- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Reconstitution of Chromatin by Stepwise Salt Dialysis
Published: Vol 11, Iss 7, Apr 5, 2021 DOI: 10.21769/BioProtoc.3977 Views: 5390
Reviewed by: Marcelo S. da SilvaChangyi ZhangAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Chromatin, rather than plain DNA, is the natural substrate of the molecular machines that mediate DNA-directed processes in the nucleus. Chromatin can be reconstituted in vitro by using different methodologies. The salt dialysis method yields chromatin that consists of purified histones and DNA. This biochemically pure chromatin is well-suited for a wide range of applications. Here, we describe simple and straightforward protocols for the reconstitution of chromatin by stepwise salt dialysis and the analysis of the chromatin by the micrococcal nuclease (MNase) digestion assay. Chromatin that is reconstituted with this method can be used for efficient homology-directed repair (HDR)-mediated gene edited with the CRISPR-Cas9 system as well as for biochemical studies of chromatin dynamics and function.
Keywords: Core histonesBackground
DNA in the eukaryotic nucleus is organized into chromatin. Therefore, the analysis of DNA-directed processes, such as replication, recombination, repair, and transcription, would ideally be performed with chromatin templates rather than with plain DNA (Kadonaga, 2019). To this end, chromatin can be reconstituted in vitro from purified components by using either ATP-dependent or ATP-independent approaches (for a review, see Lusser and Kadonaga, 2004).
Here, we describe the specific method of chromatin reconstitution that we employed in our studies of HDR-mediated gene editing with the CRISPR-Cas9 system in cells (Cruz-Becerra and Kadonaga, 2020). In this work, we found that precise HDR-mediated insertion of DNA is enhanced by the use of a chromatin donor template relative to a plain (naked) DNA donor template. We have also used this method for the biochemical analysis of chromatin, which includes the characterization of high mobility group N (HMGN) proteins (Rattner et al., 2009), chromatin dynamics (Torigoe et al., 2013), prenucleosomes (Fei et al., 2015), and nucleosome-destabilizing factor (NDF) (Fei et al., 2018).
The nucleosome, which is the repeating unit of chromatin, consists of an octamer of core histones that is associated with about 147 bp of DNA. A simple and reliable technique for reconstituting chromatin in vitro involves the ATP-independent formation of nucleosomes from purified DNA and core histones by stepwise salt dialysis (Figure 1). In this method, the core histones and the DNA are combined at high ionic strength (1 M NaCl), and nucleosomes are formed by gradually decreasing the salt concentration (Stein, 1989; Jeong et al., 1991). Some important considerations for the successful reconstitution of chromatin are as follows.
Core histones. For chromatin reconstitution, we recommend using core histones that were purified as octamers (which exist as octamers at high salt concentrations and as H2A-H2B dimers and H3-H4 tetramers at low salt concentrations) rather than individual core histones because the separate histones need to be combined in the correct stoichiometry and then purified as octamers before use (Khuong et al., 2017). Protocols for the purification of core histone octamers from different sources are described elsewhere (Laybourn and Kadonaga, 1991; Bulger and Kadonaga, 1994; Fyodorov and Levenstein, 2002; Peterson and Hansen, 2008; Khuong et al., 2017). Histones prepared by these methods have been extensively used for the efficient assembly of chromatin in vitro. We also tested a sample of commercially available native human histone octamers (catalog number 52065; BPS Bioscience, San Diego, CA) and found that they are suitable for reconstituting chromatin by the salt dialysis method.
DNA. To achieve the efficient reconstitution of chromatin, the quality of the DNA is extremely important. For the protocol described here, the DNA can be easily prepared by using the HiSpeed Plasmid Maxi Kit (catalog number 12662; QIAGEN, Hilden, Germany) or by carrying out two successive CsCl density gradient centrifugation steps. We typically use plasmid DNA prepared by these methods for reconstituting chromatin donor templates for HDR-mediated gene editing (Cruz-Becerra and Kadonaga, 2020). In addition, for mononucleosome gel shift assays, we use this salt dialysis protocol to reconstitute mononucleosomes with DNA fragments (≥ 147 bp) that are prepared by PCR amplification followed by purification with the QIAquick Gel Extraction Kit (catalog number 28704; QIAGEN, Hilden, Germany) (Chavez et al., 2019).
Histone:DNA ratio. It is essential to determine experimentally the optimal histone:DNA ratio for the desired level of nucleosome occupancy with each new preparation of DNA or histone stock. To this end, we recommend performing a titration with variations of about 10% in the histone:DNA mass ratio in a series of chromatin reconstitution reactions. For instance, in HDR-mediated gene editing with the CRISPR-Cas9 system and chromatin donor templates, we first reconstituted chromatin with histone:DNA mass ratios of 0.80:1.0, 0.90:1.0, 1.0:1.0, 1.1:1.0, and 1.2:1.0. We then evaluated the quality of the reconstituted chromatin by MNase digestion analysis and used the chromatin with the most extensive arrays of nucleosomes (Cruz-Becerra and Kadonaga, 2020).
MNase digestion assay. In this assay, the partial digestion of chromatin with different concentrations of MNase (which cleaves the linker DNA between nucleosomes) reveals the formation of arrays of nucleosomes on the DNA template. After deproteinization, the resulting DNA fragments that are derived from the oligonucleosomes show a repeating ladder pattern on an agarose gel. The detection of a periodic array of oligonucleosomes, such as tetra- and pentanucleosomes, is an indication of high quality chromatin.
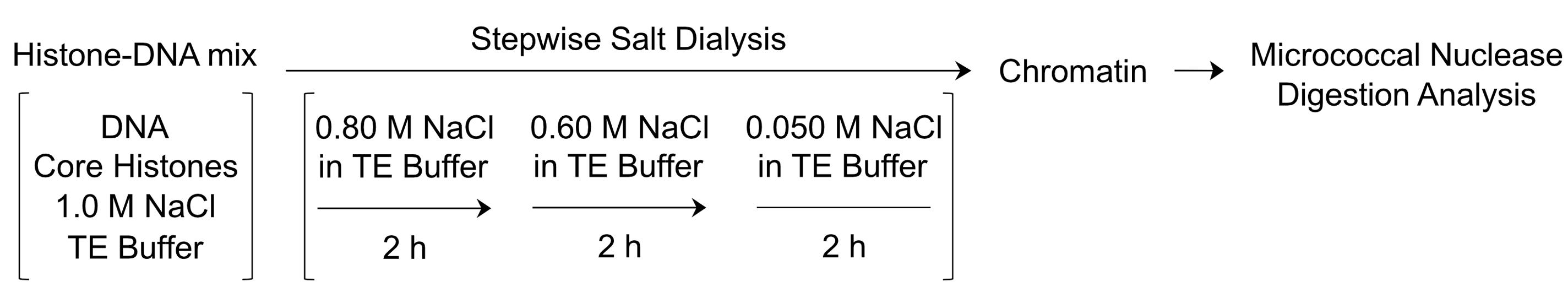
Figure 1. Schematic outline of the workflow in the reconstitution of chromatin by stepwise salt dialysis. The DNA and the histone octamers are combined in TE Buffer containing 1.0 M NaCl. Then, the histone-DNA mixture is dialyzed sequentially against TE Buffer containing 0.80 M, 0.60 M and 0.05 M NaCl. After dialysis, the quality of the resulting chromatin is assessed by subjecting a portion of the sample to micrococcal nuclease digestion analysis.
In summary, we describe a protocol for the reconstitution of chromatin by simple stepwise salt dialysis. This protocol can be completed in about 12 h (total time for chromatin reconstitution and analysis), and can be used with core histones from different sources and DNA prepared by standard laboratory procedures. In addition to the reconstitution of nucleosomes onto circular plasmid DNA, this method can be used for the preparation of mononucleosomes or polynucleosomes with linear DNA of different lengths (Chavez et al., 2019).
Materials and Reagents
200 µl barrier tips (Denville Scientific, catalog number: P1122 )
20 µl barrier tips (Denville Scientific, catalog number: P1121 )
10 µl barrier tips (Denville Scientific, catalog number: P1096-FR )
1.7 ml low binding microcentrifuge tubes (Sorenson Bioscience, catalog number: 39640T )
0.65 ml safeseal low binding microcentrifuge tubes (PGC Scientific, catalog number: 505-195 )
Note: This product has been discontinued and should be substituted by other 0.65 ml low binding microcentrifuge tubes (e.g., Sorenson Bioscience, catalog number: 11300 ).Microcentrifuge tube lid locks (VWR, catalog number: 14229-941 )
Dialysis membrane, 3.5 kDa molecular weight cutoff (Spectrum, catalog number: 132592 ) or Slide-A-Lyzer MINI dialysis device (Thermo Fisher Scientific, catalog number: 69550 )
Liquid nitrogen
Micrococcal nuclease (Sigma, catalog number: N5386 )
Proteinase K (Roche, catalog number: 03115879001)
123 bp DNA ladder (Sigma, catalog number: D5042 )
Glycogen (Sigma, catalog number: G-0885 )
Sodium chloride (Fisher Scientific, catalog number: S671-3 )
Orange G sodium salt (Sigma, catalog number: O-3756 )
Calcium chloride dihydrate (Fisher Scientific, catalog number: C79-500 )
HEPES (Fisher Scientific, catalog number: BP-310-1 )
Potassium chloride (Fisher Scientific, catalog number: P-217-500 )
Magnesium chloride hexahydrate (Fisher Scientific, catalog number: M33-500 )
EGTA (Sigma, catalog number: E-4378 )
Ammonium acetate (Fisher Scientific, catalog number: A637-500 )
Glycerol (Fisher Scientific, catalog number: G33-20 )
Sodium dodecyl sulfate (Bio-Rad, catalog number: 161-0302 )
Tris base (Fisher Scientific, catalog number: BP152-5 )
EDTA disodium salt dihydrate (Macron Fine Chemicals, catalog number: 4931-04 )
Phenol:chloroform:isoamyl alcohol (25:24:1, v/v/v) (Thermo Fisher Scientific, catalog number: J75831-AN )
Ethanol (Koptec, catalog number: V1016 )
Agarose (Promega, catalog number: V3125 )
Boric acid (Fisher Scientific, catalog number: BP-168-500 )
Ethidium bromide (Sigma, catalog number: E-8751 )
Glass distilled water
Plasmid DNA prepared with the HiSpeed Plasmid Maxi Kit (QIAGEN, catalog number: 12662 )
QIAquick Gel Extraction Kit (QIAGEN, catalog number: 28704 )
Note: This product is required only when the DNA is prepared by PCR amplification.
Histone octamer stock (BPS Bioscences, catalog number: 52065 ; or purified by the methods cited above)
TE Buffer, pH 8.0 (see Recipes)
10× TE Buffer, pH 8.0 (see Recipes)
TBE Buffer (see Recipes)
10× TBE Buffer (see Recipes)
Orange G Loading Buffer (see Recipes)
200 U/ml MNase stock (see Recipes)
MNase Buffer R (see Recipes)
Stop Buffer (see Recipes)
HEG Buffer (see Recipes)
10 mg/ml Ethidium bromide stock solution (see Recipes)
Equipment
Ice bucket
Liquid nitrogen Dewar flask
Magnetic stir bars
1 L cylinders (Pyrex, catalog number: 3022 )
1 L beakers (Pyrex, catalog number: 1000 )
Fine tip transfer pipettes (Samco Scientific, catalog number: 231 )
10 μl Micropipette (Rainin, model: SL 10-XLS)
20 μl Micropipette (Rainin, model: SL 20)
200 μl Micropipette (Rainin, model: SL 200)
Magnetic stirrer (Fisher Scientific, catalog number: 11-500-75 )
Centrifuge (Eppendorf, model: 5424 )
Minicentrifuge (RPI, model: PMC-060 )
Vortex mixer (Fisher, catalog number: 12-812 )
Horizontal gel electrophoresis apparatus (Gibco, model: H5 )
Electrophoresis power supply (Bio-Rad, catalog number: Power Pac 200)
UV transilluminator (Protein Simple, model: Alpha Imager HP)
Procedure
Reconstitution of chromatin
Here we describe the protocol for a standard chromatin reconstitution experiment. In this protocol, it is assumed that the optimal amounts of histones and DNA have been previously established.
To determine the optimal histone:DNA ratio with new preparations of histones and/or DNA, this protocol can be performed at 0.5× scale (that is, each reaction has 25 µg of DNA in 75 µl final volume instead of 50 µg of DNA in 150 µl final volume) in a series of reactions with different histone:DNA ratios. For this purpose, we typically carry out reactions with histone:DNA mass ratios of 0.80:1.0, 0.90:1.0, 1.0:1.0, 1.1:1.0 and 1.2:1.0.
For each reconstitution reaction, prepare a 150 µl histone-DNA mix that contains 50 µg of DNA and the desired mass of histone octamers in TE Buffer (pH 8.0) containing a final concentration of 1.0 M NaCl. Mix gently. Then, incubate for 15 min on ice.
It is important to consider the salt contribution from the histone stock in the calculations of the final concentration of NaCl in the histone-DNA mixture. For example, a reconstitution reaction with a desired final histone:DNA ratio of 1.0:1.0 could be prepared from 42 µl of a 1.2 mg/ml DNA stock (50 µg DNA), 42 µl of a 1.2 mg/ml histone stock containing 2.5 M NaCl (50 µg histones), 9.0 µl of 5.0 M NaCl (to give 1.0 M NaCl final concentration), 15 µl of 10× TE Buffer (i.e., 1× TE final), and 42 µl of water in a final volume of 150 µl.
Load the histone-DNA mix from Step A1 into a dialysis chamber.
The dialysis chamber consists of the sample reservoir, the dialysis membrane, and a sealing ring (Figure 2). Assemble the dialysis chamber as follows:
Cut a microcentrifuge tube (please see Note 8) such that the lid of the tube (i.e., the sample reservoir) is surrounded by a ring (i.e., the sealing ring).
Detach the sample reservoir from the sealing ring by cutting the linker between them. Then pull the two pieces apart.
Cut a square piece (about 1.5 cm × 1.5 cm) of dialysis membrane and rinse it with water to remove the storage solution. Then, immerse the membrane in TE Buffer containing 0.80 M NaCl.
Load the sample into the reservoir with a 200 µl pipette.
Drain the excess buffer from the membrane by gently tapping the edge over a paper towel.
Cover the sample with the membrane.
Close the chamber with the sealing ring (i.e., place the ring on top of the membrane, then push it down over the sample reservoir piece).
Fasten the dialysis chamber with a lid lock, and immediately immerse it into TE Buffer containing 0.80 M NaCl. The dialysis membrane should be in direct contact with the dialysis buffer (i.e., the membrane side of the dialysis chamber faces down).
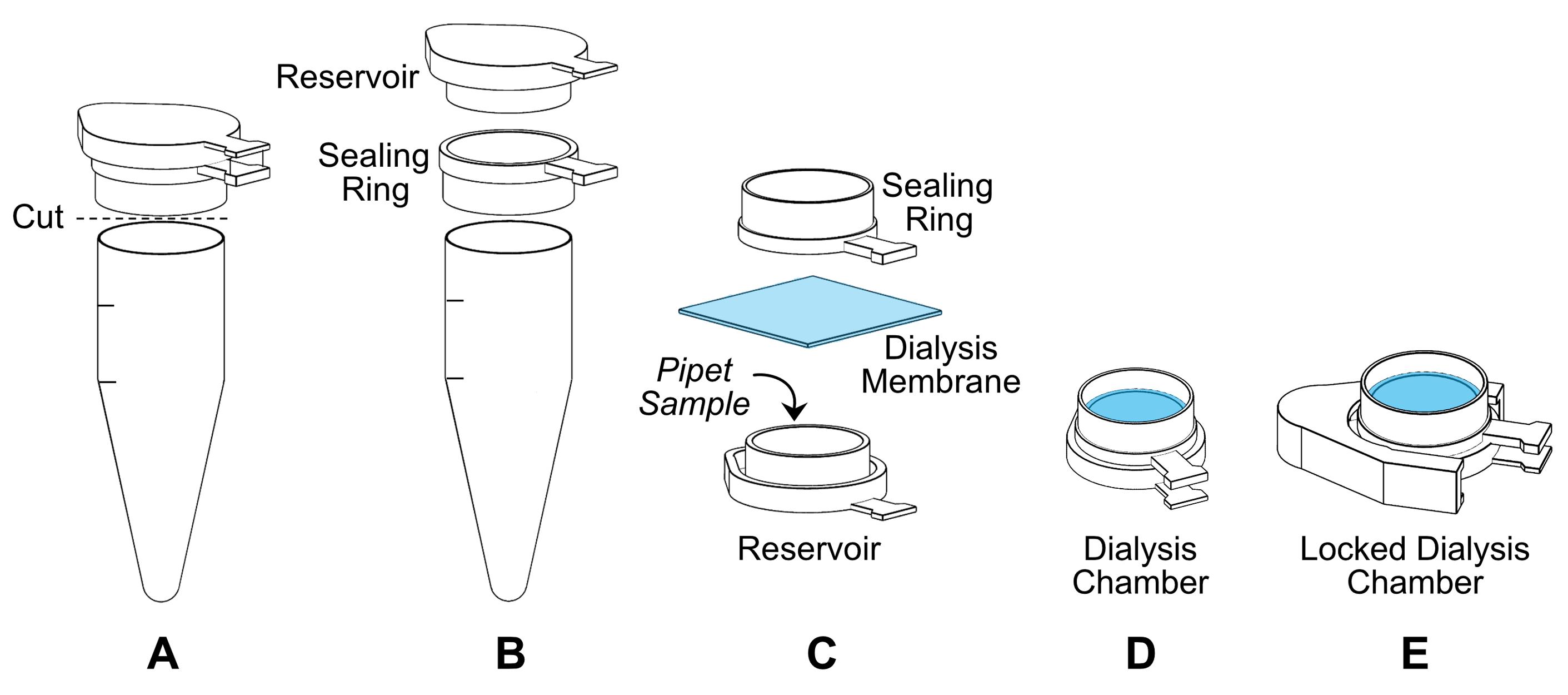
Figure 2. Diagram of the procedure for the assembly of a dialysis chamber. The dialysis chamber is prepared from a microcentrifuge tube and a small piece of dialysis membrane as follows. A. Cut the top part of a microcentrifuge tube. B. Separate the lid and the portion of the tube that remained attached. The lid serves as the sample reservoir and the cropped tube as the sealing ring. C. Pipet 150 μl of sample into the reservoir. Cover the sample with a small square of dialysis membrane (denoted in blue). Then, place the sealing ring on top of the membrane. Close the dialysis chamber by pushing the sealing ring down over the reservoir. D. Fully assembled dialysis chamber. E. Secure the dialysis chamber with a lid lock.Dialyze at room temperature (22 °C) in a stepwise manner against decreasing salt concentrations as follows:
1 L of TE Buffer containing 0.80 M NaCl for 2 h.
1 L of TE Buffer containing 0.60 M NaCl for 2 h.
1 L of TE Buffer containing 0.05 M NaCl for 2 h.
Notes:
When transferring the sealed dialysis chamber between the different buffers, it is not necessary to remove the small amount of excess buffer that is associated with the outside of the dialysis chamber.
Several reconstitution reactions can be dialyzed simultaneously in a beaker containing 1 L of dialysis buffer at medium speed on a magnetic stirrer. The dialysis chambers should be gently swirling around the top of the buffer during dialysis.
Remove the dialysis chamber from the TE Buffer containing 0.050 M NaCl and place it over a paper towel with the membrane side facing up.
Remove the remaining excess TE Buffer on the outside of the membrane with a 200 µl pipette. Then, with a new tip, carefully perforate the membrane and transfer the dialysate (i.e., reconstituted chromatin) to a microcentrifuge tube.
Store the chromatin at 4 °C. The chromatin is stable at 4 °C for several months. Do not freeze the chromatin.
Quantification of the DNA concentration in the salt dialyzed chromatin
Measure the volume of the sample after dialysis by using a 200 μl pipette. With this method, the volume increases by about 7% of the original reaction volume during the salt dialysis (i.e., in chromatin reconstitution reactions performed with initial volumes of 150 μl and 75 μl, we typically recover about 160 μl and 80 μl, respectively).
Quantify the DNA by agarose gel electrophoresis followed by analysis with ImageJ as follows.
Prepare 40, 20, and 10 ng/μl standards (in TE Buffer containing 0.05 M NaCl) of the DNA stock that was used for reconstituting the chromatin. These standards will be used to make a DNA standard curve to determine the amount of DNA in the chromatin sample (Step B2q).
Dilute 2.0 μl of the reconstituted chromatin in 14 μl of TE Buffer containing 0.050 M NaCl (i.e., 8-fold dilution). Then perform two 2-fold serial dilutions in the same buffer to give 16- and 32-fold dilutions of the reconstituted chromatin.
To 2.0 μl of the 40, 20, and 10 ng/μl DNA standards (from Step B2a), and 2.0 μl of the 8-, 16-, and 32-fold dilutions of the chromatin (from Step B2b), add 48 μl of HEG Buffer to give a final volume of 50 μl of each sample.
To each 50 μl sample, add 100 μl of Stop Buffer and 5.0 μl of 2.5 mg/ml proteinase K. Mix gently. Incubate 30 min at 37 °C.
Add 155 μl of phenol:chloroform:isoamyl alcohol and vortex vigorously for 30 s.
Centrifuge at 16,000 × g for 5 min.
Transfer 140 μl of the aqueous phase to a new microcentrifuge tube.
To the aqueous phase, add 25 μl of 2.5 M ammonium acetate and 600 μl of 100% ethanol. Mix by inverting the tube several times.
Centrifuge at 16,000 × g for 20 min.
Remove the supernatant by using a transfer pipette. Then, spin the tubes briefly in a minicentrifuge and remove any leftover liquid with a 10 μl pipette tip.
Air dry the pellet for 5 min.
To the pellet, add 5.0 μl of Orange G Loading Buffer. Incubate for 5 min. Vortex gently, then spin briefly.
Subject the DNA to electrophoresis on a 1.0% agarose-TBE gel at 3.7 V/cm until the Orange G dye reaches the bottom of the gel. The running time will vary according to the gel dimensions. As an example, a gel that is 5 cm × 6 cm × 0.5 cm (width × length × height) will take about 40 min at 100 V in a horizontal electrophoresis chamber in which the anode and the cathode are separated by 27 cm. To estimate the total voltage, multiply the distance between the electrodes in the electrophoresis chamber by the desired voltage (V) per centimeter (cm). In this example, 27 cm × 3.7 V/cm = 100 V.
Stain the gel for 10 min in EB staining solution.
Rinse the gel with water for 10 min.
Visualize the DNA with a UV transilluminator and record an image of the DNA in TIF format for quantitative analysis.
Quantify the DNA on the gel by using the Gel Analysis function of the ImageJ software (National Institutes of Health, Bethesda, MD) as described elsewhere (https://imagej.nih.gov/ij/docs/menus/analyze.html#gels).
Note: Adjust the calculated DNA amount on the gel according to the dilution factor that corresponds to each sample.
To obtain the DNA concentration in the chromatin sample, divide the calculated total amount of DNA by the volume of sample recovered after the dialysis steps (from Step B1).
With the chromatin reconstitution procedure described in Section A, we generally recover greater than 90% of the starting amount of DNA (Figure 3). For many applications, the nucleosomal DNA concentrations can be estimated (within 10% of the method described below and shown in Figure 3) by the A260nm readings (using the extinction coefficient for pure double-stranded DNA) on a NanoDrop OneC spectrophotometer (model ND-ONEC-W; Thermo Fisher Scientific, Waltham, MA). This is probably due to the low A260nm absorbance of the histones because of their low content of aromatic amino acid residues. Alternatively, the nucleosomal DNA concentrations can be determined by removal of the histones and quantification of the resulting DNA samples, as described below. In contrast, the use of a Qubit Fluorometer is not recommended because it does not provide accurate quantification of the nucleosomal DNA in the chromatin sample, probably because the binding of fluorescent dye molecules to nucleosomal DNA is less efficient than their binding to free DNA.
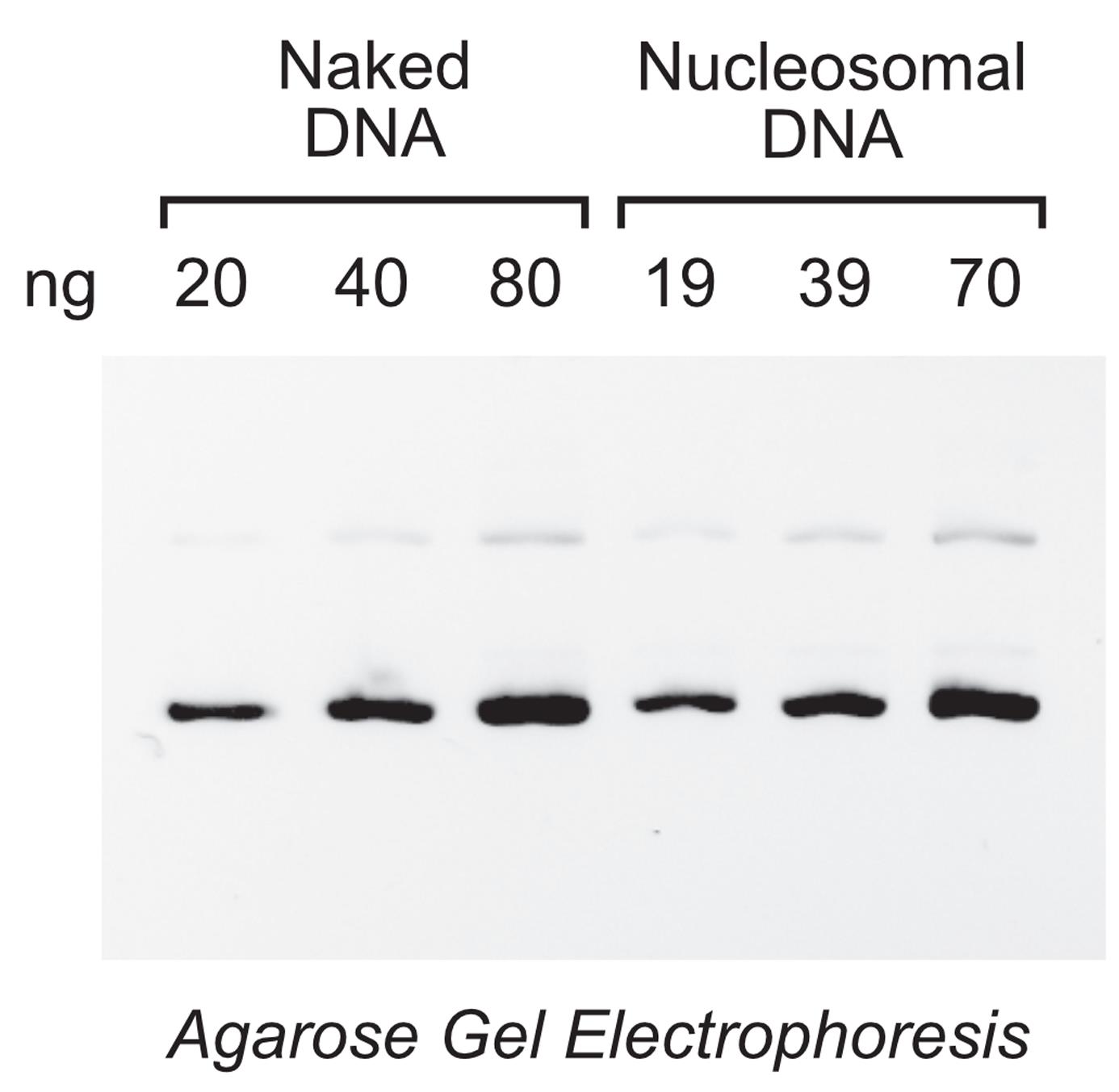
Figure 3. Quantification of the DNA in chromatin reconstituted by salt dialysis. A sample of salt dialyzed chromatin was diluted 8-, 16-, and 32- fold in TE Buffer containing 0.05 M NaCl. After deproteinization of a 2.0 μl aliquot of each of the three chromatin dilutions, the resulting DNA samples, along with DNA standards of known concentration (20, 40 and 80 ng naked DNA), were subjected to agarose gel electrophoresis analysis and visualized by staining with ethidium bromide (EB). The numbers under Nucleosomal DNA correspond to the calculated amounts of DNA in 2.0 µl of the 32-, 16-, and 8- fold dilutions (from left to right) of the chromatin sample after quantification by using the Gel Analysis function of the ImageJ software (NIH, Bethesda, MD). In this experiment, the reconstituted chromatin contained about 0.30 µg/µl DNA in 160 µl final volume (i.e., 48 µg of DNA, which corresponds to about 96% of the starting amount in the chromatin reconstitution reaction).
MNase digestion analysis of the reconstituted chromatin
The quality of the reconstituted chromatin is assessed by using the MNase digestion assay (Figures 4 and 5).
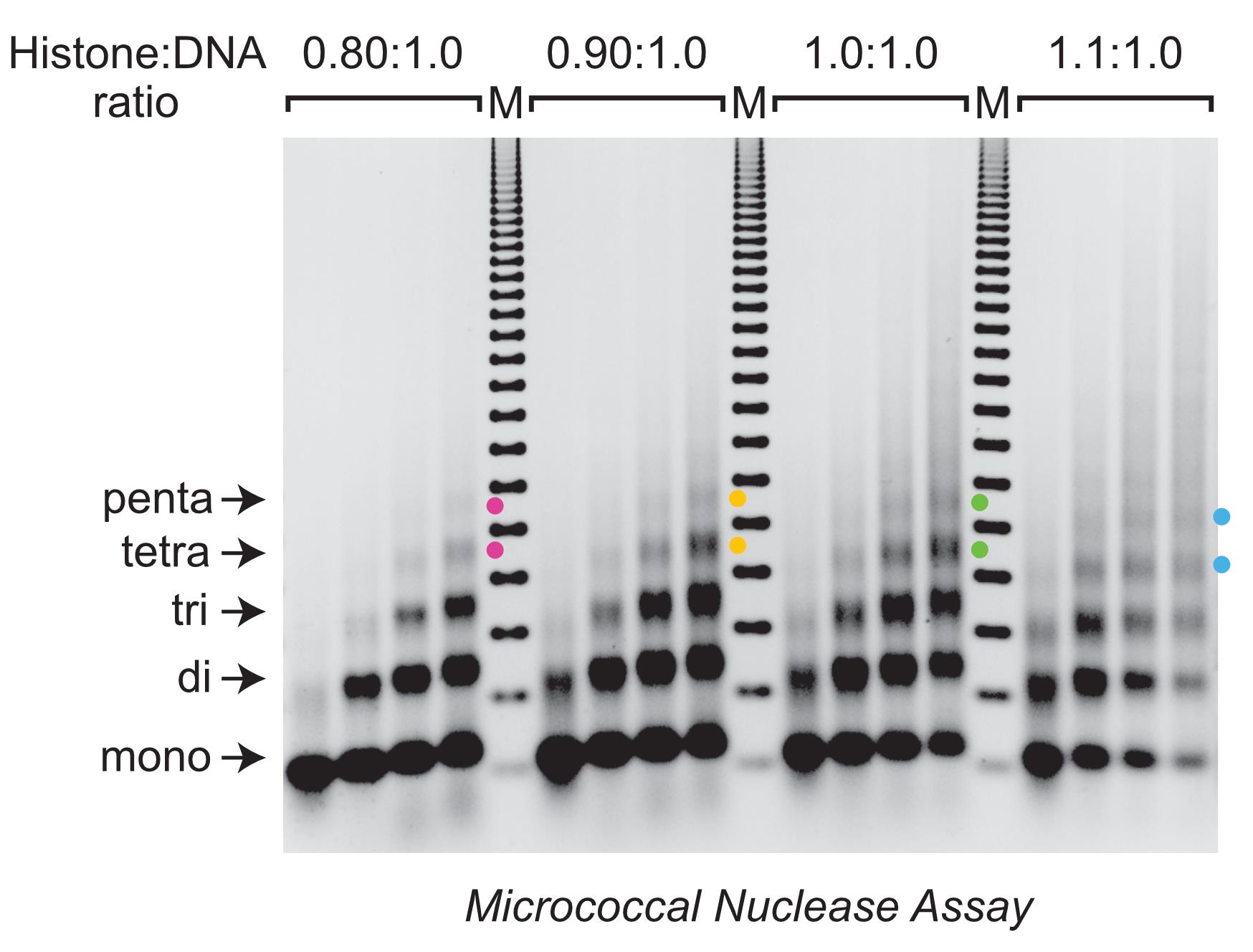
Figure 4. Determination of the optimal histone:DNA ratio for efficient chromatin reconstitution by salt dialysis. Chromatin was reconstituted with histone:DNA mass ratios of 0.80:1.0, 0.90:1.0, 1.0:1.0, and 1.1:1.0 (i.e., 25 µg of DNA with 20 µg, 22.5 µg, 25 µg, and 27.5 µg of histone octamers, respectively) by using the salt dialysis methodology outlined in Figure 1. To evaluate the quality of the reconstituted chromatin, the samples were subjected to partial digestion with four different concentrations of MNase. After deproteinization, the resulting DNA fragments were resolved by agarose gel electrophoresis and visualized by staining with ethidium bromide (EB). The arrows indicate the DNA bands that correspond to mono-, di-, tri-, tetra-, and pentanucleosomes. In this experiment, there are higher proportions of tetra- and pentanucleosomal DNA fragments at histone:DNA ratios of 0.90:1.0 (yellow dots) and 1.0:1.0 (green dots) than at 0.80:1.0 (pink dots) or 1.1:1.0 (blue dots). For many applications, such as HDR-mediated gene editing (Cruz-Becerra and Kadonaga, 2020), it is best to use chromatin with the lowest histone:DNA ratio that yields the most extensive nucleosome arrays. In this case, the 0.90:1.0 chromatin and the 1.0:1.0 chromatin are of comparable quality and are better than the 0.80:1.0 chromatin and the 1.1:1.0 chromatin, as assessed by the MNase analysis. Therefore, the recommended histone:DNA ratio is 0.90:1.0. The DNA size markers (M) are the 123 bp ladder (Millipore Sigma), which consists of bands that are integral multiples of 123 bp. The smallest 123 bp band migrates slightly faster than the mononucleosome DNA band.
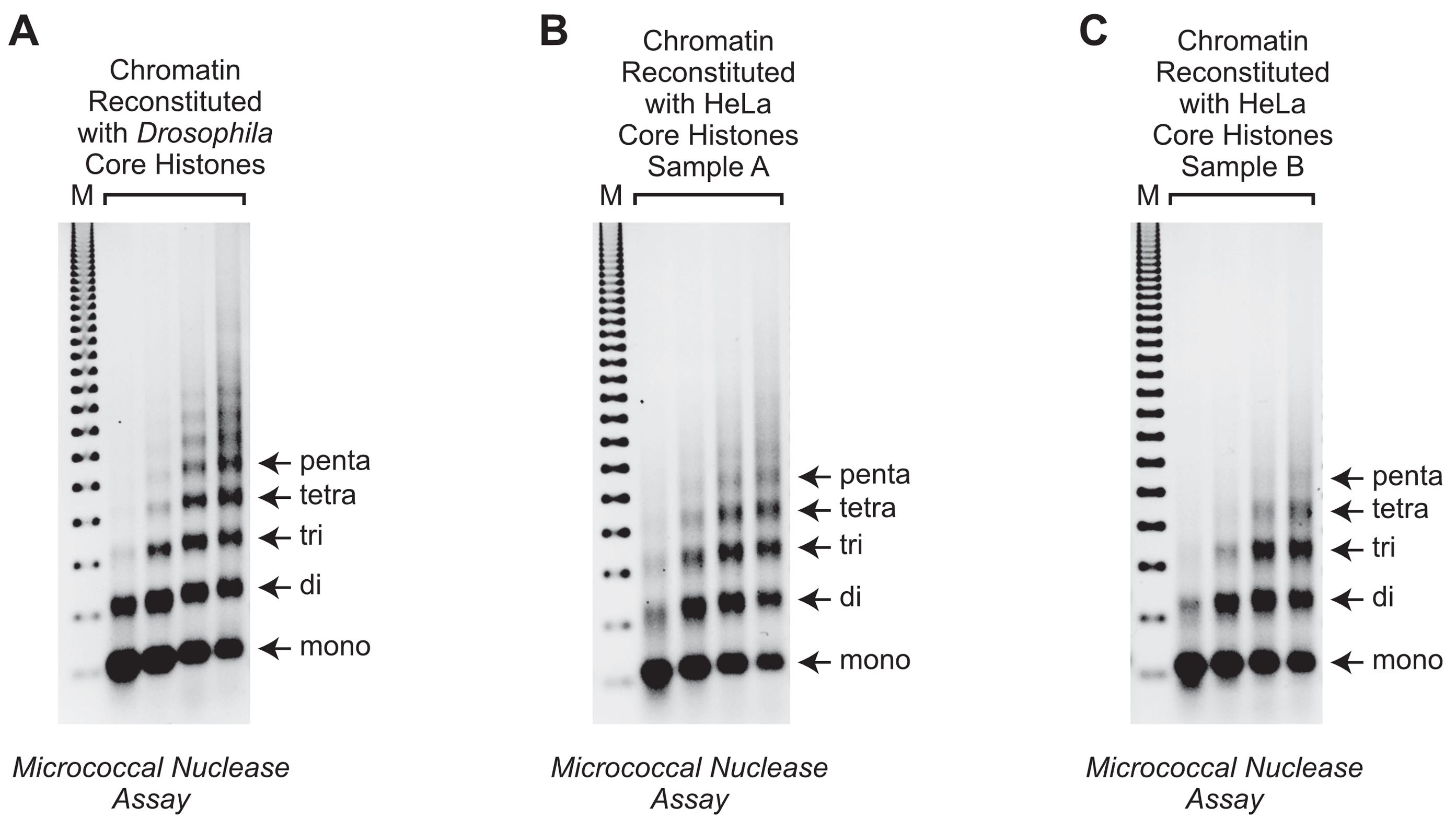
Figure 5. MNase digestion analysis of salt dialyzed chromatin. Chromatin was reconstituted with three different preparations of core histones and the same plasmid DNA template (pBKS-GAPDH-HDR; 5.6 kbp; Cruz-Becerra and Kadonaga, 2020) by using the salt dialysis methodology outlined in Figure 1. A. Chromatin prepared with core histones from Drosophila melanogaster (Dm) 0-12 h embryos. The histones were purified by using the method of Fyodorov and Levenstein (2002). B. Chromatin reconstituted with HeLa core histones prepared as described in Khuong et al. (2017) (denoted as Sample A). C. Chromatin assembled with HeLa core histones obtained from BPS Bioscience (San Diego, CA) (denoted as Sample B). After partial digestion of the chromatin samples with four different concentrations of MNase, the samples were deproteinized, and the resulting DNA fragments were resolved by agarose gel electrophoresis and visualized by staining with ethidium bromide (EB). The arrows indicate the DNA bands that correspond to mono-, di-, tri-, tetra-, and pentanucleosomes. The DNA size markers (M) are the 123 bp ladder (Millipore Sigma), which consists of bands that are integral multiples of 123 bp. The smallest 123 bp band migrates slightly faster than the mononucleosome DNA band.
Perform partial MNase digestion of the reconstituted chromatin (from Step A5) with four different concentrations of enzyme. Each MNase digestion reaction contains 35 µl of chromatin (5.7 ng/µl DNA; 200 ng total DNA), 9.0 µl of 10 mM CaCl2, and 5.0 µl of MNase at various concentrations (e.g., 1.3, 0.67, 0.33, and 0.17 mU/μl; the optimal concentrations will vary with different preparations of MNase) in a final volume of 49 µl. The reactions are set up as follows:
Dilute an aliquot of chromatin to 5.7 ng/μl DNA in HEG Buffer in 175 µl final volume.
Thaw an aliquot of a 200 U/ml MNase stock in a container with water at 4 °C. After thawing, keep the enzyme stock on ice.
Make MNase working stocks that contain 1.3, 0.67, 0.33, and 0.17 mU/μl in MNase Buffer R as follows:
Dilute the 200 U/ml enzyme stock to 13 mU/μl in cold MNase Buffer R (e.g., 2.0 ul of 200 U/ml MNase in 30 μl final volume).
Make a 10-fold dilution of 13 mU/μl MNase to obtain 1.3 mU/μl.
Perform three 2-fold serial dilutions starting with 1.3 mU/μl MNase to obtain 0.67, 0.33, and 0.17 mU/μl.
Immediately before use, combine 5.0 μl of the MNase working stock with 9.0 μl of 10 mM CaCl2 to give a final volume of 14 μl MNase-CaCl2 mix. Note that it is necessary to prepare a different MNase-CaCl2 mix for each of the four MNase working stocks from Step C1c in this section. If several chromatin samples will be analyzed simultaneously, prepare the required volume of each MNase-CaCl2 mix for the total number of samples plus one (e.g., for four chromatin samples, prepare 5 × 14 µl = 70 μl of each MNase-CaCl2 mix).
Add 14 μl of MNase-CaCl2 mix to 35 μl of chromatin (5.7 ng/μl DNA; from Step C1a in this section) to give a final volume of 49 µl. Mix gently, then spin briefly. Incubate exactly 10 min. Repeat this step with each MNase-CaCl2 mix (and each chromatin sample).
Stop the MNase digestion reactions by adding 7.0 μl of 0.50 M EDTA (to 63 mM EDTA final concentration) to each tube. Mix gently.
Recover the DNA as follows.
Add 100 μl of Stop Buffer and 5.0 μl of 2.5 mg/ml proteinase K. Mix gently. Incubate 40 min at 37 °C.
Add 155 μl of phenol:chloroform:isoamyl alcohol and vortex vigorously for 30 s.
Centrifuge at 16,000 × g for 5 min.
Transfer 140 μl of the aqueous phase to a new microcentrifuge tube.
To the aqueous phase, add 25 μl of 2.5 M ammonium acetate and 600 μl of 100% ethanol. Incubate for 10 min.
Centrifuge at 16,000 × g for 20 min.
Remove the supernatant by using a transfer pipette. Then, spin the tubes briefly in a minicentrifuge and remove any leftover liquid with a pipette tip.
Air dry the pellet for 5 min.
Resolve the DNA fragments by agarose gel electrophoresis.
To the pellet, add 5.0 μl of Orange G Loading Buffer. Incubate for 5 min. Vortex gently, then spin briefly.
Subject the DNA to electrophoresis on a 1.3% agarose-TBE gel at 3.3 V/cm until the Orange G dye reaches the bottom of the gel (this will take about 2.5 h with a gel that is 11 cm, 14 cm, and 0.6 cm in width, length, and height, respectively).
Visualize the DNA by EB staining.
Stain the gel for 30 min in EB staining solution.
Remove the excess EB with water (typically, two 10 min rinses in water).
Visualize the DNA with a UV transilluminator.
Notes
The catalog numbers, models, and manufacturers that are given in the Materials and Reagents and Equipment sections are those of specific items that we have used to perform this method. This protocol could be performed with comparable items from other sources.
Before performing chromatin reconstitution experiments, we recommend analyzing the quality of the histone and DNA preparations by SDS-polyacrylamide and agarose gel electrophoresis, respectively. For the histones, an equimolar stoichiometry of all four histones should be observed with no detectable degradation or contamination. The plasmid DNA should be mostly supercoiled DNA and should not contain bacterial genomic DNA.
DNA and histone stocks of ≥ 1 mg/ml concentration are recommended.
Avoid contamination of the materials and reagents with nucleases and proteases. Use nuclease- and protease-free laboratory plasticware and glassware, and wear clean gloves throughout the entire procedure.
Use only low binding microcentrifuge tubes to reduce the adhesion of the histones and DNA to the plastic.
Mix the reaction reagents by flicking the tube, except when vortexing is indicated.
Perform all the steps at room temperature (22 °C) except when stated otherwise.
Chromatin can be reconstituted in reaction volumes of 150 µl and 75 µl with similar results. We use dialysis chambers prepared from 1.7 ml or 0.65 ml low binding tubes for 150 µl or 75 µl reconstitution reactions, respectively. Do not use smaller reaction volumes with these dialysis chambers.
As an alternate option, the Slide-A-Lyzer MINI dialysis units from Thermo Fisher Scientific (catalog number: 69550 , Thermo Fisher Scientific, Waltham, MA) could be used instead of the dialysis chambers described here.
We recommend that the conductivity of the reconstituted chromatin sample is measured to determine the completion of dialysis. The conductivity of the reconstituted chromatin at the end of dialysis should be within 10% of the conductivity of TE Buffer containing 0.05 M NaCl.
Recipes
TE Buffer, pH 8.0
10 mM Tris
1.0 mM EDTA
10× TE Buffer, pH 8.0
100 mM Tris
10 mM EDTA
TBE Buffer
89 mM Tris base
89 mM Boric acid
2.0 mM EDTA
10× TBE Buffer
0.89 M Tris base
0.89 M Boric acid
20 mM EDTA
Orange G Loading Buffer
6.25% (v/v) glycerol with Orange G (add Orange G dye until the solution is medium-dark orange)
1× TBE Buffer
200 U/ml MNase stock
Resuspend the lyophilized enzyme in 5.0 mM sodium phosphate, pH 7.0 containing 2.5 µM of CaCl2
Make small aliquots
Quick-freeze in liquid nitrogen and store at -80 °C
MNase Buffer R
10 mM K+-HEPES, pH 7.6
10 mM KCl
1.5 mM MgCl2
0.50 mM EGTA
10% (v/v) glycerol
Stop Buffer
20 mM EDTA
0.20 M NaCl
1.0% (w/v) SDS
0.25 mg/ml glycogen
HEG Buffer
25 mM K+-HEPES, pH 7.6
0.10 mM EDTA
10% (v/v) glycerol
10 mg/ml Ethidium bromide stock solution
Dissolve 100 mg of ethidium bromide in 10 ml of water
Store at 4 °C; protect from light
1.0 µg/ml Ethidium bromide staining solution
Perform a 10,000-fold dilution of 10 mg/ml ethidium bromide stock solution in water
Acknowledgments
We thank all of the previous members of the Kadonaga laboratory whose biochemical work on chromatin assembly has served as the reference for the protocol described here. We are also grateful to Sharon Torigoe, George Kassavetis, Long Vo ngoc, and Claudia Medrano for critical reading of the manuscript, and to Daniel Zamorano for helping with Figure 2. J.T.K is the Amylin Chair in the Life Sciences. G.C.-B. is a recipient of a Pew Latin American Postdoctoral Fellowship, and the University of California at San Diego Molecular Biology Cancer Fellowship. This work was supported by a grant from the National Institutes of Health (R35 GM118060) to J.T.K. The protocol described here was used to reconstitute chromatin donor templates for HDR-mediated gene editing via the CRISPR-Cas9 system in Cruz-Becerra and Kadonaga (2020).
Competing interests
The authors declare no competing financial interests.
References
- Bulger, M. and Kadonaga, J. T. (1994). Biochemical reconstitution of chromatin with physiological nucleosome spacing. Methods Mol Gen 5: 241-262.
- Chavez, C., Cruz-Becerra, G., Fei, J., Kassavetis, G.A. and Kadonaga, J. T. (2019). The tardigrade damage suppressor protein binds to nucleosomes and protects DNA from hydroxyl radicals. Elife 8: e47682.
- Cruz-Becerra, G. and Kadonaga, J. T. (2020). Enhancement of homology-directed repair with chromatin donor templates in cells.Elife 9: e55780.
- Fei, J., Ishii, H., Hoeksema, M. A., Meitinger, F., Kassavetis, G. A., Glass, C. K., Ren, B. and Kadonaga, J. T. (2018). NDF, a nucleosome-destabilizing factor that facilitates transcription through nucleosomes. Genes Dev 32(9-10): 682-694.
- Fei, J., Torigoe, S.E., Brown, C.R., Khuong, M.T., Kassavetis, G.A., Boeger, H. and Kadonaga, J.T. (2015). The prenucleosome, a stable conformational isomer of the nucleosome. Genes Dev 29(24): 2563-2575.
- Fyodorov, D. V. and Levenstein, M. E. (2002). Chromatin assembly using Drosophila systems. Curr Protoc Mol Biol Chapter 21: 7.
- Jeong, S. W., Lauderdale, J. D. and Stein, A. (1991). Chromatin assembly on plasmid DNA in vitro. Apparent spreading of nucleosome alignment from one region of pBR327 by histone H5 . J Mol Biol 222(4): 1131-1147.
- Kadonaga, J. T. (2019). The transformation of the DNA template in RNA polymerase II transcription: a historical perspective.Nat Struct Mol Biol 26(9): 766-770.
- Khuong, M. T., Fei, J., Cruz-Becerra, G. and Kadonaga, J. T. (2017). A simple and versatile system for the ATP-dependent assembly of chromatin.J Biol Chem 292(47): 19478-19490.
- Laybourn, P. J. and Kadonaga, J. T. (1991). Role of nucleosomal cores and histone H1 in regulation of transcription by RNA polymerase II.Science 254(5029): 238-245.
- Lusser, A. and Kadonaga, J.T. (2004). Strategies for the reconstitution of chromatin. Nat Methods 1(1): 19-26.
- Peterson, C. L. and Hansen, J. C. (2008). Chicken erythrocyte histone octamer preparation. CSH Protoc: pdb.prot 5112.
- Rattner, B. P., Yusufzai, T. and Kadonaga, J. T. (2009). HMGN proteins act in opposition to ATP-dependent chromatin remodeling factors to restrict nucleosome mobility. Mol Cell 34(5): 620-626.
- Stein, A. (1989). Reconstitution of chromatin from purified components. Methods Enzymol 170: 585-603.
- Torigoe, S. E., Patel, A., Khuong, M. T., Bowman, G. D. and Kadonaga, J. T. (2013). ATP-dependent chromatin assembly is functionally distinct from chromatin remodeling.Elife 2: e00863.
Article Information
Copyright
![]() Cruz-Becerra and Kadonaga. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Cruz-Becerra and Kadonaga. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Cruz-Becerra, G. and Kadonaga, J. T. (2021). Reconstitution of Chromatin by Stepwise Salt Dialysis. Bio-protocol 11(7): e3977. DOI: 10.21769/BioProtoc.3977.
- Cruz-Becerra, G. and Kadonaga, J. T. (2020). Enhancement of homology-directed repair with chromatin donor templates in cells.Elife 9: e55780.
Category
Molecular Biology > DNA
Biochemistry > DNA
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.