- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Novel Method to Construct Binary CRISPR Vectors for Plant Transformation by Single Round of PCR Amplification
Published: Vol 11, Iss 7, Apr 5, 2021 DOI: 10.21769/BioProtoc.3971 Views: 6005
Reviewed by: Alessandro DidonnaKangquan YinJonathan R Gilkerson
Original research article
The authors used this protocol in:

Jan 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
CRISPR/Cas9 is an established and flexible tool for genome editing. However, most methods used to generate expression clones for the CRISPR/Cas9 are time-consuming. Hence, we have developed a one-step protocol to introduce sgRNA expression cassette(s) directly into binary vectors (Liu et al., 2020). In this approach, we have optimized the multiplex PCR to produce an overlapping PCR product in a single reaction to generate the sgRNA expression cassette. We also amplified two sgRNA expression cassettes through a single round of PCR. Then, the sgRNA expression cassette(s) is cloned into the binary vectors in a Gateway LR or Golden gate reaction. The system reported here provides a much more efficient and simpler procedure to construct expression clones for CRISPR/Cas9-mediated genome editing. In this protocol, we describe the detailed step-by-step instructions for using this system.
Keywords: Cloning systemBackground
Bacteria defend against viruses through a protein system, consisting of the clustered regularly interspaced short palindromic repeat (CRISPR), the CRISPR-associated (Cas) protein, CRISPR RNAs (crRNAs) and trans-encoded crRNA (tracrRNA). Researchers have now developed this system into a key tool for targeted genome editing. CRISPR – binary vectors express two elements – the sgRNAs with a target sequence (target-sgRNAs) and Cas9 protein – to cleave target genomic regions. Feng et al. (2013) have constructed gateway vectors to co-express Cas9 and sgRNAs in plants through Agrobacterium sp.-mediated transformation. In a restriction-ligation reaction, a gene-specific sgRNA spacer substitutes the target region in the entry clone, which encodes attL recombination sites. Then, an “LR Clonase” reaction transfers the target-sgRNA cassette into a destination clone, which contains a Cas9 expression cassette (Feng et al., 2013). Ma et al. (2016) have developed this basic system into a CRISPR/Cas9 system for multiplex genome editing in rice. In this approach, a restriction-ligation reaction inserts a spacer into intermediate vectors to produce an sgRNA expression cassette, which fuses with adaptors for Golden Gate cloning or Gibson Assembly (Ma et al., 2016). In an alternative approach, an overlapping PCR, with two rounds of reactions, can also establish a sgRNA expression cassette with adaptors. The sgRNA expression cassette can then be introduced into a binary vector via Golden Gate cloning or Gibson Assembly (Ma et al., 2016). However, traditional cloning, based on restriction-ligation reactions or two-round overlapping PCRs, is time-consuming.
Herein, we report a novel method to construct the binary vectors with one or two targets by a single round of PCR and a single LR reaction or Golden Gate cloning (Liu et al., 2020). Using this system, an expression clone can be constructed within 36 hours, which significantly improves efficiency and reduces costs.
Materials and Reagents
200 µl PCR tubes (Biosharp, catalog number: BS-02-P )
1.5 ml microcentrifuge tubes (Biosharp, catalog number: BS-15-M )
Pipette tips (Biosharp, catalog numbers: BS-10-T , BS-200-T , BS-1000-T )
Competent E. coli T1 cells (TransGen, catalog number: CD501-02 )
LR clonase (GatewayTM LR ClonaseTM Enzyme Mix, catalog number: 11791-043 )
NEB Cutsmart buffer (New England BioLabs, catalog number: B7204S )
NEBuffer 3.1 (New England BioLabs, catalog number: B7203S )
EcoRV (New England BioLabs, catalog number: R0195L )
BsaI-HF (New England BioLabs, catalog number: R3733L )
T4 DNA ligase (New England BioLabs, catalog number: M0202L )
PCR SuperMix (TransGen, catalog number: AS111-11 )
KOD FX (Toyobo, catalog number: KFX-101 )
dNTPs Mixture (2mM) (Toyobo, catalog number: NTP-201 )
Sterilized double distilled H2O (Phygene, catalog number: PH0727 )
DreamTaq or other equivalent DNA polymerase (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: K1072 )
Agarose (Biowest, catalog number: 111860 )
Plasmid prep mini kit (OMEGA, catalog number: D3350-01 )
Gel Extraction Kit (OMEGA, catalog number: D2500-01 )
Spectinomycin (Sigma-Aldrich, catalog number: PHR1441 )
Tris (Solarbio Life Scientific, catalog number: T8060 )
Acetic acid (MERCK, catalog number: M10006307 )
0.5 M EDTA (Solarbio Life Scientific, catalog number: E1170 )
Tryptone (Oxoid, catalog number: LP0042 )
Yeast extract (Oxoid, catalog number: LP0021 )
Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S5886 )
Ethidium bromide (EB) (Sigma-Aldrich, catalog number: E8751 )
Universal primers for PCR screening (see Table 1)
Table 1. Universal primers
Primers Sequences (5′ → 3′) OJP001 TCGCGTTAACGCTAGCATGGATCTC OJP002 GTAACATCAGAGATTTTGAGACAC OJP008 ACCACCTCGGCTATCCACA OJP026 ATAGCCTTATGCAGTTGCTCT OJP065 CGACTCGGTGCCACTTTTTC PJF997 (the donor vector containing OsU3-sgRNA expression cassette), PJF999 (the donor vector containing OsU6-sgRNA expression cassette), PJG090 (the donor vector for amplification of two spacers), PJG097 (the destination vector for one target), PJG112 (the destination vector for two targets). All the vectors were developed in our previous work (Liu et al., 2020)
50× TAE electrophoresis buffer (see Recipes)
LB medium (see Recipes)
LB agar medium (see Recipes)
Equipment
Pipettes (Eppendorf)
Microcentrifuge (Eppendorf, model: Centrifuge 5424 )
Heating block (Hangzhou Allsheng Instruments, model: MK-20 )
Thermal cycler (Thermo Fisher Scientific, Applied BiosystemsTM, model: Veriti® 96 well thermal cycler )
Water bath (Shanghai Binglin, model: BLHH-6N )
NanoDrop (Thermo Fisher Scientific, Thermo ScientificTM, model: NanoDropTM 2000 )
Gel Imaging System (The ChemiDoc XRS+ System, BIO-RAD, model: 1708265 )
Software
SnapGene (GSL Biotech LLC, https://www.snapgene.com/)
WPS Excel (Kingsoft Office, https://www.wps.cn/)
Procedure
Design sgRNA primers
Design sgRNA spacers using any online tools, such as CRISPR-P (http://crispr.hzau.edu.cn/cgi-bin/CRISPR2/CRISPR).
Note: The transcription of sgRNAs is derived by OsU3 or OsU6 promoters, which have different transcription initiation sites (the first nucleotide of spacer). The transcription initiation site of OsU3 promoter is nucleotide A, while that of OsU6 promoter is nucleotide G.
To construct binary vectors harboring one spacer starting with an ‘A’, add the following nucleotides to 3′ downstream of the sense (SS) and antisense (AS) sgRNA spacers to obtain sgRNA primers:
SS: 5′-gttttagagctatgctgaaa-3′
AS: 5′-tgccacggatcatctgcac-3′
To construct binary vectors harboring one target starting with a ‘G’, add the following nucleotides to 3′ downstream of the sense (SS) and antisense (AS) sgRNA spacers to obtain sgRNA primers:
SS: 5′-gttttagagctagaaatag-3′
AS: 5′-ggcagccaagccagcaccc-3′
To construct binary vectors harboring two targets, starting with an ‘A’ and a ‘G’, respectively:
Add the following nucleotides to 5′ upstream of the sense of ‘A’-started sgRNAs (A-sgRNA-SS) and antisense of ‘G’-started sgRNAs (G-sgRNA-AS) to obtain sgRNA primers:
A-sgRNA-SS: 5′-agGGTCTCAggca-3′
G-sgRNA-AS: 5′-agGGTCTCAaaac-3′
Add the following nucleotides to 3′ downstream of the sense of ‘A’-started sgRNAs (A-sgRNA-SS) and antisense of ‘G’-started sgRNAs (G-sgRNA-AS) oligonucleotides:
A-sgRNA-SS: 5′-gttttagagctatgc-3′
G-sgRNA-AS: 5′-ggcagccaagccagc-3′
Order the primers from any qualified company.
To construct binary vectors harboring one target, dilute the primers to a work concentration of 1 μM with sterilized double distilled H2O.
To construct binary vectors harboring two targets, dilute the primers to a work concentration of 10 μM with sterilized double distilled H2O.
Notes:
When you are designing sgRNAs to binary vectors harboring two targets, it is important but unnecessary to exclude sgRNA sequences containing BsaI restriction sites.
The concatenate function in WPS Excel is very useful in obtaining the correct sequences of sgRNA primers. For example, in order to obtain sgRNA forward primer in Excel, add spacer sequence in cell A1 and SS sequence in cell B1. Set cell C1=CONCATENATE(A1, B1), and C1 is sgRNA forward primer.
Prepare PCR templates
Linearize the donor vectors with EcoRV.
PJF997 or PJF999 2-3 μg
10× Cutsmart buffer 5 μl
EcoRV 1 μl
ddH2O to 50 μl
Incubate at 37 °C for 3 h
Recycle the two fragments produced by digestion reaction into the same EP tube and purify them together using a Gel Extraction Kit according to the manufacturer’s instructions.
Measure the DNA concentration using the NanoDrop then dilute the DNA product to a work concentration of 3 ng/μl.
Construct a binary vector harboring one target (Figure 1)
Set up an optimized multiplex PCR reaction:
2× KOD-FX buffer 5 μl
dNTPs 2 μl
KOD-FX 0.2 μl
Linearized donor vectors (3 ng/μl) 0.2 μl
OJP001 (10 μM) 0.3 μl
OJP002 (10 μM) 0.3 μl
sgRNA-primer-F (1 μM) 0.2 μl
sgRNA-primer-R (1 μM) 0.2 μl
ddH2O to 10 μl
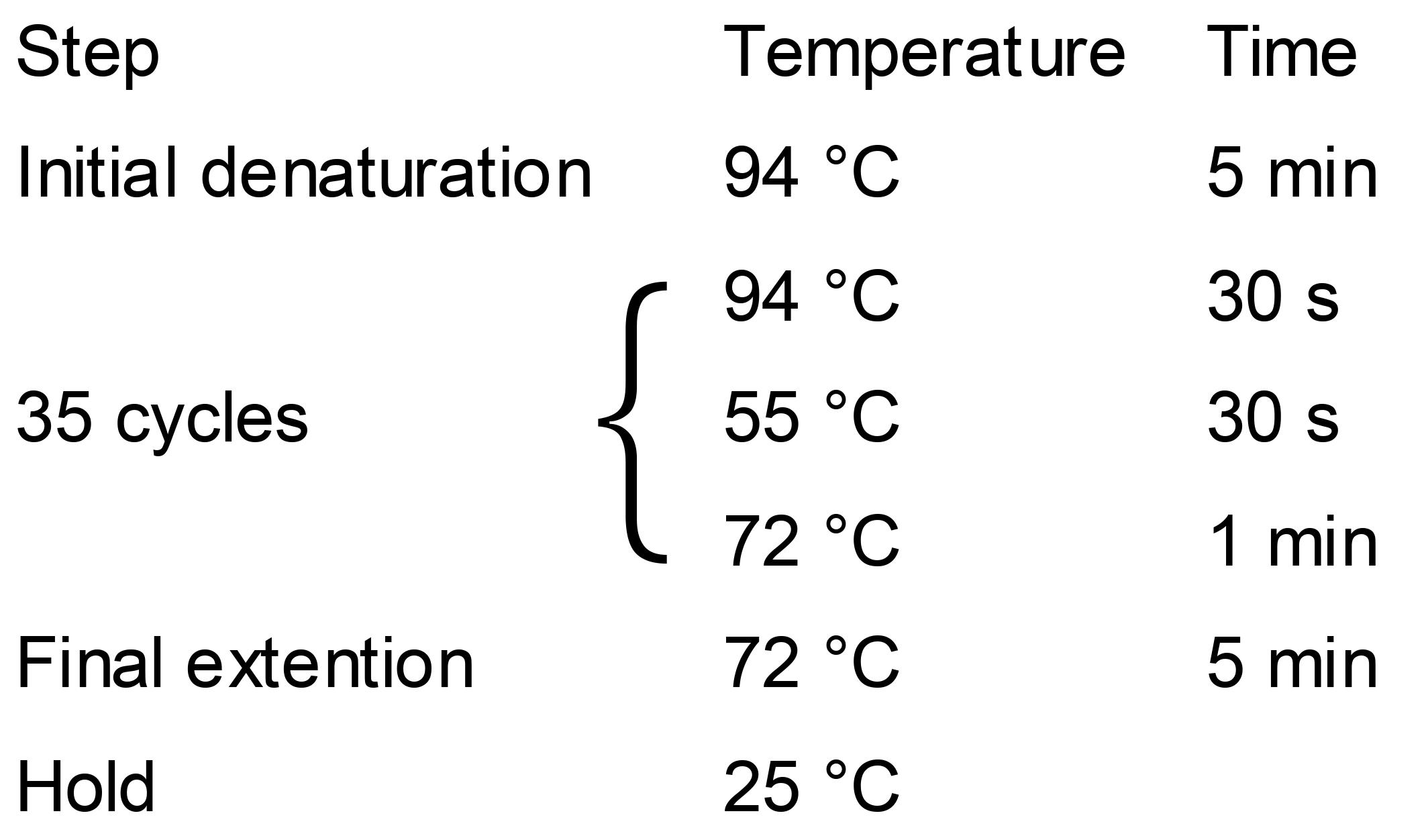
Run PCR in a thermal cycle with the following program:
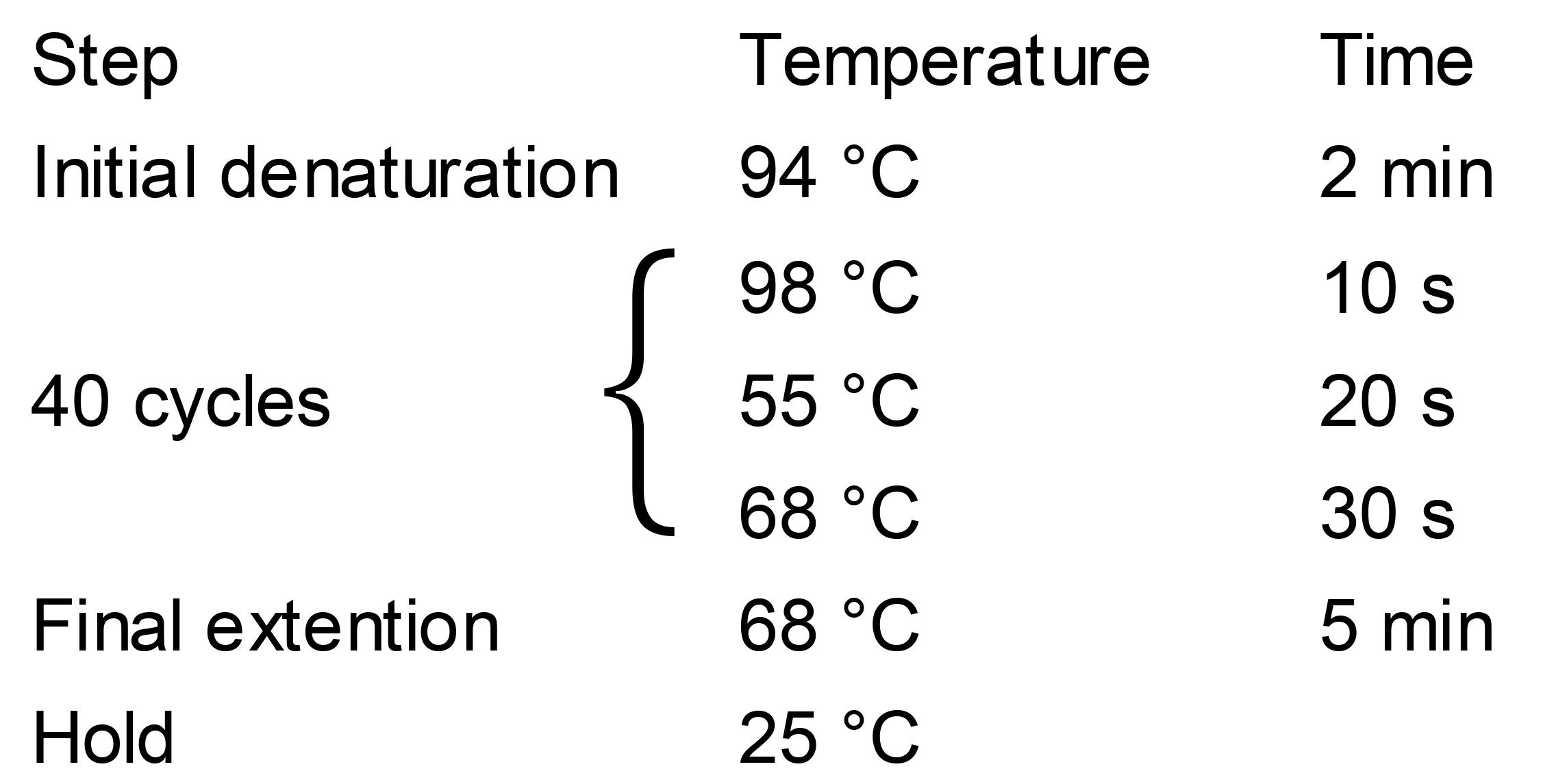
Load 2 μl PCR products onto 1% agarose gel for electrophoresis.
Notes:
The size of the PCR product is 774bp (linearized PJF997 as template) or 872bp (linearized PJF999 as template).
The rest of the PCR product does not need to be purified.
Set up an LR reaction to clone one sgRNA into the destination vector.
PCR product 0.4 μlLR clonase 0.4 μl
PJG097 0.2 μl
ddH2O 1 μl
Incubate at 25 °C for 3 h
Note: For the method to prepare PJG097 for the LR reaction, refer to Liu et al. (2020).
Transform 1 μl of the reaction products into 20 μl competent T1 cells. Spread the transformed cells on LB agar plate containing 50 μg/ml spectinomycin. Then, incubate the plates at 37 °C overnight.
The steps of transformation:
T1 chemically competent cells were taken out from -80 °C and quickly inserted into the ice. After about 5 min, the target reaction products were added into the melted competent cells and gently mixed. The competent cells were left for 25 min in the ice.
The competent cells were heated in a water bath at 42 °C for 45 s and then quickly placed on ice for 2 min.
700 μl sterilized LB medium without antibiotics was added into the centrifuge tube, and shaken for 60 min at 200 rpm at 37 °C.
Note: This step needs to be carried out in aseptic conditions.
Set up a colony PCR reaction to verify the positive clones. Pick two to four colonies from each plate. A portion of each clone is directly diluted into the PCR mix and the remainder of the each clone is retained.
PCR Supermix 5 μl
OJP008 0.3 μl
OJP026 0.3 μl
ddH2O to 10 μl
Note: If the linearized PJF999 was used as the template of the multiplex PCR, use OJP065 instead of OJP008.
Colony PCR Conditions:
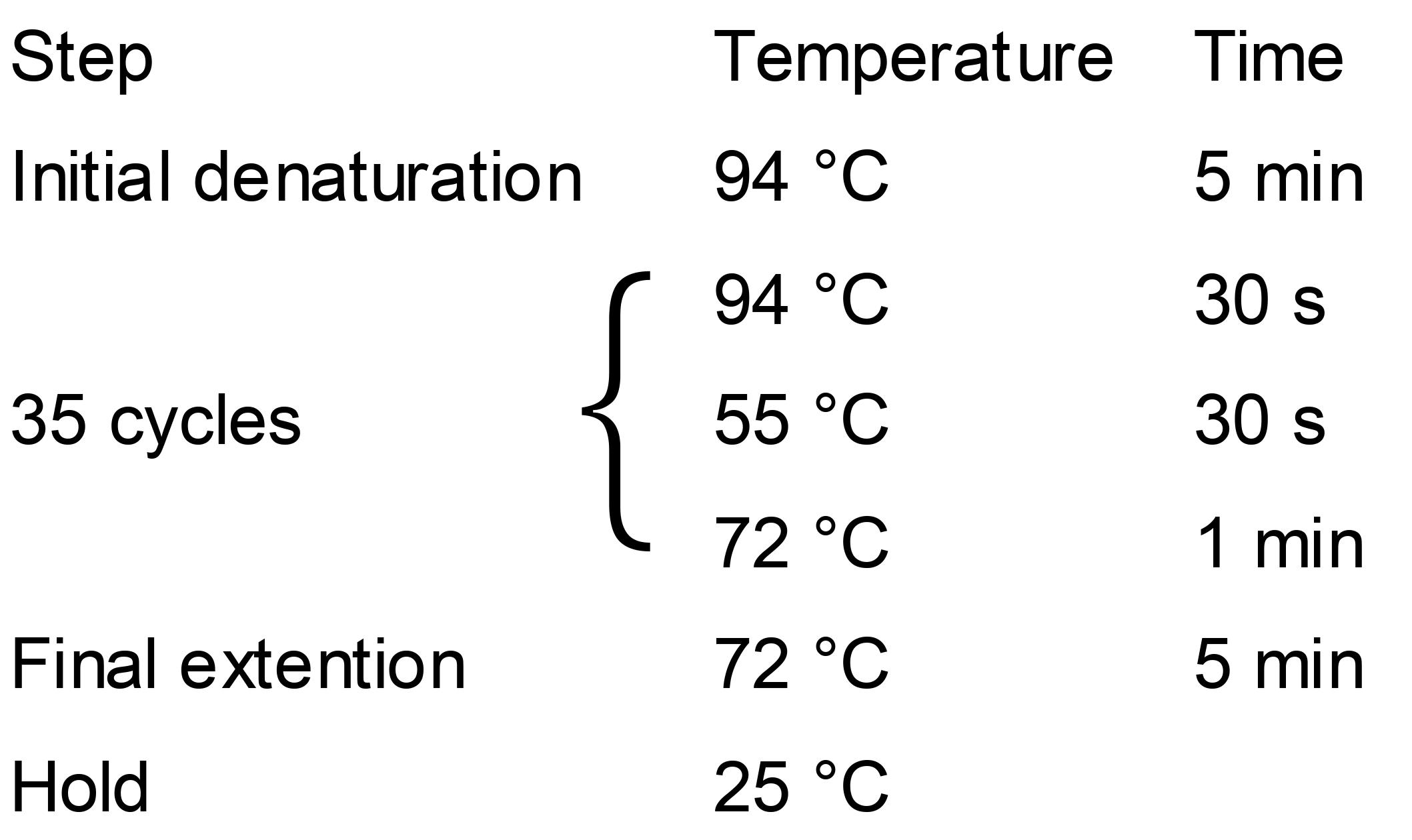
Load 5 μl PCR products onto 1% agarose gel for electrophoresis.
Select one to three positive colony from each transformation and transfer them into 6 ml LB medium containing 50 μg/ml spectinomycin. Then, incubate overnight at 37 °C in an orbital shaker at 220 rpm.
Isolate the plasmid DNA from the overnight cultures using a commercial miniprep kit according to the manufacturer’s instructions.
Validate the plasmids by Sanger sequencing using OJP026.
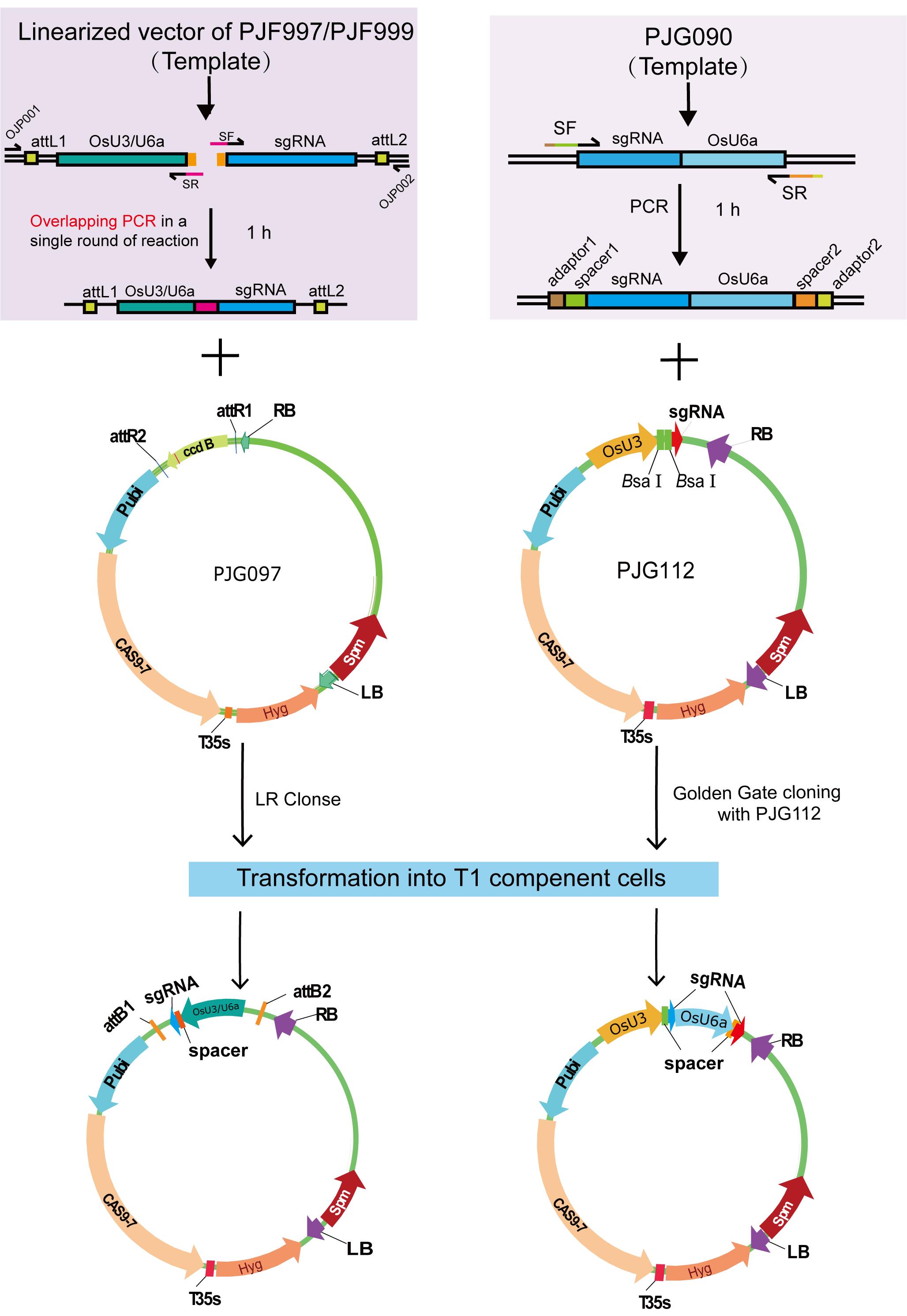
Figure 1. Schematic illustration of the cloning procedure described in the protocol
Construct a binary vector harboring two targets (Figure 1)
Set up an optimized multiplex PCR reaction.
2× KOD-FX buffer 5 μl
dNTPs 2 μl
KOD-FX 0.2 μl
PJG090 (3 ng/μl) 0.1 μl
sgRNA-primer-F (10 μM) 0.2 μl
sgRNA-primer-R (10 μM) 0.2 μl
ddH2O to 10 μl
Run PCR in a thermocycling with the following program:
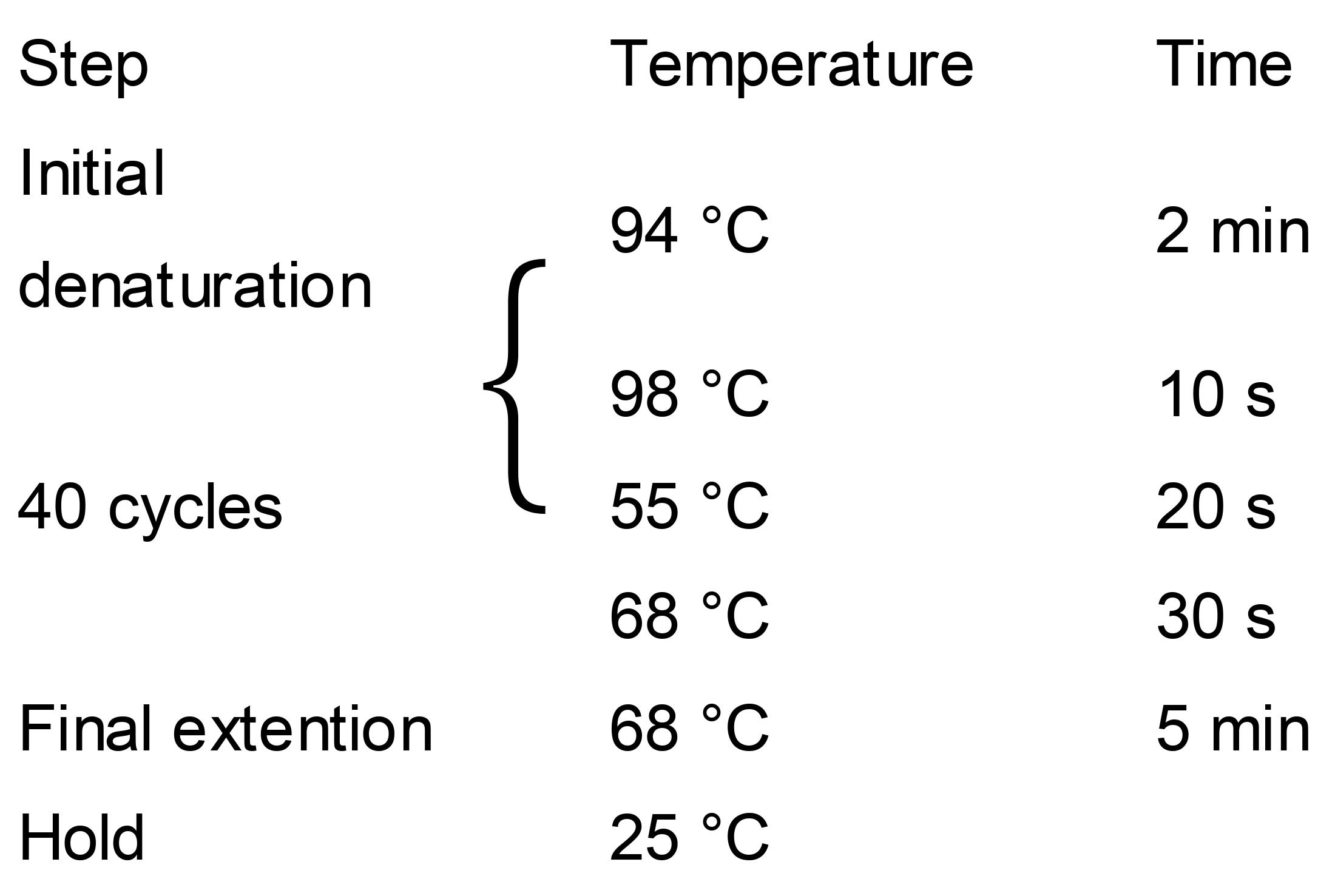
Load 2 μl PCR products onto 1% agarose gel for electrophoresis.
Set up a Golden Gate reaction to clone two sgRNAs into the destination vector.
Non-purified PCR product 1 μl
PJG112 50 ng
Cutsmart Buffer (NEB) 1 μl
T4 ligase buffer (NEB) 0.4 μl
BsaI (NEB) 5 U
T4 DNA ligase (NEB) 20 U
ddH2O to 15 μl
Golden Gate reaction Conditions:
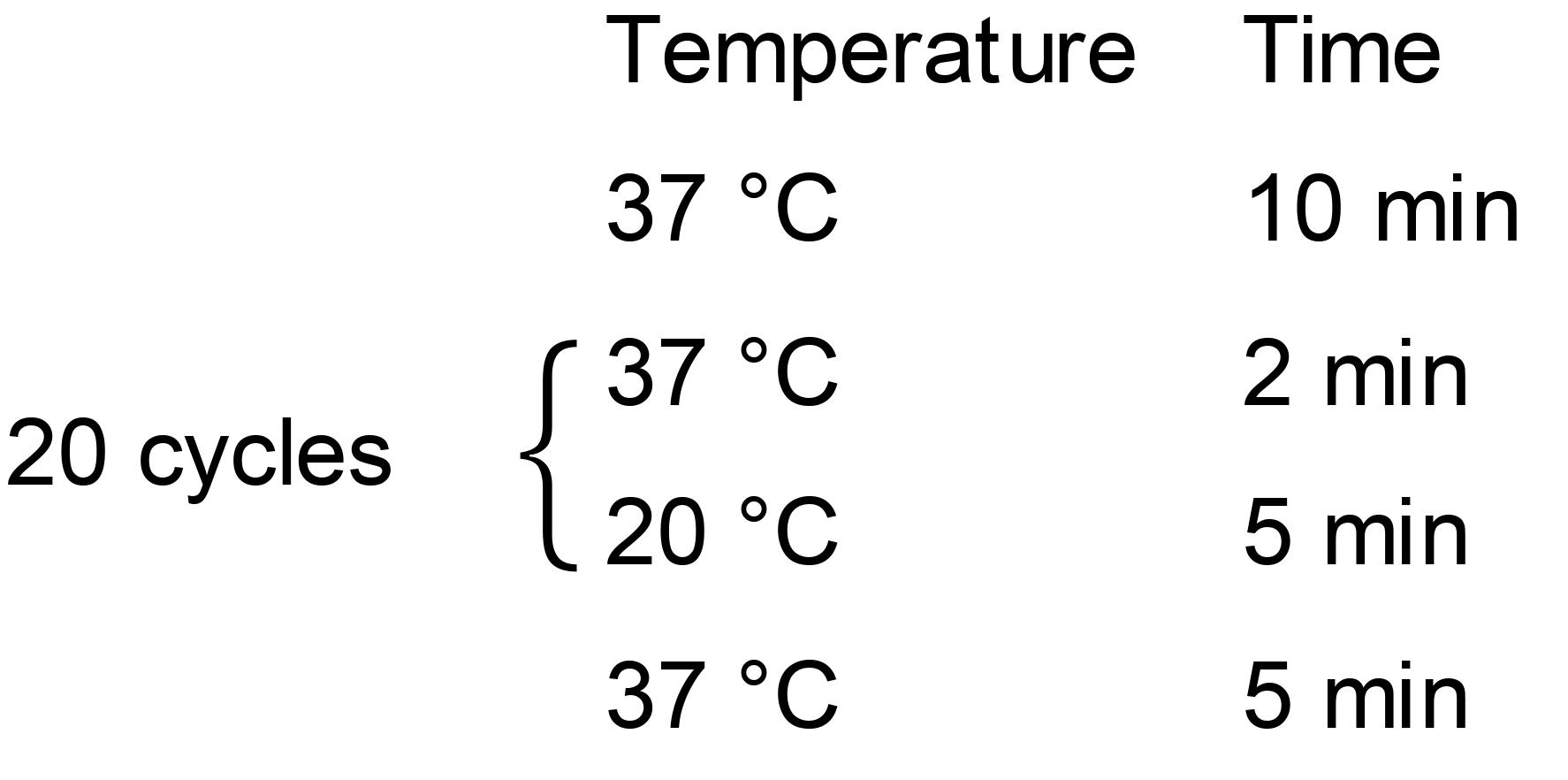
Transform 4 μl of the reaction products into 20 μl competent T1 cells. Spread the transformed cells on LB agar plate containing 50 μg/ml spectinomycin. Then, incubate the plates at 37 °C overnight.
Note: This step needs to be carried out under aseptic conditions.
Set up a colony PCR reaction to verify the positive clones. Pick two to four colonies from each plate.
PCR mix 5 μl
OJP008 0.3 μl
OJP026 0.3 μl
ddH2O 4.4 μl
Colony PCR Conditions:
Load PCR products onto 1% agarose gel for electrophoresis.
Select one to three positive colony from each transformation and transfer them into 6 ml LB medium containing 50 μg/ml spectinomycin under aseptic conditions. Then, incubate overnight at 37 °C in an orbital shaker.
Isolate the plasmid DNA from the overnight cultures using a commercial miniprep kit according to the manufacturer’s instructions.
Validate the plasmids by Sanger sequencing using OJP026.
Recipes
50× TAE electrophoresis buffer
Tris 242 g/L
Acetic acid 57.1 ml/L
0.5 M EDTA (pH 8.0) 100 ml/L
LB medium
Tryptone 10 g/L
NaCl 10 g/L
Yeast extract 5 g/L
Autoclave sterilization for 15 min at 121 °C
LB agar medium
Tryptone 10 g/L
NaCl 10 g/L
Yeast extract 5 g/L
Agar 12 g/L
Atoclave sterilization for 15 min at 121 °C
Acknowledgments
We thank Luis Alejandro José Mur (Aberystwyth University) for his critical reading and editing on the manuscript. C.F. was supported by grants from the National Science Foundation of China (31800250 and 31960063). This protocol is developed based on our previous study published in PeerJ (Liu et al., 2020).
Competing interests
The authors declare no conflict of interest.
References
- Feng, Z. Y., Zhang, B. T., Ding, W. N., Liu, X. D., Yang, D. L., Wei, P. L., Cao, F. Q., Zhu, S. H., Zhang, F., Mao, Y. F. and Zhu, J. K. (2013). Efficient genome editing in plants using a CRISPR/Cas system. Cell Res 23(10): 1229-1232.
- Liu, X., Zhou, X., Li, K., Wang, D., Ding, Y., Liu, X., Luo, J. and Fang, C. (2020). A simple and efficient cloning system for CRISPR/Cas9-mediated genome editing in rice. PeerJ 8: e8491.
- Ma, X. L., Zhu, Q. L., Chen, Y. L. and Liu, Y. G. (2016). CRISPR/Cas9 Platforms for Genome Editing in Plants: Developments and Applications.Molecular Plant 9(7): 961-974.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Li, K., Wang, Y. and Fang, C. (2021). A Novel Method to Construct Binary CRISPR Vectors for Plant Transformation by Single Round of PCR Amplification. Bio-protocol 11(7): e3971. DOI: 10.21769/BioProtoc.3971.
Category
Plant Science > Plant molecular biology > Genetic analysis
Molecular Biology > DNA > DNA cloning
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.