- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

FRET-based Microscopy Assay to Measure Activity of Membrane Amino Acid Transporters with Single-transporter Resolution
Published: Vol 11, Iss 7, Apr 5, 2021 DOI: 10.21769/BioProtoc.3970 Views: 7030
Reviewed by: Pooja VermaRenuka KudvaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Secondary active transporters reside in cell membranes transporting polar solutes like amino acids against steep concentration gradients, using electrochemical gradients of ions as energy sources. Commonly, ensemble-based measurements of radiolabeled substrate uptakes or transport currents inform on kinetic parameters of transporters. Here we describe a fluorescence-based functional assay for glutamate and aspartate transporters that provides single-transporter, single-transport cycle resolution using an archaeal elevator-type sodium and aspartate symporter GltPh as a model system. We prepare proteo-liposomes containing reconstituted purified GltPh transporters and an encapsulated periplasmic glutamate/aspartate-binding protein, PEB1a, labeled with donor and acceptor fluorophores. We then surface-immobilize the proteo-liposomes and measure transport-dependent Fluorescence Resonance Energy Transfer (FRET) efficiency changes over time using single-molecule Total Internal Reflection Fluorescence (TIRF) microscopy. The assay provides a 10-100 fold increase in temporal resolution compared to radioligand uptake assays. It also allows kinetic characterization of different transport cycle steps and discerns kinetic heterogeneities within the transporter population.
Keywords: Single-molecule FRETBackground
Membrane resident secondary active transporters or solute carriers (SLC) mediate cellular uptake of amino acids, hormones, neurotransmitters, vitamins, and drugs, among other solutes. They couple concentrative substrate uptake to energetically favorable dissipation of electrochemical gradients of ions maintained primarily through the work of Na+/K+ ATPases (Lingrel and Kuntzweiler, 1994). The structural mechanisms of many secondary active transporters have come to light over the last decades (Grewer and Rauen, 2005; Shi, 2013; Vandenberg and Ryan, 2013; Drew and Boudker, 2016).
Measuring radioligand uptake into cells, membrane vesicles, or proteoliposomes is a traditional means to assay transport (Nimigean, 2006; Geertsma et al., 2008; Volpe, 2016). The cells and membrane vesicles provide the most native environment. By contrast, proteoliposomes allow precise control over compositions of membranes and luminal solutions. Broadly, purified transporters are reconstituted into liposomes with internal and external buffers providing the necessary ionic gradients. Gradient-dependent accumulation of the radiolabeled substrates in proteoliposomes is then measured. Specifically, a radiolabeled substrate is added to the external buffer, aliquots are taken at different time points and filtered. Finally, radioactivity trapped in the filters within the vesicles is measured. Initial rates of transport are measured as functions of substrate concentrations, lipid compositions, or temperature. These ensemble-based measurements provide mean uptake rates averaged over the population of transporters and over time. A shortcoming of the method relies on estimating what fraction of the transporters is reconstituted in an active form. Thus, accurate turnover times may be challenging to determine. Furthermore, the manual pipetting of aliquots limits the temporal resolution to 5 s or more, as reported in studies with various channels and transporters, including primary and secondary solute transporters and potassium channels (Knol et al., 1996; Heginbotham et al., 1998; Jung et al., 1998; van der Heide and Poolman, 2000; Enkvetchakul et al., 2004; Borths et al., 2005; Yamashita et al., 2005; Boudker et al., 2007).
We describe a single-molecule Fluorescence Resonance Energy Transfer (smFRET)-based uptake assay of liposome-reconstituted purified transporters (Figure 1). The current embodiment of this assay provides a temporal resolution of approximately 50 ms, a 100-fold increase compared to traditional radioligand uptake assays. The number and orientation of transporters in vesicles can be controlled. The assay detects the time it takes for each transporter to transport the first substrate molecule (during this time, the protein binds, translocates and releases the substrate into the vesicle lumen) and allows estimation of the mean turnover time of each transporter. A single measurement follows the activity of ca. 100-500 individual transporters simultaneously. The need for radioactive materials, which might be difficult and expensive to procure and are harmful to researchers and the environment, is eliminated.
We recently used this assay to study archaeal sodium and aspartate symporter, GltPh (Ciftci et al., 2020). We engineered a glutamate/aspartate FRET sensor based on a periplasmic glutamate/aspartate binding protein PEB1a by introducing two cysteine mutations for labeling with donor and acceptor fluorophores (ccPEB1a). The FRET efficiency of the labeled ccPEB1a increases as it binds an amino acid. We further mutated ccPEB1a to tune its aspartate affinity to allow tracking single- or up to 5-30 rounds of transport. GltPh containing a single cysteine mutation on the transporter's extracellular side was biotinylated and reconstituted into liposomes at 1:1,000 protein to lipid ratio (PLR) to enrich the population of proteoliposomes containing one transporter (Figure 1A). ccPEB1a was encapsulated by freeze-thaw cycles, and proteoliposomes were extruded through 0.1 µm filters, immobilized in smFRET imaging chambers, equipped with a rapid perfusion system, and imaged using TIRF microscopy (Figure 1B). The first half-cycle times for each transporter are determined manually from the onset of the FRET efficiency increase in a single-molecule trajectory following the substrate injection into the imaging chamber. The consequent continuous increase in FRET efficiency is fitted to a time-dependent aspartate binding equation (Ciftci et al., 2020) to obtain mean transporter turnover times (Figure 1C).
The assay was inspired by and, in part, based on a single-molecule assay developed for a neutral amino acid transporter MhsT utilizing periplasmic leucine, isoleucine, valine-binding protein LIV-BP (Fitzgerald et al., 2019). We expect analogous single-molecule transport assays to be developed for various other transporters, taking advantage of the natural diversity of periplasmic binding proteins (Berntsson et al., 2010) or other types of sensor proteins (Deuschle et al., 2005; Hou et al., 2011; Chen et al., 2013; Liu et al., 2015).
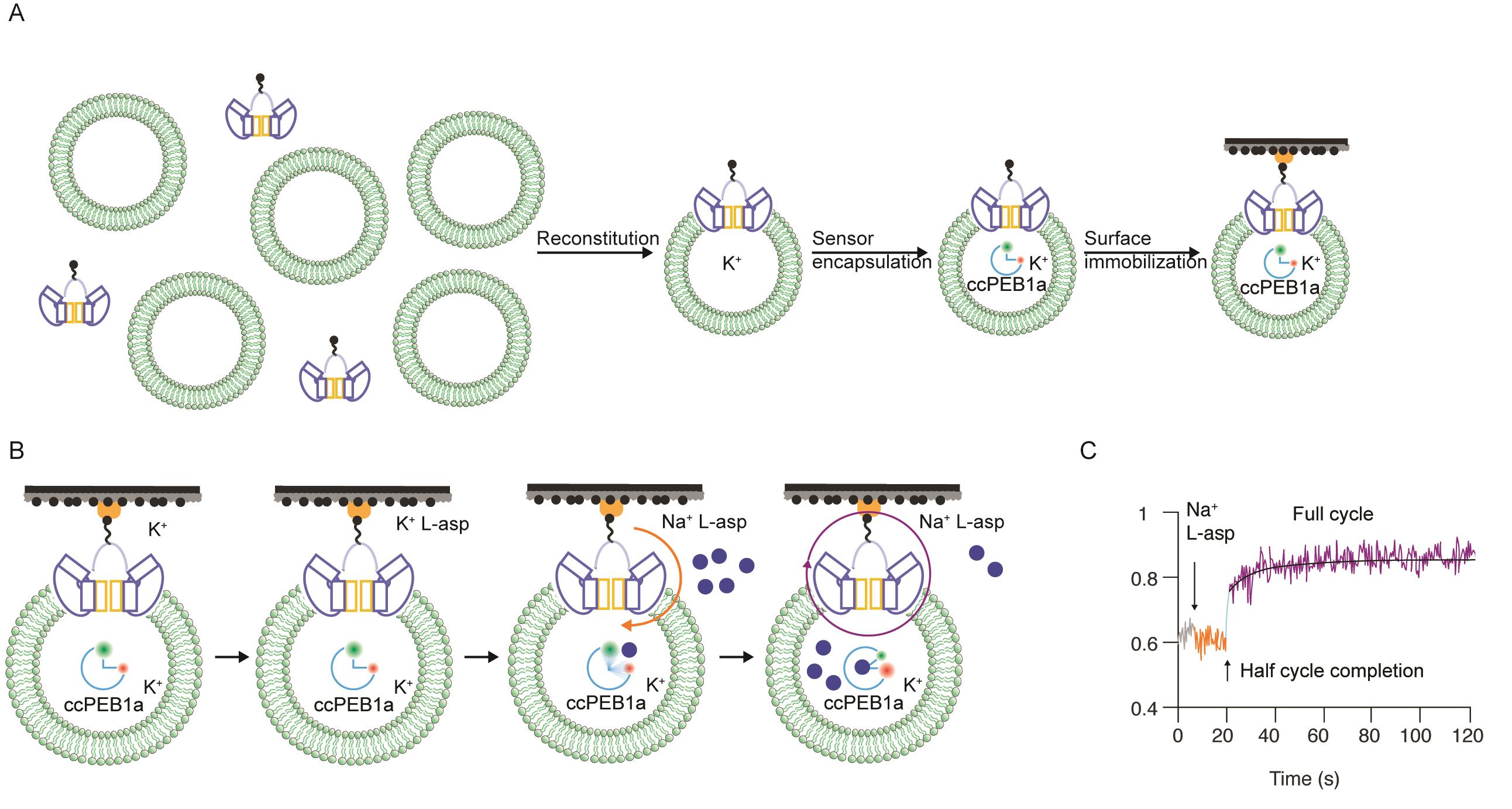
Figure 1. Single-molecule transport assay. A. Sample preparation flowchart. Biotinylated transporters are reconstituted into liposomes, the FRET sensor encapsulated, and proteoliposomes are immobilized in imaging chambers. B. Proteoliposomes are first imaged in a Resting buffer (K+), then a Non-activating buffer (K+/L-Asp) to test leakage, followed by an Activating buffer (Na+/L-Asp, blue circles) to initiate transport. Orange and purple arrows indicate the first half-cycle and consecutive turnovers, respectively. C. A representative single-molecule FRET efficiency trajectory.
Materials and Reagents
50 ml Syringe (BD, catalog number: 309654 )
Amicon Ultra-15 Centrifugal filters 100K (EMD Millipore, catalog number: UFC910096 )
Amicon Ultra-4 Centrifugal filters 100K (EMD Millipore, catalog number: UFC810096 )
Amicon Ultra-15 Centrifugal filters 10K (EMD Millipore, catalog number: UFC901024 )
Amicon Ultra-4 Centrifugal filters 10K (EMD Millipore, catalog number: UFC801024 )
Whatman Nucleopore Track-Etched membrane, 0.4 µm, 19 mm (Sigma-Aldrich, catalog number: WHA800282 )
Whatman Nucleopore Track-Etched membrane, 0.1 µm, 19 mm (Sigma-Aldrich, catalog number: WHA800309 )
Tube revolver/rotator for 1.5 ml reaction tubes
General Long-Term Storage Cryogenic Tubes (NalgeneTM, Thermo Fisher Scientific, catalog number: 5000-0020 )
50 ml tubes (Fisher Scientific, catalog number: 1443222 )
Glycerol (Fisher Scientific, catalog number: G334 )
Liquid N2
Chloroform (Fisher Scientific, catalog number: 507517453 )
Triton X-100 (Sigma-Aldrich, catalog number: T9284 )
LB broth, miller (Fisher Bioreagents, catalog number: BP1426-2 )
Ampicillin, sodium salt (GoldBio, catalog number: A-301-100 )
Ni-NTA superflow (QIAGEN, catalog number: 30430 )
Thiol reactive dyes (LD555, LD655) (Lumidyne Technologies, catalog numbers: LD555-MAL , LD655-MAL )
Dimethyl sulfoxide (DMSO) (Fisher Bioreagents, catalog number: BP231-100 )
Aspartate-glutamate binding protein, PEB1a (UniProt ID: M9QLL2. Mutations for site-specific labeling: C18S/N73C/K149C; mutations to tune affinity: A64F, D102L, Y198F, R89L)
Escherichia coli (E. coli) Polar Lipid Extract (Avanti Polar Lipids, catalog number: 100600C-100mg )
Egg phosphatidylcholine (PC) (Avanti Polar Lipids, catalog number: 840051C-25mg )
Bio-beads SM2 absorbent media (Bio-Rad, catalog number: 152-3920 )
Protocatechuic acid (PCA) (Sigma-Aldrich, catalog number: 37580-25G-F )
Protocatechuate-3,4-dioxygenase (PCD) (Sigma-Aldrich, catalog number: P8279 )
L-arabinose (GoldBio, catalog number: A-300-1 )
HEPES (Fisher Bioreagents, catalog number: BP310-100 )
Sodium chloride (NaCl) (Fisher Scientific, catalog number: S271-3 )
Potassium chloride (KCl) (EMD Millipore, catalog number: PX1405-1 )
Calcium chloride dihydrate (Sigma-Aldrich, catalog number: 223506 )
Tris (2-carboxyethyl) phosphine hydrochloride, 98% (TCEP) (Alfa Aesar, catalog number: 40587-09 )
Ethylenediaminetetraacetic acid disodium salt, dihydrate 100% (EDTA) (Alfa Aesar, catalog number: C1201100-500A5 )
Lysozyme, egg white (GoldBio, catalog number: L-040-10 )
Phenylmethylsulfonyl fluoride (PMSF) (MP Biomedicals, catalog number: 195381 )
Sucrose, ultrapure, RNase Free (MP Biomedicals, catalog number: 04821713 )
L-Aspartic acid, monopotassium salt (Research Products International, catalog number: 1115-63-5 )
n-Dodecyl-β-D-maltoside (DDM) (Anagrade, Anatrace, catalog number: D310 25 GM )
Thrombin from bovine plasma (Sigma-Aldrich, catalog number: T4648 )
β-D-1-thiogalactopyranoside (IPTG) (GoldBio, catalog number: I2481C100 )
Bovine serum albumin (BSA) (Fraction V, OmniPur, catalog number: 2930 )
Streptavidin (Invitrogen, catalog number: S888 )
Thermo ScientificTM EZ-LinkTM Maleimide-PEG11-Biotin (Fisher Scientific, catalog number: PI21911 )
N-ethyl maleimide (Sigma-Aldrich, catalog number: E3876 )
QIAprep spin miniprep kit (50×) (QIAGEN, catalog number: 27104 )
T7 express lysY competent E. coli (High efficiency) (New England Biolabs, catalog number: C3010I )
Thermo ScientificTM FastDigest DpnI (Fisher Scientific, catalog number: FERFD1704 )
PfuUltra II Hotstart PCR master mix (Agilent Technologies, catalog number: 600852 )
LB agar plates, ampicillin-100 (Teknova, catalog number: L1004 )
Pet21(+) vector (Genscript)
Resuspension buffer (see Recipes)
Wash buffer (see Recipes)
Elution buffer (see Recipes)
Size Exclusion Chromatography (SEC) buffer (see Recipes)
cysGltPh SEC buffer (see Recipes)
Resting buffer (see Recipes)
Non-activating buffer (see Recipes)
Activating buffer (see Recipes)
T50 buffer (see Recipes)
cysGltPh Resuspension buffer (see Recipes)
Equipment
Spectrophotometer (Horiba Photon Technology PTI, Quantamaster 8000 )
NanoDrop 2000c (Fisher Scientific, catalog number: ND-2000C )
2,800 ml Fernbach-style culture flasks with baffles (Fisher Scientific, catalog number: 09-552-39 )
Glass beaker
Magnetic stir bars
Superdex 200 Increase 10/300 GL column (GE Healthcare, catalog number: 28-9909-44 )
AKTA pure chromatography system (GE Lifesciences)
Aldrich® single-neck round-bottom flask, 100 ml capacity, Joint: ST/NS 24/40 (Sigma-Aldrich, catalog number: Z414492 )
BUCHI RotavaporTM R-300 Rotary Evaporator with Controller and V-300 Pump
Dry-seal vacuum desiccator (Fisher Scientific, catalog number: 086427)
Avanti Mini Extruder kit (Avanti Polar Lipids, catalog number: 610000 )
Filter Support (Avanti Polar Lipids, catalog number: 610014 )
MAXQ 8000 Incubated/Refrigerated Stackable Shaker (Thermo Fisher Scientific)
Drummond Portable Pipet-Aid (Thomas Scientific, catalog number: 13-661-17E )
Pipetman Kit, P1000, P200, P20, P10 (Gilson, catalog number: F167370 )
Avanti JXN-26 Centrifuge (Beckman Coulter)
Avanti JXN-30 Centrifuge (Beckman Coulter)
Optima MAX-XP Tabletop Ultracentrifuge (Beckman Coulter)
Optima L-100XP Ultracentrifuge (Beckman Coulter)
J-LITE JLA-8.1000 Fixed-Angle Aluminum Rotor, 6 × 1,000 ml, 8,000 rpm, 15,970 × g (Beckman Coulter, catalog number: 363688 )
JA-20 Fixed-Angle Aluminum Rotor, 8 × 50 ml, 20,000 rpm, 48,400 × g (Beckman Coulter, catalog number: 334831 )
Type 45 Ti Fixed-Angle Titanium Rotor (Beckman Coulter, catalog number: 339160 )
TLA-100.3 Fixed-Angle Rotor (Beckman Coulter, catalog number: 349481 )
1 L (1,000 ml) Polycarbonate Bottle with Cap Assembly, 95 × 191 mm (Beckman Coulter, catalog number: A98812 )
70 ml, Polycarbonate Bottle Assembly, 38 × 102 mm (Beckman Coulter, catalog number: 355622 )
26.3 ml, Polycarbonate Bottle with Cap Assembly, 25 × 89 mm (Beckman Coulter, catalog number: 355618 )
50 ml, Polycarbonate Bottle with Cap Assembly, 29 × 104 mm (Beckman Coulter, catalog number: 357000 )
Avestin EmulsiFlex C3 cell disruptor (Avestin)
Dounce tissue grinder set
XK 16/20 column (Cytivalifesciences, catalog number: 28988937 )
SpectrumTM Spectra/PorTM 4 RC Dialysis Membrane Tubing 12,000 to 14,000 Dalton MWCO (Fisher Scientific, catalog number: 08-667D )
SpectrumTM Dialysis Tubing Closures: Spectra/PorTM Standard Type (Fisher Scientific, catalog number: 08-670-11A )
Software
SPARTAN (Scott Blanchard laboratory, https://www.scottcblanchardlab.com/software)
MATLAB (MathWorks, https://www.mathworks.com/products/matlab.html)
Prism (GraphPad Software, Inc., https://www.graphpad.com/)
Origin (OriginLab Corporation, Northampton, MA, USA, https://www.originlab.com/)
Procedure
ccPEB1a sensor construction
Clone PEB1a with C terminal 6x His-tag into pet21a (+) vector.
Mutagenize PEB1a to remove native cysteine and introduce two cysteines for site-specific labeling with fluorophores (C18S, N73C, K149C), yielding ccPEB1a construct.
C18S:
Forward primer: GGGTGCGAGCGTTGCGTTTAG
Reverse primer: CGCAACGCTCGCACCCAGCGC
N73C:
Forward primer: CTGGACTGTGGCAGCGTGGATGCGGTTATC
Reverse primer: GATAACCGCATCCACGCTGCCACAGTCCAG
K149C:
Forward primer: CGGCATTGATGTGTGCTTCAGCGAATTTCC
Reverse primer: GGAAATTCGCTGAAGCACACATCAATGCCG
Introduce additional mutations at or near the aspartate/glutamate-binding site to tune affinity. ccPEB1a binds aspartate with a dissociation constant of ~79 nM. ccPEB1a Y198F variant has 5.6 µM affinity allowing visualization of up to 5 transport cycles in 100 nm liposomes.
Mix 100 ng of DNA with the above mutations and 50 µl of T7 Express lysY competent E. coli cells in a 1.5 ml reaction tube and incubate on ice for 30 min.
Heat shock at 42 °C for 45 s and immediately place the tube on ice for 5 min and add 750 µl of autoclaved LB.
Incubate for 1 h at 37 °C with shaking at 250 rpm and plate on agar plates supplemented with 100 µg/ml ampicillin.
Grow overnight for ~16 h at 37 °C with shaking at 250 rpm and prepare a glycerol stock by mixing 500 µl culture with 500 µl of autoclaved 50% glycerol, store at -80 °C.
ccPEB1a purification
Prepare 4 L of LB according to manufacturer’s protocol in two 2,800 ml Fernbach-style culture flasks with baffles and separately 300 ml in 1 L flask. Autoclave and let cool to 37 °C or room temperature.
Supplement all LB with 100 µg/ml ampicillin before use.
Inoculate 300 ml LB with bacterial glycerol stock of ccPEB1a and grow overnight for ~16 h at 37 °C with shaking at 250 rpm.
Inoculate LB with the overnight culture from Step B3 to a final absorbance of 0.1 at 600 nm. Dilute 200 µl of cell culture 5 times with LB for absorbance measurements.
Induce cells with 1 mM freshly prepared IPTG when the absorbance reaches 0.8.
Let cultures grow for 3 h at 37 °C and 16 h at 18 °C with shaking at 250 rpm.
Harvest cells by centrifuging for 15 min at 3,326 × g at 4 °C in Avanti JXN-26 centrifuge using JLA-8.1000 fixed-angle rotor. Use 10 ml of Resuspension buffer (see Recipe 1) per 2 L bacterial cell cultures to resuspend the pellets.
Add 100 µg/ml lysozyme.
Incubate on ice for 30 min and dilute 1:3 (v:v) with deionized water.
Centrifuge for 30 min at 3,578 × g in Avanti JXN-26 centrifuge using JA-20 fixed-angle rotor at 4 °C to pellet the debris. The supernatant still has some particulate matter and is very viscous. A second identical centrifugation step can further clarify the supernatant, prevent clogging the column, and extend the life of the resin.
Mix supernatant with 3-5 ml of Ni-NTA resin washed with 3 × 5 column volumes (CV) of water and 1 × 5 CV of wash buffer (see Recipe 2) in a beaker and stir gently on a stirrer plate for 1 h at 4 °C using a magnetic stir bar.
Assemble the XK20/16 column as described in the manufacturer’s instructions and slowly pour the resin slurry in, pulling the flow-through with a syringe.
Wash with 5 CV of Wash buffer by pulling the flow-through with a syringe.
Elute proteins using 3 CV Elution buffer (see Recipe 3) by gravity.
Concentrate the sample in an Amicon Ultra-15 Centrifugal filter with a 10K molecular weight cutoff until the volume reaches < 500 µl. Determine the protein concentration by measuring absorbance at 280 nm in a spectrophotometer. Calculate protein’s extinction coefficient and molecular weight using the online Expasy Protparam tool from Expasy Bioinformatics Resource Portal.
Centrifuge the protein solution at 49,192 × g at 4 °C for 15 min in Optima MAX-XP Tabletop Ultracentrifuge using TLA-100.3 fixed-angle rotor. Aspirate the protein solution using a 1 ml pipette without disturbing the pellet of protein aggregates.
Polish the protein by Size Exclusion Chromatography (SEC) using Superdex 200 Increase 10/300 GL column, following pre-equilibration of Superdex 200 Increase 10/300 GL column in 50 ml of SEC buffer (see Recipe 4). Set the flow rate and pressure limit of the column according to the manufacturer’s instructions. A flow rate of 0.3 ml/min and 2 MPa pressure limit are ideal for this column. Monitor protein elution by following absorbance at 280 nm and collect 0.4 ml fractions.
Collect the peak fractions and immediately proceed with the labeling procedure. The protein should come out as a monomer (Figure 2).
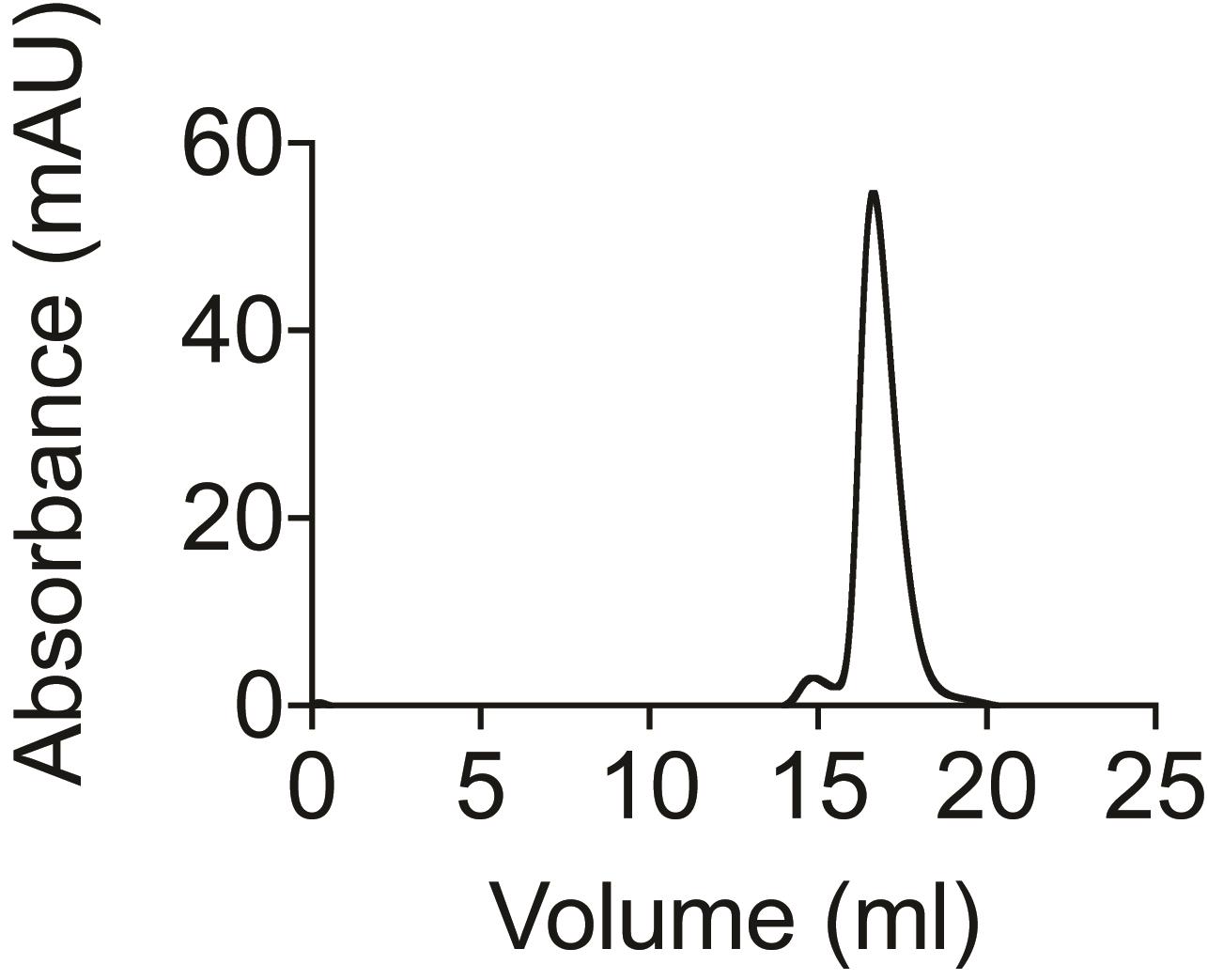
Figure 2. SEC profile of monomeric PEB1a eluting from the Superdex 200 Increase 10/300 GL column
ccPEB1a labeling
Resuspend 1,000 nmol dried LD555 or LD655 in 100 µl DMSO to a 10 mM final concentration. Vortex until the dye chunks disappear. Aliquot in 10 µl aliquots and store at -20 °C.
Thaw aliquots of LD555 and LD655 dyes and mix with freshly SEC-purified ccPEB1a protein at 1:1:1 (mol:mol:mol) ratio. The molar ratio can be empirically optimized depending on the labeling efficiency. Note that there are no reducing agents in the SEC buffer because they can reduce the labeling efficiency. Thus, solvent-exposed cysteine residues may oxidize over time and it is critical to mix the protein solution with maleimide reagents immediately after elution from SEC.
Wrap the Eppendorf tube in aluminum foil and incubate on a rotator for 45 min-1 h.
Purify labeled monomeric ccPEB1a from the unreacted dye by SEC, following pre-equilibration of Superdex 200 Increase 10/300 GL as described in Step B17 of the ‘ccPEB1a purification’ section. Use the same flow rate and pressure limits as in Step B17.
Concentrate labeled protein to ~5 µM using Amicon Ultra-4 Centrifugal filter with 10K molecular weight cutoff.
If the ccPEB1a protein variant binds substrate with dissociation constant < 5 µM, dialyze the protein against 4,000-fold SEC buffer in a 4 L bucket using dialysis membrane tubing with a 12-14 kDa molecular weight cutoff to remove bound L-aspartate. Incubate 15 cm tubing in distilled water for 30 min. Rinse the tubing with distilled water and close one end with a dialysis tubing clip. After making sure the clip is tight and there are no leaks, load the protein solution. Fold the excess tubing over itself, leaving a 2 cm space on top of the protein solution, and close it with a clip. Invert the assembly 1-2 times to ensure no leaks and place it into a beaker filled with SEC buffer. Replace the buffer three times every 4 h.
Measure the protein sample absorption at 280 nm, 555 nm (LD555), and 655 nm (LD655). Use the extinction coefficients of the protein and the dyes: 150,000 cm-1 M-1 (LD555) and 250,000 cm-1 M-1 (LD655) to calculate their respective concentrations and determine labeling efficiency.
Aliquot labeled protein in 20 µl aliquots and flash freeze in liquid N2. Store in liquid N2 until use.
Preparation of the liposome stocks
Mix 4 ml (100 mg) of E. coli Polar Lipid Extract and 1.33 ml (33 mg) of Egg PC (stored at -80 °C in chloroform) at a final 3:1 (w:w) ratio in a 100 ml round bottom flask. Use glass pipettes to transfer chloroform solutions. Dry the mixture using a rotary evaporator, rotating the flask for ~30 min under vacuum; lipids will form a thin film on the flask’s walls.
Cover the flask opening loosely with foil and leave in a dry-seal vacuum desiccator connected to vacuum overnight.
To hydrate the lipid film, add 33.25 ml of Resting buffer (see Recipe 6) to the round bottom flask to achieve a final concentration of 4 mg/ml lipids. To facilitate the lipid film hydration process, perform freeze/thaw cycles as follows: freeze the lipid suspension by immersing the flask into the liquid nitrogen. You can prepare a liquid nitrogen bath in a thermal flask or an ice bucket. Use a thermal glove to hold the flask by its neck or use a vapor tube extender as you lower it into the liquid nitrogen. The lipid suspension will make a sound akin to an egg on a frying pan as it freezes. Once the noise subsides in 1-2 min, the suspension is fully frozen. Transfer the flask into a water bath at room temperature to thaw the suspension. Video 1 showcases the setup we use. Repeat freeze/thaw cycles 10 times until the lipid film on the flask sides disappears, and the suspension is turbid white color.
 Video 1. Preparation of liposomes stocks for protein reconstitution
Video 1. Preparation of liposomes stocks for protein reconstitutionAliquot liposomes in 1 ml aliquots, flash-freeze in liquid N2, and store at -80 °C until further use.
Reconstitution of sodium/aspartate symporter, GltPh, into liposomes and encapsulation of ccPEB1a
Purify N278C/C321A GltPh single cysteine mutant (cysGltPh), as previously described (Akyuz et al., 2013), with a few modifications below.
Repeat Steps B1-B4 with 12 L of LB instead of 4 L and inoculate LB with a cysGltPh glycerol stock.
Induce with 20 mM of L-arabinose for 3 h at 37 °C with shaking at 250 rpm.
Lyse the cells in cysGltPh Resuspension buffer (see Recipe 10) using the EmulsiFlex C3 cell disruptor. 4 passes are usually enough to disrupt the majority of the cells.
Centrifuge the cell suspension for 1 h at 100,000 × g and 4 °C in Optima L-100XP Ultracentrifuge using Type 45 Ti Fixed-Angle Titanium Rotor to pellet the membranes.
Homogenize crude cell membranes in Solubilization buffer (see Recipe 11) supplemented with 40 mM n-dodecyl β-D-maltopyranoside (DDM) for 1 h at 4 °C and remove the insoluble material by centrifuging for 1 h at 100,000 × g and 4 °C in Optima L-100XP Ultracentrifuge using Type 45 Ti Fixed-Angle Titanium Rotor.
Mix the supernatant with 3-5 ml of Ni-NTA resin and assemble the column as described in Steps B11-B12.
Wash with 5 CV of Solubilization buffer supplemented with 1 mM DDM and 40 mM imidazole and elute in the presence of 250 mM imidazole.
Incubate for ~16 h at 21 °C with thrombin at 10 units per mg protein to cleave the His-tag.
Purify cleaved cysGltPh by SEC using Superdex 200 Increase 10/300 GL column pre-equilibrated in 50 ml cysGltPh SEC buffer (see Recipe 5).
Thaw aliquots of maleimide-PEG11-biotin and NEM and mix with freshly purified cysGltPh protein at 1:2:4 (mol:mol:mol) protein:maleimide-PEG11-biotin:NEM ratio. Note that there are no reducing agents in the SEC buffer, and cysteine residues may oxidize over time. Thus, it is critical to mix the protein solution with maleimide reagents immediately after elution from SEC.
Purify labeled cysGltPh from the unreacted reagents by SEC, using pre-equilibrated Superdex 200 Increase 10/300 GL column.
Concentrate labeled transporter to 4 mg/ml using Amicon Ultra-4 Centrifugal filter with 100K molecular weight cutoff.
Take out 1 ml liposome aliquot (4 mg/ml) and extrude through 400 nm membranes using a mini-extruder as shown in the video (https://www.sigmaaldrich.com/video/life-science/avanti-mini-extruder.html). 21 passes should yield uniformly sized unilamellar liposomes.
Destabilize liposomes by adding Triton X-100 at 1:2 (w:w) detergent to lipid ratio.
Add cysGltPh to liposomes at a 1:1000 (w:w) PLR and incubate on a rotator at room temperature for 30 min. Adjust PLR to enrich the liposome population with one transporter per vesicle. We detail calculations in Ciftci et al. (2020).
Wash Bio-Beads extensively with 10 CV ethanol and 20 CV water before use in a 50 ml tube. Invert the slurry 2-3 times, let the beads settle at the bottom, and aspirate the supernatant with a pipettor. Add the beads at a 100 mg/ml final concentration to the liposome suspension and incubate on a rotator at room temperature for 2 h. It is critical not to add too many beads to avoid lipid loss, which would reduce the yield of proteoliposomes.
Transfer the liposome solution from the beads using a 1 ml pipettor into a new cryovial. Repeat Steps C17 and C18 for5 more times using fresh beads at 4 °C.
Pellet proteoliposomes by centrifugation at 49,192 × g at 4 °C for 1 h in Optima MAX-XP Tabletop Ultracentrifuge using TLA-100.3 fixed-angle rotor.
Remove supernatant and resuspend proteoliposomes in an equal volume of the Resting buffer, freeze in liquid N2 and thaw in a water bath at room temperature.
Repeat Steps C20 and C21 twice more.
Pellet proteoliposomes as above and resuspend at 25 mg/ml in 160 µl of the Resting buffer.
Add fluorescently labeled ccPEB1a from Step C8 to a final concentration of 0.6 µM and encapsulate by 2 freeze/thaw cycles as above. Two freeze/thaw cycles should result in up to 30 % encapsulation efficiency (Colletier et al., 2002; Chaize et al., 2004; Costa et al., 2014). Increasing the number of the freeze/thaw cycles might lead to improved encapsulation efficiency but might also result in an increased fraction of the denatured ccPEB1a. We found that the current procedure allows us to image up to 500 vesicles in a single sm-FRET recording, sufficient for efficient data collection. Proteoliposomes can be stored at -80 °C at this step until further use.
Pellet proteoliposomes by centrifugation as above to remove unencapsulated PEB1a. Replace the supernatant with an equal volume of the Resting buffer.
Extrude proteoliposomes (21 passages) through 100 nm membranes using a mini-extruder (Avanti Polar Lipids). Proteoliposomes can be stored at 4 °C for up to 5-6 h before use. You can verify the liposome integrity and size distribution by negative-stain or cryo-EM. Representative images are shown in Figure S7 in Ciftci et al. (2020).
Single-molecule experiments
The preparation of quartz microscope slides and glass coverslips functionalized with PEG-biotin for TIRF microscopy is based on published methods (Joo and Ha, 2012).
We performed all single-molecule FRET imaging experiments using a home-built prism-based TIRF microscope constructed around a Nikon Eclipse Ti inverted microscope body, as described previously (Juette et al., 2016). Step-by-step guides on building a prism-based single-molecule TIRF microscopy setup are available (Selvin and Ha, 2008; Axelrod, 2008; Roy et al., 2008).
Passivate the quartz slides using a solution of 1 µM BSA and 1 µM duplex DNA in 150 µl of T50 buffer (see Recipe 9) by flowing the mixture through the injection port and incubating for 5 min. Further details can be found in Blanchard et al. (2004) and Joo and Ha (2012). Wash away excess BSA and DNA by flowing 150 µl of T50 buffer through the injection port.
To image non-specifically attached liposomes, flow no more than 10 µl of the proteoliposome suspension from Step E25. Keep a record of the sample quantity you have used in this step.
Immediately wash the surface with 150 µl of Resting buffer to remove unattached proteoliposomes.
Image attached molecules at 100 mW laser power at 10 Hz temporal resolution for a total of 300 s.
Photobleach non-specifically attached molecules by imaging the entire immobilization surface at 600 mW laser power and wash the chamber with 150 µl of T50 buffer.
Flow 150 µl of pre-mixed 0.8 µM streptavidin and 1 µM duplex DNA solution in T50 buffer and incubate for 5-10 min.
Wash away excess streptavidin-duplex DNA with 150 µl of T50 buffer and 150 µl of the Resting buffer before attaching proteoliposomes.
Flow the same volume of the liposomes as in Step F4. Immediately wash the surface with 150 µl Resting buffer to remove unattached proteoliposomes. If necessary, flow a more concentrated sample or increase the sample volume to reach the appropriate single-molecule density. At optimal density, the fraction of molecules rejected in gettraces module of SPARTAN should be ~40% (see Data analysis and the online SPARTAN manual). If injecting larger sample amounts, re-check non-specific attachment (Steps F4-F7) using a matching sample.
Image attached molecules at 100 mW laser power at 10 Hz temporal resolution for a total of 300 s and determine the number of molecules that specifically and non-specifically attach to the surface (Data analysis). Ideally, the non-specifically attached fluorescent particles imaged in Step F6 should be < 25% of the specifically attached molecules.
Supplement the Non-activating and Activating buffers (see Recipes 7-8) with an oxygen-scavenging system consisting of 2 mM PCA and 50 nM PCD (Aitken et al., 2008).
Image proteoliposomes to test for leakage by flowing 50 µl of the Non-activating buffer approximately 3 s after the start of the movie. Trace amounts of detergent left after Steps E17-E18 can allow L-asp to spontaneously diffuse into liposomes and lead to changes in FRET efficiency of encapsulated ccPEB1a in the absence of the active sodium-driven uptake. If a significant number of single-molecule trajectories (>10%) show FRET efficiency changes at this step, discard the liposomes and increase the amount of bio-beads or the number of bio-beads incubation cycles in your next preparation. Do not exceed 200 mg/ml of bio-beads in each round. If 200 mg/ml of bio-beads is still not enough, increase the number of incubation rounds. Carefully observe the turbidity level of liposome preparations after each round. A significant reduction in turbidity indicates the adsorption of liposomes to the excess bio-beads. We have observed that 6 rounds using 100 mg/ml bio-beads are enough to remove Triton X-100 detergent from our liposome preparations. This number can differ depending on the nature of detergent used to solubilize liposomes and incubation temperature. Detergents with higher critical micelle concentration (CMC) are easier to remove. One can check the remaining detergent in liposome preparations using a colorimetric assay (Urbani and Warne, 2005). We have observed that slight temperature variations (by a temperature-controlled chamber) can lead to significant differences in the detergent removal efficiency.
Image proteoliposomes to measure transport by flowing 50 µl of the Activating buffer approximately 3 s after the start of the movie. Adjust the time resolution and the total duration of movies depending on the transporter’s turnover rate. For cysGltPh variants with turnover times of ~30-100 s, we use the laser power of 20 mW at 2.5 Hz temporal resolution. At this laser power, the mean photobleaching time is ~300 s. For variants with turnover times of ~1 s, we use laser power of 75 mW at 10 Hz, resulting in the mean photobleaching time of ~60 s.
Wash the sample chamber with 2 ml of Resting buffer and photobleach any remaining fluorescent molecules as in Step F7 before attaching new proteoliposomes for the next round of transport recordings. Repeat Steps F10- F15.
Data analysis
Perform smFRET data analysis using SPARTAN, a freely available custom-built software package written in MATLAB (Juette et al., 2016). This provides a detailed explanation of particle detection and generation of fluorescence trajectories implemented in SPARTAN (https://www.scottcblanchardlab.com/software).
Process data files collected in TIFF format using the ‘gettraces’ module to detect single molecules and extract time-dependent fluorescence changes. Correct for crosstalk. This module creates an output file with .rawtraces extension. The crosstalk value can be calculated using the ‘crosstalkcorrect’ function implemented in SPARTAN, which calculates the mean acceptor intensity observed after the acceptor photobleaches across all trajectories in a given dataset, where acceptor photobleached before the donor. This function requires a .rawtraces file and returns the crosstalk value along with the percentage of trajectories used to calculate it.
Process the .rawtraces files from step 2 in ‘autotrace’ module. Apply the default parameters for FRET lifetime, background noise, number of donor blinking events, and signal-to-background ratio described in Supplementary Table 1 in Juette et al. (2016). We also apply a lower threshold of 0.4 and an upper threshold of 0.7 for FRET efficiency of the first frame of each single-molecule trajectory. We chose these thresholds based on the analysis of thousands of ccPEB1a single-molecule trajectories described in the ‘Engineering L-asp sensor’ section and shown in Figure 1B in Ciftci et al. (2020). FRET efficiency values outside of the 0.4-0.7 range likely correspond to denatured ccPEB1a.
Manually inspect the selected trajectories from ‘autotrace’ module using ‘sorttraces’ module. Discard trajectories with multiple donor or acceptor photobleaching steps which indicates the presence of more than one sensor per vesicle or non-specific labeling of the sensor. Discard also trajectories that show a visually apparent lack of anti-correlation between donor and acceptor intensities (Figure 4).
Classify trajectories into responding and nonresponding in ‘sorttraces’ by using the bins designated as ‘No’, ‘All’, or ‘Best’. Save responding and nonresponding trajectories separately. Figure S5 in Supplemental Information in Ciftci et al. (2020), shows examples of responding trajectories (Figure 4).
Manually record the time points at which FRET efficiency starts increasing in each single-molecule trajectory in the first column of an EXCEL file response_times.xlsx such that the order of the trajectories in responding_molecules.traces file matches the order of response time entries in response_times.xlsx. Thus, the response time detected in the 1st trajectory is in the 1st row, the response time detected in the 2nd trajectory is in the 2nd row, etc. Record the shortest response time observed in any trajectory to the first row of the second column; this number is a good approximation for the Activating buffer injection time. Tracking the order is only important if one uses the MATLAB script TurnoverTime.m in step 7.
Fit the gradual increases in FRET efficiencies following the initial step-wise increases to a time-dependent binding equation to obtain mean turnover times. Materials and Methods section ‘Analysis of the smFRET transport trajectories’ in Ciftci et al. (2020) explains the derivation of this equation. Optionally, use a MATLAB script TurnoverTime.m (https://github.com/hdciftci/SingleMoleculeTransportAssay.git) to fit the data. This script is modified based on avgFretTime.m script found in SPARTAN software (https://www.scottcblanchardlab.com/software) and requires two input files; the responding_molecules.traces and response_times.xlsx. Rate_constants.csv outputted by this script will have fitted turnover times along with other optimized parameters used in the equation.
Plot the distributions of the response times after subtracting the Activating buffer injection time, which correspond to the first half-cycle times, and the mean turnover times. We used PRISM (GraphPad Software, Inc.) for all histograms and Origin (OriginLab Corp., Northampton, MA, USA) for population contour plots in Ciftci et al. (2020). Data in Ciftci et al. (2020) are averages of at least 3 independent experiments.
For a more detailed explanation of the analysis approach, please refer to the Materials and Methods ‘Analysis of the smFRET trajectories’ section in Ciftci et al. (2020). A summarized view of steps and example data are shown in Figure 3 and Figure 4, respectively.
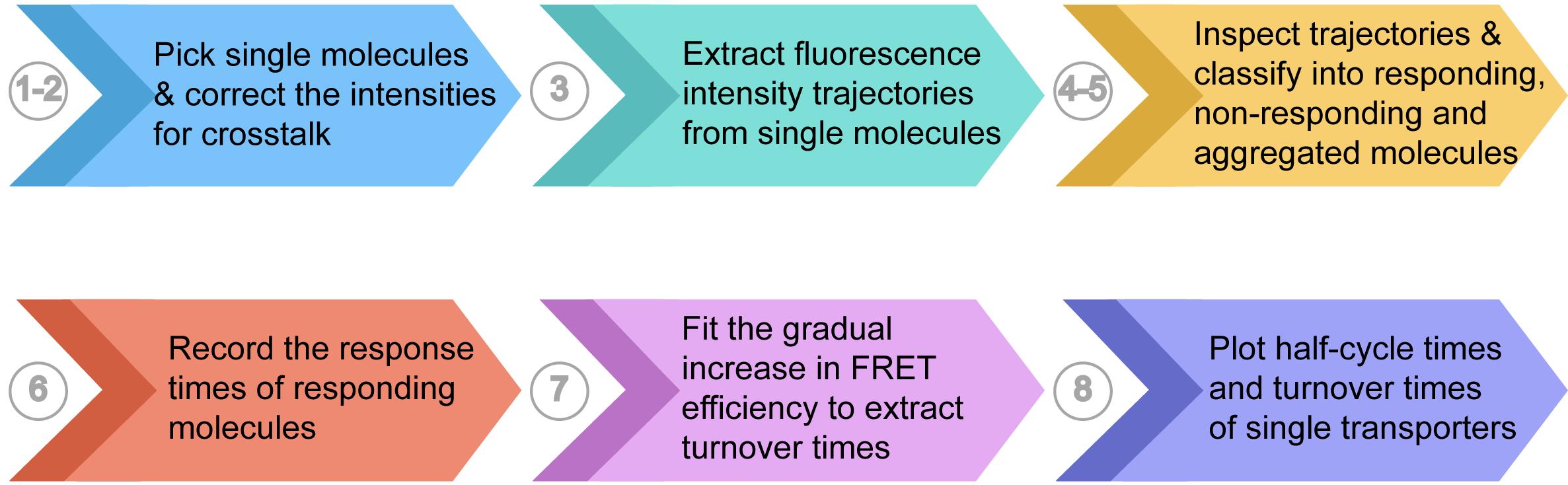
Figure 3. Flowchart of the data analysis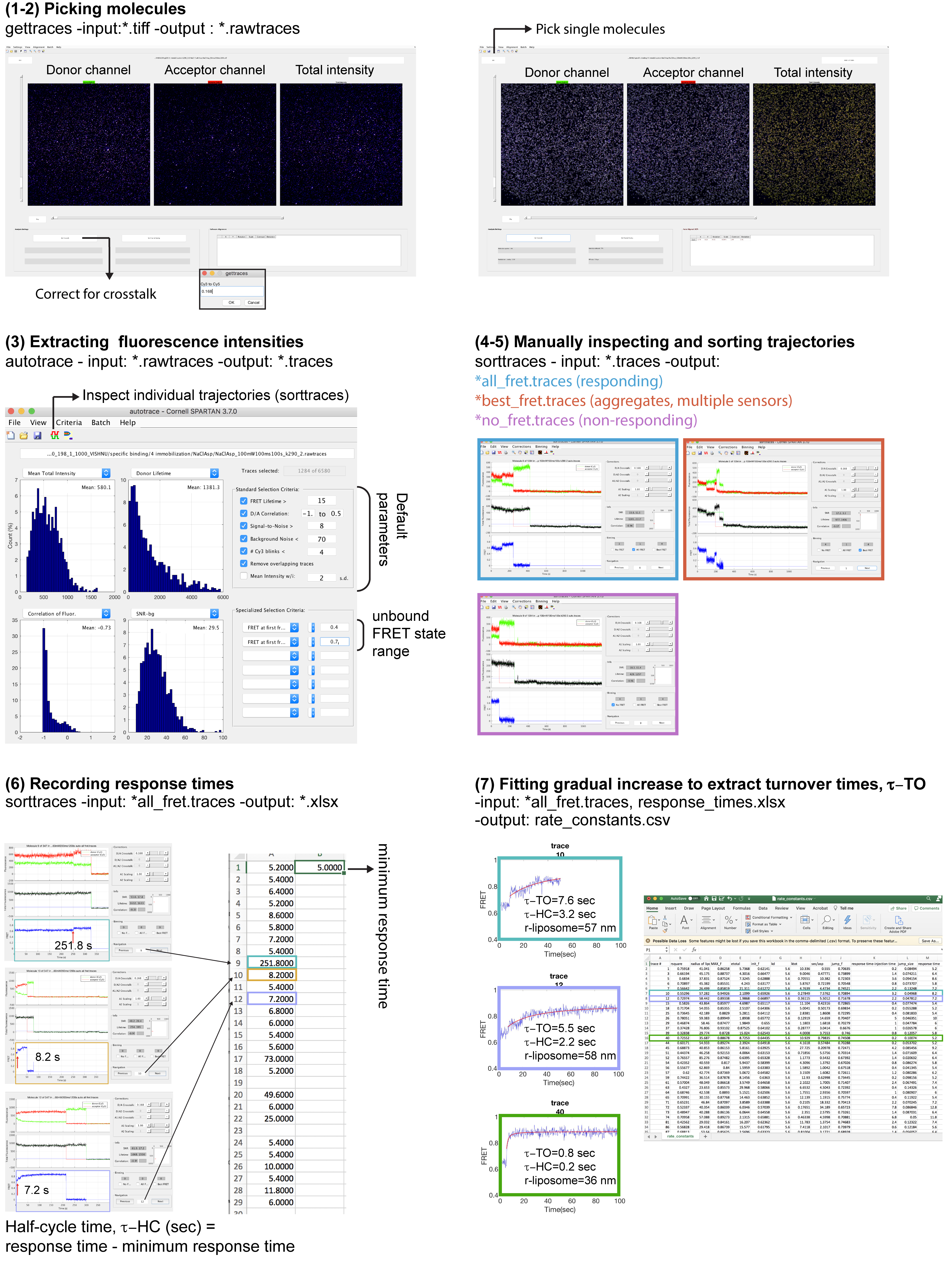
Figure 4. Example data generated from steps described in ‘Data Analysis’. (Steps 1-2) Fluorescence intensities are corrected for crosstalk, and single molecules are picked using ‘gettraces’ module. (Step 3) Fluorescence intensities of donor and acceptor molecules and the corresponding FRET efficiencies of picked molecules are extracted using ‘autotrace’ module. (Steps 4-5) Fluorescence trajectories are inspected for anti-correlation and response pattern using ‘sorttraces’ module and classified accordingly as responding or non-responding molecules. Trajectories without anti-correlation and/or showing multiple donor or acceptor photobleaching steps are excluded from further analysis. (Step 6) Response times are manually detected and recorded in an excel sheet. (Step 7) Average turnover times are calculated using the ‘Turnover Time’ script.
Recipes
Resuspension buffer
10 mM Tris-HCl, pH 8.0
2 mM EDTA
0.5 M sucrose
Wash buffer
10 mM Tris-HCl, pH 7.4
200 mM NaCl
1 mM Tris(2-carboxyethyl) phosphine (TCEP)
40 mM imidazole
Elution buffer
10 mM Tris-HCl, pH 7.4
200 mM NaCl
1 mM Tris(2-carboxyethyl) phosphine (TCEP)
250 mM imidazole
Size Exclusion Chromatography (SEC) buffer
50 mM HEPES/Tris, pH 7.4
200 mM KCl
cysGltPh SEC buffer
20 mM HEPES/Tris, pH 7.4
200 mM NaCl
0.1 mM L-aspartic acid
1 mM DDM
Resting buffer
50 mM HEPES/Tris, pH 7.4
200 mM KCl
Non-activating buffer
50 mM HEPES/Tris pH 7.4
200 mM KCl
1 µM L-aspartic acid
Activating buffer
50 mM HEPES/Tris, pH 7.4
200 mM NaCl
1 µM L-aspartic acid
T50 buffer
10 mM Tris/HCl, pH 7.4
50 mM KCl
cysGltPh Resuspension buffer
20 mM HEPES/Tris, pH 7.4
200 mM NaCl
1 mM EDTA, pH 7.4
Solubilization buffer
50 mM HEPES/Tris, pH 7.4
200 mM NaCl
1 mM TCEP
1 mM L-asp
Acknowledgments
We thank Roger Altman for the preparation of smFRET devices. Funding: NINDS R37NS085318 to O.B. and S.C.B., AHA 19PRE34380215 to H.D.C. 7R01GM098859 to S. C. B. This project has received funding from the European Union's Horizon 2020 research and innovation program under the Maire Sklodowska-Curie grant agreement MEMDYN No 660083 (G.H.M.H). This work is originally published as Ciftci et al. (2020).
Competing interests
S. C. B holds equity interest in Lumidyne Technologies.
References
- Aitken, C. E., Marshall, R. A. and Puglisi, J. D. (2008). An oxygen scavenging system for improvement of dye stability in single-molecule fluorescence experiments.Biophys J 94(5): 1826-1835.
- Akyuz, N., Altman, R. B., Blanchard, S. C. and Boudker, O. (2013). Transport dynamics in a glutamate transporter homologue. Nature 502(7469): 114-118.
- Axelrod, D. (2008). Chapter 7: Total internal reflection fluorescence microscopy. Methods Cell Biol 89: 169-221.
- Blanchard, S. C., Gonzalez, R. L., Kim, H. D., Chu, S. and Puglisi, J. D. (2004). tRNA selection and kinetic proofreading in translation. Nat Struct Mol Biol 11(10): 1008-1014.
- Borths, E. L., Poolman, B., Hvorup, R. N., Locher, K. P. and Rees, D. C. (2005). In vitro functional characterization of BtuCD-F, the Escherichia coli ABC transporter for vitamin B12 uptake. Biochemistry 44(49): 16301-16309.
- Boudker, O., Ryan, R. M., Yernool, D., Shimamoto, K. and Gouaux, E. (2007). Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature 445(7126): 387-393.
- Chaize, B., Colletier, J. P., Winterhalter, M. and Fournier, D. (2004). Encapsulation of enzymes in liposomes: high encapsulation efficiency and control of substrate permeability. Artif Cells Blood Substit Immobil Biotechnol 32(1): 67-75.
- Chen, T. W., Wardill, T. J., Sun, Y., Pulver, S. R., Renninger, S. L., Baohan, A., Schreiter, E. R., Kerr, R. A., Orger, M. B., Jayaraman, V., Looger, L. L., Svoboda, K. and Kim, D. S. (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499(7458): 295-300.
- Ciftci, D., Huysmans, G. H. M., Wang, X., He, C., Terry, D., Zhou, Z., Fitzgerald, G., Blanchard, S. C. and Boudker, O. (2020). Single-molecule transport kinetics of a glutamate transporter homolog shows static disorder. Sci Adv 6(22): eaaz1949.
- Colletier, J. P., Chaize, B., Winterhalter, M. and Fournier, D. (2002). Protein encapsulation in liposomes: efficiency depends on interactions between protein and phospholipid bilayer. BMC Biotechnol 2: 9.
- Costa, A. P., Xu, X. and Burgess, D. J. (2014). Freeze-anneal-thaw cycling of unilamellar liposomes: effect on encapsulation efficiency. Pharm Res 31(1): 97-103.
- Deuschle, K., Okumoto, S., Fehr, M., Looger, L. L., Kozhukh, L. and Frommer, W. B. (2005). Construction and optimization of a family of genetically encoded metabolite sensors by semirational protein engineering. Protein Sci 14(9): 2304-2314.
- Enkvetchakul, D., Bhattacharyya, J., Jeliazkova, I., Groesbeck, D. K., Cukras, C. A. and Nichols, C. G. (2004). Functional characterization of a prokaryotic Kir channel. J Biol Chem 279(45): 47076-47080.
- Drew, D. and Boudker, O. (2016). Shared Molecular Mechanisms of Membrane Transporters. Annu Rev Biochem 85: 543-572.
- Fitzgerald, G. A., Terry, D. S., Warren, A. L., Quick, M., Javitch, J. A. and Blanchard, S. C. (2019). Quantifying secondary transport at single-molecule resolution. Nature 575(7783): 528-534.
- Geertsma, E. R., Nik Mahmood, N. A., Schuurman-Wolters, G. K. and Poolman, B. (2008). Membrane reconstitution of ABC transporters and assays of translocator function.Nat Protoc 3(2): 256-266.
- Grewer, C. and Rauen, T. (2005). Electrogenic glutamate transporters in the CNS: molecular mechanism, pre-steady-state kinetics, and their impact on synaptic signaling. J Membr Biol 203(1): 1-20.
- Hou, B. H., Takanaga, H., Grossmann, G., Chen, L. Q., Qu, X. Q., Jones, A. M., Lalonde, S., Schweissgut, O., Wiechert, W. and Frommer, W. B. (2011). Optical sensors for monitoring dynamic changes of intracellular metabolite levels in mammalian cells. Nat Protoc 6(11): 1818-1833.
- Joo, C. and Ha, T. (2012). Preparing sample chambers for single-molecule FRET. Cold Spring Harb Protoc 2012(10): 1104-1108.
- Juette, M. F., Terry, D. S., Wasserman, M. R., Altman, R. B., Zhou, Z., Zhao, H. and Blanchard, S. C. (2016). Single-molecule imaging of non-equilibrium molecular ensembles on the millisecond timescale. Nat Methods 13(4): 341-344.
- Jung, H., Tebbe, S., Schmid, R. and Jung, K. (1998). Unidirectional reconstitution and characterization of purified Na+/proline transporter of Escherichia coli. Biochemistry 37(31): 11083-11088.
- Knol, J., Veenhoff, L., Liang, W. J., Henderson, P. J., Leblanc, G. and Poolman, B. (1996). Unidirectional reconstitution into detergent-destabilized liposomes of the purified lactose transport system of Streptococcus thermophilus. J Biol Chem 271(26): 15358-15366.
- Lingrel, J. B. and Kuntzweiler, T. (1994). Na+,K+-ATPase.J Biol Chem 269(31): 19659-19662.
- Liu, D., Evans, T. and Zhang, F. (2015). Applications and advances of metabolite biosensors for metabolic engineering. Metab Eng 31: 35-43.
- Nimigean, C. M. (2006). A radioactive uptake assay to measure ion transport across ion channel-containing liposomes. Nat Protoc 1(3): 1207-1212.
- Roy, R., Hohng, S. and Ha, T. (2008). A practical guide to single-molecule FRET. Nat Methods 5(6): 507-516.
- Selvin, P. R., and Ha, T. (2008). Single-molecule techniques. Cold Spring Harbor Laboratory Press.
- Shi, Y. (2013). Common folds and transport mechanisms of secondary active transporters. Annu Rev Biophys 42: 51-72.
- Urbani, A. and Warne, T. (2005). A colorimetric determination for glycosidic and bile salt-based detergents: applications in membrane protein research. Anal Biochem 336(1): 117-124.
- van der Heide, T. and Poolman, B. (2000). Osmoregulated ABC-transport system of Lactococcus lactis senses water stress via changes in the physical state of the membrane. Proc Natl Acad Sci U S A 97(13): 7102-7106.
- Vandenberg, R. J., and Ryan, R. M. (2013). Mechanisms of glutamate transport. Physiol Rev 93 (4):1621-57.
- Volpe, D. A. (2016). Transporter assays as useful in vitro tools in drug discovery and development. Expert Opin Drug Discov 11 (1):91-103.
- Yamashita, A., Singh, S. K., Kawate, T., Jin, Y. and Gouaux, E. (2005). Crystal structure of a bacterial homologue of Na+/Cl‑-dependent neurotransmitter transporters. Nature 437(7056): 215-223.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Ciftci, D., Huysmans, G. H. M., Wang, X., He, C., Terry, D., Zhou, Z., Fitzgerald, G., Blanchard, S. C. and Boudker, O. (2021). FRET-based Microscopy Assay to Measure Activity of Membrane Amino Acid Transporters with Single-transporter Resolution. Bio-protocol 11(7): e3970. DOI: 10.21769/BioProtoc.3970.
- Ciftci, D., Huysmans, G. H. M., Wang, X., He, C., Terry, D., Zhou, Z., Fitzgerald, G., Blanchard, S. C. and Boudker, O. (2020). Single-molecule transport kinetics of a glutamate transporter homolog shows static disorder. Sci Adv 6(22): eaaz1949.
Category
Biophysics > Microscopy
Biochemistry > Protein > Single-molecule Activity > Imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.