into the muscle. Pull the needle backwards until it is in the vein.
Press the PicoPump foot pedal twice to eject 1 nl.
After a successful injection, the immediate detection of fluorescence will happen in the heart (Figure 2E) and from there the injected substance will be widely circulated throughout the body vasculature which is entirely visible within seconds to 1 min after your injection (see Figure 2D-E).
Note: Remove any larvae that are not injected correctly. Intravenous injection requires more technical practice than a brain injection since the target tissue (caudal vein plexus) is far thinner and smaller in area than the tectum.
Post-injection larvae recovery
Recover injected embryos by removing them from the agarose.
Use the side of the forceps to create a break in the agarose beginning underneath the head.
Drag the forceps along the body until the tip of the tail is reached.
Larvae should be able to squirm out easily and swim into the fish water above the gel.
Use transfer pipette to place injected larvae into a clean Petri dish with fresh fish water supplemented with PTU.
Place larvae into 28.5 °C incubator to recover.
At desired time points after injection, monitor the health of the larvae before beginning additional analysis.
Note: Larvae should show no overt signs of change and should be indistinguishable from non- injected stage-matched controls. Healthy larvae will have a consistent rhythmic heartbeat with apparent blood flow, have intermittent bursts of movement, and be straight bodied.
Data analysis
Examples of data analysis using this method can be found in previous publications (Earley et al., 2018; Yang et al., 2020) where the experiments have used brain or intravenous microinjection of LPS or bacteria to examine various types of immune response. Downstream analyses include determining changes in expression of immune response genes (see Figure 5 in Earley et al., 2018 and Figure 5 in Yang et al., 2020), dynamic cellular behaviors and mobilization of macrophages (see Figure 1 in Yang et al., 2020), and routes of molecular drainage from the brain to the periphery (see Figure 2 in Yang et al., 2020).
Recipes
50× PTU stock solution (1L)
Add 1.5 g N-Phenylthiourea (PTU) to 1 L of distilled water and stir overnight at room temperature.
Store at -20 °C.
Note: To inhibit pigmentation in developing embryos use at a 1× concentration in fish water for a final concentration of 0.003% PTU and replace daily. The liquid 50× stock can be stored at room temperature for several months.
25× Tricaine stock solution (100 ml)
Add 400 mg tricaine powder (3-amino benzoic acid ethyl ester) to 100 ml distilled water.
Store at -20 °C.
Note: To administer tricaine as an anaesthetic use at 0.5×-1× concentration. The liquid 25× stock can be stored at room temperature for several months.
1.5% low melt agarose (100 ml)
Add 1.5 g low melt agarose powder to 100 ml distilled water.
Heat for 20 s to less than 1 min in microwave until solution is clear and fluid.
Store at room temperature in a glass bottle with a lid and can be repeatedly re-used by re-melting the agarose in the microwave.
Note: To prevent liquid from boiling over, stand nearby to monitor. Multiple rounds of heating can result in evaporation of liquid and increase the gel percentage.
Acknowledgments
This protocol accompanies the publications (Earley et al., 2018; Yang et al., 2020). The work was funded by NIH NIGMS (Grant Number 1R35G M1 24719 to C.E.S.)
Competing interests
The authors declare no competing interests.
Ethics
Animal experimentation: This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All of the animals were handled according to approved institutional animal care and use committee (IACUC) protocols (#16-160 and #19-132) of the UNC Chapel Hill.
References
- Avdesh, A., Chen, M., Martin-Iverson, M.T., Mondal, A., Ong, D., Rainey-Smith, S., Taddei, K., Lardelli, M., Groth, D. M., Verdile, G., et al. (2012). Regular care and maintenance of a zebrafish (Danio rerio) laboratory: an introduction. J Vis Exp (69): e4196.
- Davis, J. M., Clay, H., Lewis, J. L., Ghori, N., Herbomel, P. and Ramakrishnan, L. (2002). Real-Time Visualization of Mycobacterium-Macrophage Interactions Leading to Initiation of Granuloma Formation in Zebrafish Embryos. Immunity 17(6): 693-702.
- Earley, A. M., Dixon, C. T. and Shiau, C. E. (2018). Genetic analysis of zebrafish homologs of human FOXQ1, foxq1a and foxq1b, in innate immune cell development and bacterial host response. PloS One 13(3): e0194207.
- Herbomel, P., Thisse, B. and Thisse, C. (1999). Ontogeny and behaviour of early macrophages in the zebrafish embryo. Development 126(17): 3735-3745.
- Kanther, M. and Rawls, J. F. (2010). Host-microbe interactions in the developing zebrafish. Curr Opin Immunol 22(1): 10-19.
- Kimmel, C. B. (1989). Genetics and early development of zebrafish. Trends Genet 5(8): 283-288.
- Kimmel, C.B., Ballard, W.W., Kimmel, S.R., Ullmann, B., and Schilling, T. F. (1995). Stages of embryonic development of the zebrafish. Dev Dyn 203(3): 253-310.
- Kimmel, C.B., Sepich, D.S., and Trevarrow, B. (1988). Development of segmentation in zebrafish. Development 104 (Suppl): 197-207.
- Menudier, A., Rougier, F.P., and Bosgiraud, C. (1996). Comparative virulence between different strains of Listeria in zebrafish (Brachydanio rerio) and mice. Pathol Biol (Paris) 44(9): 783-789.
- Neely, M.N., Pfeifer, J.D., and Caparon, M. (2002). Streptococcus-zebrafish model of bacterial pathogenesis. Infect Immun 70(7): 3904-3914.
- O'Toole, R., Von Hofsten, J., Rosqvist, R., Olsson, P. E., and Wolf-Watz, H. (2004). Visualisation of zebrafish infection by GFP-labelled Vibrio anguillarum. Microb Pathog 37(1): 41-46.
- Phelan, P. E., Pressley, M. E., Witten, P. E., Mellon, M. T., Blake, S., and Kim, C. H. (2005). Characterization of snakehead rhabdovirus infection in zebrafish (Danio rerio). J Virology 79(3): 1842-1852.
- Phelps, H. A. and Neely, M. N. (2005). Evolution of the zebrafish model: from development to immunity and infectious disease. Zebrafish 2(2): 87-10.
- Pressley, M. E., Phelan, P. E., 3rd, Witten, P. E., Mellon, M. T. and Kim, C. H. (2005). Pathogenesis and inflammatory response to Edwardsiella tarda infection in the zebrafish. Dev Comp Immunol 29(6): 501-513.
- Prouty, M. G., Correa, N. E., Barker, L. P., Jagadeeswaran, P. and Klose, K. E. (2003). Zebrafish-Mycobacterium marinum model for mycobacterial pathogenesis. FEMS Microbiol Lett 225(2): 177-182.
- Sullivan, C. and Kim, C. H. (2008). Zebrafish as a model for infectious disease and immune function. Fish Shellfish Immunol 25(4): 341-350.
- Traver, D., Herbomel, P., Patton, E. E., Murphey, R. D., Yoder, J. A., Litman, G. W., Catic, A., Amemiya, C. T., Zon, L. I. and Trede, N. S. (2003). The zebrafish as a model organism to study development of the immune system. Adv Immunol 81: 253-330.
- Trede, N. S., Langenau, D. M., Traver, D., Look, A. T. and Zon, L. I. (2004). The use of zebrafish to understand immunity. Immunity 20(4): 367-379.
- van der Sar, A. M., Musters, R.J., van Eeden, F. J., Appelmelk, B. J., Vandenbroucke-Grauls, C. M. and Bitter, W. (2003). Zebrafish embryos as a model host for the real time analysis of Salmonella typhimurium infections. Cell Microbiol 5(9): 601-611.
- Yang, L., Jiménez, J. A., Earley, A. M., Hamlin, V., Kwon, V., Dixon, C. T. and Shiau, C. E. (2020). Drainage of inflammatory macromolecules from the brain to periphery targets the liver for macrophage infiltration. eLife 9: e58191.
- Yang, L.-L., Wang, G.-Q., Yang, L.-M., Huang, Z.-B., Zhang, W.-Q., and Yu, L.-Z. (2014). Endotoxin molecule lipopolysaccharide-induced zebrafish inflammation model: a novel screening method for anti-inflammatory drugs. Molecules 19(2): 2390-2409.
Article Information
Copyright
![]() Rojas and Shiau. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Rojas and Shiau. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Rojas, A. M. and Shiau, C. E. (2021). Brain-localized and Intravenous Microinjections in the Larval Zebrafish to Assess Innate Immune Response. Bio-protocol 11(7): e3978. DOI: 10.21769/BioProtoc.3978.
- Yang, L., Jiménez, J. A., Earley, A. M., Hamlin, V., Kwon, V., Dixon, C. T. and Shiau, C. E. (2020). Drainage of inflammatory macromolecules from the brain to periphery targets the liver for macrophage infiltration. eLife 9: e58191.
Category
Immunology > Immune cell function
Microbiology > Microbe-host interactions > In vivo model
Cell Biology > Tissue analysis > Tissue imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.