- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Generation of Mouse Primary Hypothalamic Neuronal Cultures for Circadian Bioluminescence Assays
Published: Vol 11, Iss 5, Mar 5, 2021 DOI: 10.21769/BioProtoc.3944 Views: 4348
Reviewed by: Chiara AmbrogioSamuel S HinmanKuldeep Singh Attri
Original research article
The authors used this protocol in:

Jul 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
An endogenous circadian clock system enables organisms to adapt to time-of-day dependent environmental changes. In consequence, most physiological processes exhibit daily rhythms of, e.g., energy metabolism, immune function, sleep, or hormone production. Hypothalamic circadian clocks have been identified to play a particular role in coordinating many of these processes. Primary neuronal cultures are widely used as a physiologically relevant model to study molecular events within neurons. However, as circadian rhythms include dynamic molecular changes over longer timescales that vary between individual cells, longitudinal measurement methods are essential to investigate the regulation of circadian clocks of hypothalamic neurons. Here we provide a protocol for generating primary hypothalamic neuronal cultures expressing a circadian luciferase reporter. Such reporter cells can be used to longitudinally monitor cellular circadian rhythms at high temporal resolution by performing bioluminescence measurements.
Keywords: Primary hypothalamic neuronsBackground
To adapt to recurring time-of-day dependent changes in their environment, many organisms have developed an endogenous circadian clock system that regulates 24-h rhythms of behavioral and physiological processes (Sharma, 2003). In mammals, a master circadian pacemaker resides in the hypothalamic suprachiasmatic nucleus (SCN). It coordinates cellular clock regulation throughout the body with external time. Daily patterns of sleep, appetite, and metabolism are regulated by cellular circadian clocks residing in hypothalamic neurons (Cedernaes et al., 2019).
In mammalian cells, circadian clocks consist of interlocked transcriptional-translational feedback loops (TTFLs). In the core TTFL, the transcription factors circadian locomotor output cycles kaput (CLOCK) and brain and muscle aryl hydrocarbon receptor nuclear translocator-like protein 1 (BMAL1 or ARNTL) activate the expression of their own repressors, period (PER1-3) and cryptochrome (CRY1/2), which leads to circadian oscillations of gene expression and abundance of protein products (Ko and Takahashi, 2006).
Methods to directly quantify mRNA and protein levels, such as quantitative PCR or Western blotting, require repetitive sampling from different individuals or preparations at different time points, which is labor-intensive, and, in case of short time intervals or low amounts of cellular material, highly impractical (Yu and Hardin, 2007). To overcome these problems, circadian reporters, where the expression of the firefly luciferase enzyme is under the control of a clock gene promoter (e.g., Bmal1 or Per2), have been developed (Brown et al., 2005; Ramanathan et al., 2012; Fang et al., 2017). They allow real-time tracking of cellular circadian rhythms by performing bioluminescence measurements and therefore are widely used to study circadian clock function (Ramanathan et al., 2012).
Immortalized cell lines are valuable tools for molecular biological research, as they are readily available and can be expanded without limitations (Pan et al., 2009). In recent years, they have become established in-vitro models for the investigation of hypothalamic clocks (Fick et al., 2010 and 2011; Tsang et al., 2020). However, since immortalized cell lines are genetically and phenotypically different from their tissue origins, data obtained from such systems has to be interpreted critically and a verification with a more physiologically relevant model system is advised (Pan et al., 2009).
Primary neuronal cultures are obtained directly from the animal’s central nervous system and maintain many physiological and biochemical characteristics of their tissue origin (Gordon et al., 2013; Verma et al., 2020). Therefore, they are useful model systems to address physiological relevance. Here we provide a protocol for the generation of primary hypothalamic neuron cultures stably transduced with a circadian Bmal1-luciferase reporter. Creation of these reporter cells allows observation and quantitative description of hypothalamic neuronal circadian rhythms in real-time, avoiding labor-intensive and material-consuming biochemical experiments. These cells can further be used to investigate clock resetting effects of various factors such as hormones or metabolites.
Materials and Reagents
96-well plate (Corning, catalog number: 3610 )
BD Falcon cell strainer, 70 µm (BD Biosciences, catalog number: 352350 )
100-mm culture dish
35-mm dish
15-ml Falcon tube
10-ml serological pipette
Fire-polished glass pipette
2-8-month-old male and female C57BL/6J mice
Bmal1-luciferase lentivirus (Brown et al., 2005)
Poly-D-lysine hydrobromide (Millipore Sigma, catalog number: P6407 )
Laminin (BD Biosciences, catalog number: 354232 )
Hank’s balanced salt solution, HBSS (PAA, catalog number: H15-008 )
Earle’s balanced salt solution, EBSS, with Phenol Red (Thermo Fisher Scientific, Gibco, catalog number: 24010043 )
Papain, suspension (Worthington Biochemical Corporation, catalog number: LS003126 )
Deoxyribonuclease I, DNaseI (Worthington Biochemical Corporation, catalog number: LS002058 )
Neurobasal medium, minus phenol red (Thermo Fisher Scientific, Gibco, catalog number: 12348017 )
B-27 supplement (Thermo Fisher Scientific, Gibco, catalog number: 17504044 )
GlutaMAX supplement (Thermo Fisher Scientific, Gibco, catalog number: 35050061 )
Ovomocoid (Worthington Biochemical Corporation, catalog number: LS003085 )
Bovine serum albumin (MilliporeSigma, catalog number: A7030 )
L-cysteine (MilliporeSigma, catalog number: C7352 )
Fetal bovine serum, FBS (Thermo Fisher Scientific, Gibco, catalog number: 10500-064 )
Trypan blue solution (MilliporeSigma, catalog number: T8154 )
Cytosine β-D-arabinofuranoside, AraC (Millipore Sigma, catalog number: C1768 )
Pasteur pipettes (Th. Geyer, catalog number: 7691061 )
2-Mercaptoethanol (Millipore Sigma catalog number: M3148 )
Adhesive clear PCR seal (Biozym, catalog number: 600208 )
D-luciferin sodium salt (Applichem, catalog number: A1006 )
Polybrene, Hexadimethrine bromide (Millipore Sigma, catalog number: H9268 )
Equipment
Sterile laminar flow hood (Thermo Fisher Scientific, model: MSC-Advantage, catalog number: 51025411 )
Multimode microplate reader (Berthold Technologies, model: Tristar LB941 )
Fluorescence microscope (Nikon, model: Eclipse Ts2R )
LED unit (Nikon, model: C-LEDFL470 )
Fluorescence filter cube (Nikon, model: C-LED470 , Excitation: 470/40 nm, Dichroic: 500 nm, Emission: 535/55 nm)
10× objective (Nikon, model: CFI Achromat ADL-10× , NA: 0.4)
20× objective (Nikon, model: CFI Achromat LWD ADL-20× , NA: 0.4)
Hemocytometer (Laboroptik, model: Neubauer )
Water bath (GFL, model: 1002 )
Dissecting microscope (Leica, model: MZ6 )
Dissection scissors (Fine Science Tools, catalog number: 91402-14 )
Dissection forceps (Fine Science Tools, catalog number: 11000-12 )
No.11 scalpel (Feather, catalog number: 200210011 )
No.5 sharp forceps (Fine Science Tools, catalog number: 11252-20 )
2 × curved forceps (Fine Science Tools, catalog number: 11274-20 )
Student Fine Scissors (Fine Science Tools, catalog number: 91460-11 )
Software
MicroWin 2000 (Labsis, https://labsis.de)
Procedure
Schedule matings
To generate E15-16 embryos, schedule the mating day of the adult mice 15-16 d before dissection. We use 2-8-month-old adult C57BL/6J mice for mating.
On the next morning, confirm successful mating by vaginal plug check as described previously (Behringer et al., 2016).
Confirm pregnancy by palpitation or visually before dissection.
Reagent preparation
Note: Avoid repeated freeze-thaw cycles for labile reagents.
Prepare poly-D-lysine (PDL) stock solution (500 µg/ml). Divide into 5-ml aliquots and store at -20 °C.
Prepare laminin stock solution (0.1 µg/µl) in PBS. Divide into 250-µl aliquots and store at -20 °C.
Prepare ovo/albumin inhibition solution, containing 6 mg ovomucoid and 6 mg bovine serum albumin in 6 ml EBSS. Store at 4 °C until use.
Prepare DNaseI solution, with 1,000 U DNase I in 500 µl EBSS and store at -20 °C.
Prepare plating medium (Neurobasal + 2% (v/v) B27 + 2 mM Glutamax + 10% (v/v) FBS + 1× penicillin/streptomycin) and store at 4 °C until use. Equilibrate at 37 °C before adding it to the culture.
Prepare feeding medium (same as plating medium, but without FBS) and store at 4 °C until use. Equilibrate at 37 °C before adding it to the culture.
Prepare digestion solution (100 U Papain, 5 ml EBSS, 1.1 mM EDTA (11 µl 0.5 M EDTA), 5.5 mM cysteine (3.3 mg), 0.067 mM 2-Mercaptoethanol (2.34 µl 1 % (v/v)/14.3 M pure liquid) and 250 µl DNaseI solution), sterile filtrate with a 0.2-µm filter. Activate at 37 °C for 20-30 min before use.
Prepare resuspension medium (2.7 ml EBSS, 300 µl ovo/albumin inhibition solution, 150 µl DNaseI solution).
PDL and laminin double coating
One day before dissection coat 96-well plates with PDL and laminin.
Dilute PDL stock solution with PBS to working concentration 50 µg/ml. Mix 0.5 ml PDL stock solution (500 µg/ml) with 4.5 ml PBS to prepare 5 ml diluted PDL solution (50 µg/ml).
Filter with 0.2-µm filter before use.
Coat the culture surface of a 96-well plate with 7.5 µg/cm2 PDL. Cover the wells of a 96-well plate with 48 µl diluted PDL solution (50 µg/ml).
Incubate overnight at room temperature (or at least for 2 h at 37 °C).
Wash 3 times with 100 µl sterile H2O.
Allow to dry completely under a sterile cell culture hood.
Thaw laminin stock solution slowly at 2-8 °C.
Dilute laminin stock solution with PBS to a working concentration of 6.4 µg/ml. Mix 320 µl laminin stock solution (0.1 µg/µl) with 4.68 ml PBS to prepare 5 ml diluted laminin solution (6.4 µg/ml).
Cover the PDL-coated wells with 50 µl diluted laminin solution (6.4 µg/ml). The final coating concentration will be around 1 µg/cm2.
Incubate for 2 h at 37 °C.
Wash 3 times with sterile 100 µl PBS, add 100 µl plating medium and store at 37 °C and 5 % CO2 in the incubator.
Dissection
Note: A swiftly executed preparation procedure is crucial for cell viability. Consider practicing the procedure several times before preparing experimental samples. Before starting the dissection, ensure that required materials are in place and that all equipment is disinfected.
Use embryonic day 15 to 16 (E15-16) old embryos from a pregnant mother (C57BL/6J mothers usually have 8-10 pups).
Work under a laminar flow hood and always apply aseptic techniques to reduce the risk of contamination with bacteria, fungi, and mycoplasma.
Sacrifice mother by cervical dislocation, open the abdominal cavity with dissection scissors and forceps. The embryos are located at the posterior part of the abdominal cavity.
Carefully remove the uterus horns with two curved forceps with gentle opposite pulling motions and transfer them into a 100-mm culture dish filled with ice-cold HBSS.
Extract embryos with dissection scissors and curved forceps and transfer them into a new 100-mm culture dish with ice-cold HBSS.
Decapitate the embryos with dissection scissors.
Hold the head in position by piercing No. 5 sharp forceps into orbital cavities and by pushing the rostral part of the brain down. Simultaneously use curved forceps to remove the outer skin and skull, by peeling them gently off from caudal to rostral. Start with the left hemisphere and then repeat that step with the right hemisphere. Carefully remove the brain with curved forceps and deposit it into a 35-mm Petri dish with ice-cold HBSS. Repeat Steps D5 to D7 for the other embryos.
Note: Avoid applying pressure onto the tissue while extracting the brains, to maintain brain integrity. Furthermore, take care that the brains are always covered with medium, do not let them dry out.
Under a dissecting microscope, isolate the hypothalami with forceps and scalpel (see Figure 1 for details). Collect them in a 35-mm dish with ice-cold HBSS and keep on ice. Use a cut 1,000-µl pipette tip or a dropper with a wide opening to transfer the dissected tissue.

Figure 1. Steps for hypothalamus dissection from intact E16 brains. A. Turn the brain over so that the ventral part is facing upwards. Remove the caudal part of the brain by making a coronal cut at the posterior border of the mammillary bodies. Make a second coronal cut ~1.5 mm anterior from the first to remove the rostral part of the brain. B. Rotate the remaining brain to get a coronal orientation and make sure that the rostral part is facing upwards. Dissect the hypothalamic tissue block: make two lateral cuts ~0.5 mm each from the midline and one additional cut ventral to the anterior commissure (ac). Red dashed lines indicate the cutting positions. The dissected area is highlighted in blue. Further abbreviations: LV – lateral ventricle, Thal – thalamus, Hypo – hypothalamus, Teg – tegmentum, Med – medulla oblongata, VP – ventral pallidum, OCh – optic chiasm, 3V – 3rd ventricle, Cx – cortex, CPu – caudate putamen.
Dissociation and plating
Transfer dissected hypothalami into a 15-ml Falcon tube, remove remaining HBSS and add 5 ml digestion solution.
Digest the tissue pieces for 30-60 min at 37 °C while stirring gently every 4-5 min.
Note: A longer digestion period may increase cell yield, but it will also decrease viability. The incubation time must be determined empirically. Start with 30 min, determine yield and viability as described in Step E13. Increase incubation time if necessary.
Triturate 13 times with a 10-ml serological pipette.
Carefully and slowly triturate 13 times with a fire-polished glass pipette. Avoid creating bubbles during trituration.
Wait 2 min for remaining undissociated tissue to settle and transfer supernatant into a new 15- ml Falcon tube.
Centrifuge at 300 × g for 5 min.
Remove the clear supernatant and resuspend the cell pellet with 3 ml resuspension medium.
Gently triturate 7 times with a fire-polished glass pipette.
Remove remaining tissue clumps by using a 70 µm cell strainer.
Carefully and slowly transfer the cell suspension to 5 ml ovo/albumin inhibition solution in a 15-ml Falcon tube and centrifuge at 70 × g for 5 min to prepare a discontinuous density gradient.
Remove supernatant.
Add 2-3 ml plating medium and resuspend 7 times with a fire-polished glass pipette.
Quantify the number of viable cells by trypan blue exclusion assay (e.g., with a Neubauer chamber).
Seed 3.25 × 105 viable cells/cm2 in plating medium into a 96-well plate double coated with PDL and laminin.
Feeding and lentiviral transduction with Bmal1-luciferase reporter
On the next day, transduce the cells with Bmal1-luciferase lentivirus (Brown et al., 2005). Details for lentiviral particle production are described in Tsang et al. (2020).
Note: It is recommended to use a GFP-expressing control virus to determine transduction efficiency.
Thaw lentiviral aliquots at room temperature immediately before use. Avoid keeping them for prolonged times at ambient temperature and avoid unnecessary freeze-thaw cycles.
Prepare several lentiviral dilutions (e.g., 0, 1:5, 1:50. 1:250) in feeding medium containing 16 µg/ml polybrene.
Note: It is recommended testing at least three different concentrations to determine the optimal transduction conditions. We transduced the cells with ~ 1 × 108 infection units (IFUs) per 1 ml in the presence of 8 µg/ml polybrene.
Replace half of the volume of the plating medium by half of the volume of lentiviral particle-containing feeding medium.
24 h later, refresh half of the volume of the old medium with fresh feeding medium containing 5 µM AraC.
Feed the cells every 3 d with feeding medium, as above, but without AraC.
Note: It is recommended to perform quality control experiments to ensure that the prepared culture is free from microbial contamination (e.g., bacteria, fungi, or mycoplasma). Mycoplasma contamination can be tested by using PCR-based detection kits (LookOut Mycoplasma PCR Detection Kit, MilliporeSigma). Although cell identification and integrity tests are commonly performed in cell culture, they are not necessary for these primary cultures, since isolated cells are directly used for the experiment and not maintained for longtime.
Synchronization and bioluminescence measurements
On day 9 in vitro (DIV9), synchronize the cells for 2 h with 100 nM dexamethasone. Therefore, pipette 25 µl pre-warmed feeding medium containing 900 nM dexamethasone into the wells containing 200 µl medium and incubate for 2 h at 37 °C and 5% CO2.
During incubation prepare the feeding medium containing 0.5 mM D-luciferin and place it into a water bath at 37 °C.
Note: D-luciferin is light sensitive. Protect from light.
After incubation aspirate medium and change to pre-warmed feeding medium with 0.5 mM D-luciferin.
Seal the plate with transparent adhesive foil, place it into the microplate reader and start the measurement.
Perform the luminescence measurement without filter at 34 °C with an integration time of 1 min per well.
Normalize all bioluminescence traces by subtracting the 24-h running average and analyze circadian parameters as described previously (Landgraf et al., 2015).
Note: Synaptic formation starts to be evident at DIV7 (Figure 2). Neurons are considered to be mature at DIV14 (Biffi et al., 2013; Kos et al., 2016). We use DIV9 neurons for standard circadian luciferase experiments. Bmal1-luciferase reporter rhythms are stable for a week without further medium refreshing during the recording.
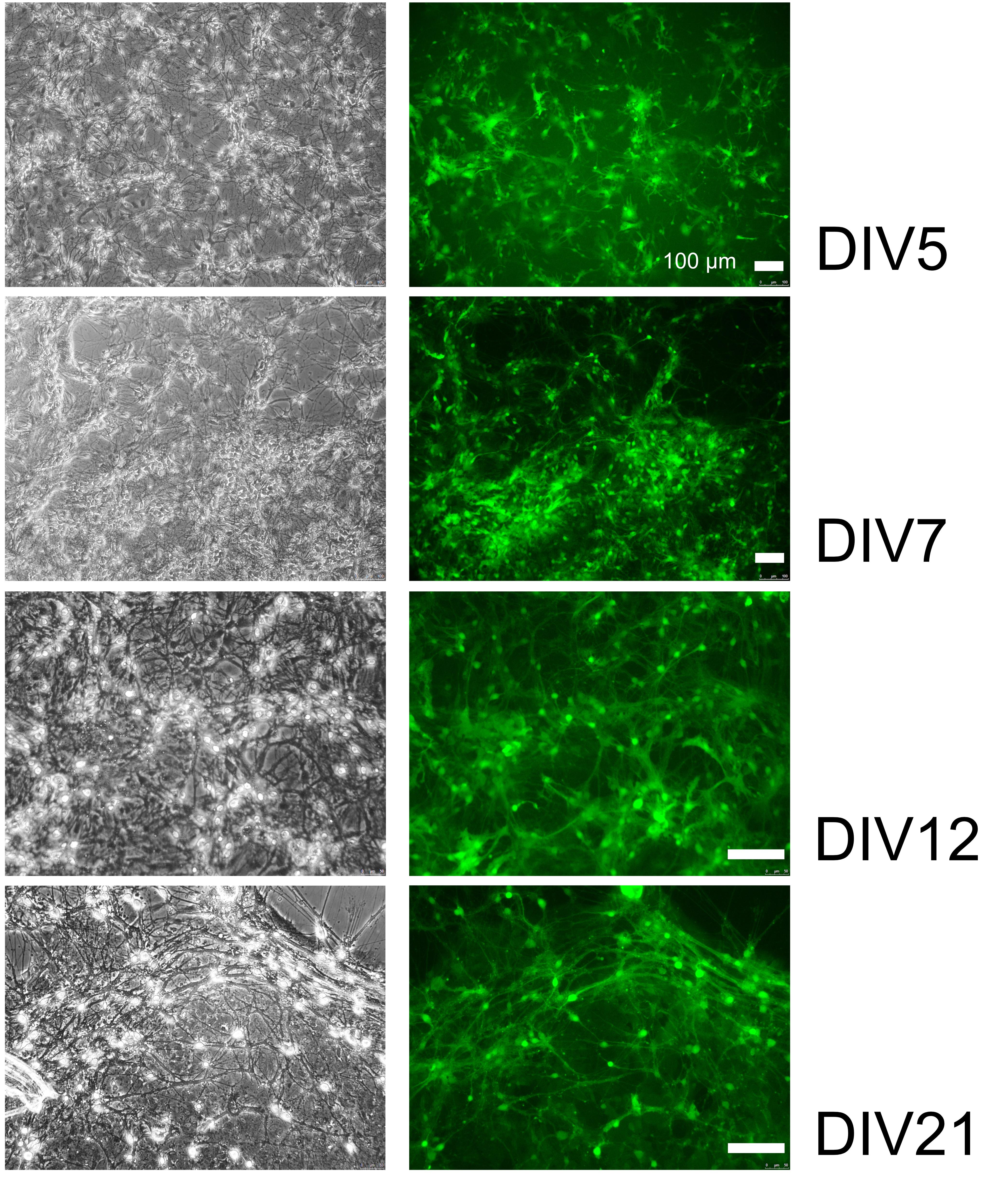
Figure 2. Synaptic connectivity increases with days in vitro. Representative bright-field (left) and fluorescence (right) images of primary hypothalamic neurons transduced with a GFP-expressing lentivirus. 10× and 20× magnification.
Data analysis
The Mikrowin2000 software allows real-time monitoring of bioluminescence signals. Bmal1-luciferase rhythms are plotted and displayed. During the measurement, wells of interest can be selected and examined. Mikrowin2000 continuously saves the experiment as a *.dat file, which can be opened by the software once the measurement procedure has been completed. To analyze the data, export the raw data to Excel. Calculate the 24-h running average and subtract that from baseline readings for normalization (Figure 3). We use GraphPad Prism for plotting, sine wave fitting, rhythm parameter determination and statistical analyses. Statistical assessment of rhythmicity is done with JTK_cycle or CircaCompare (Hughes et al., 2010; Parsons et al., 2020).
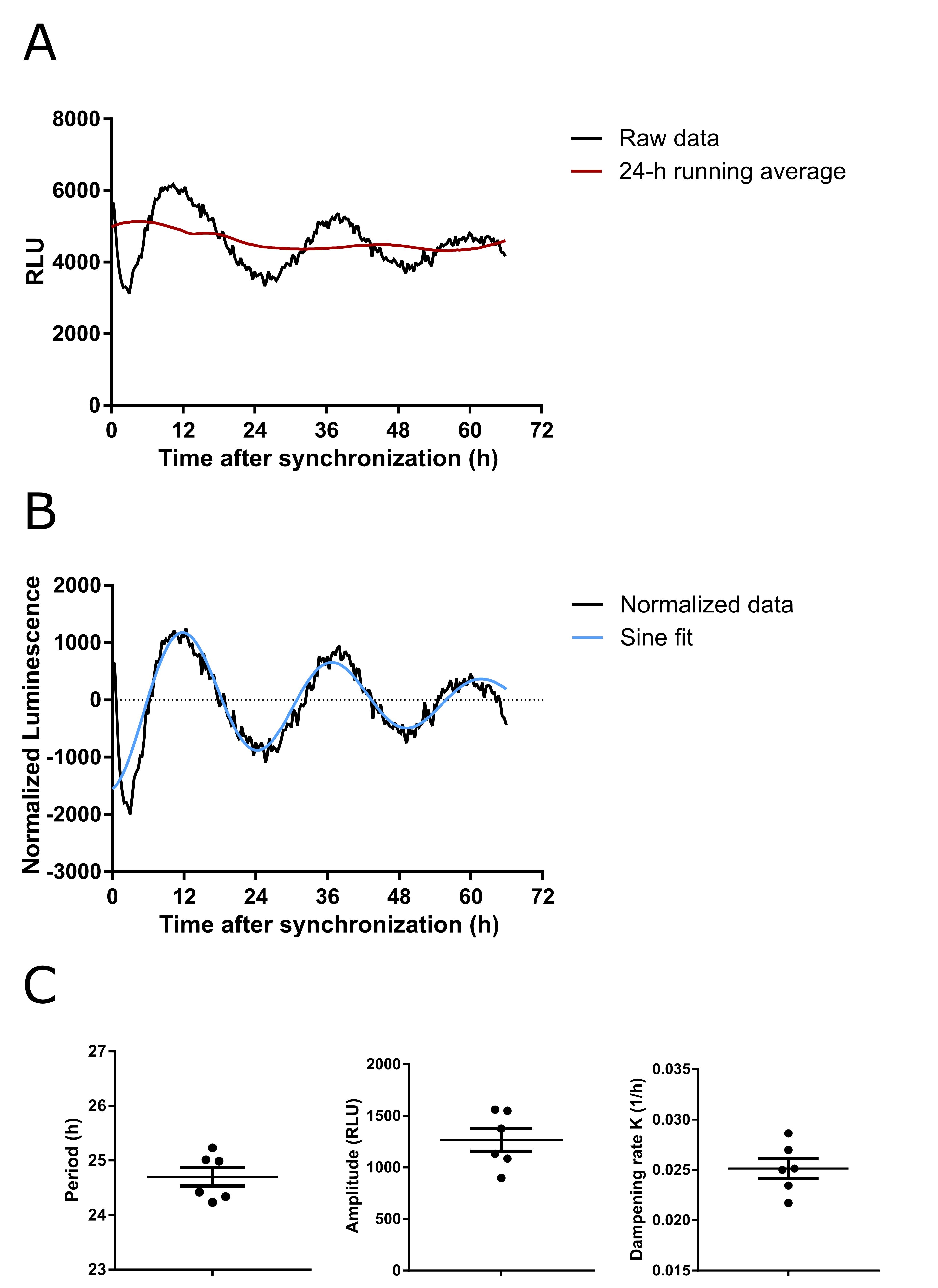
Figure 3. Bioluminescence measurements of synchronized primary hypothalamic neurons expressing Bmal1-luciferase. Bioluminescence traces are normalized by subtracting their 24-h running average. Circadian parameters, such as amplitude, period (wavelength), and dampening rate (K) are determined by fitting a damped sine wave function (Y = Amplitude*exp(-K*X)*sin((2*pi*X/Wavelength) + Phase shift). A. Representative raw data (black) and the calculated 24-h running average (red). B. Representative normalized data (black) and damped sine wave fit (blue). C. Quantification of period, amplitude and dampening rate. Data are presented as mean ± SEM (n = 6).
Acknowledgments
This study was supported by funds of the German Research Foundation (DFG; OS353-7/1, OS353-10/1 and GRK-1957). The protocol was used for Tsang et al. (2020).
Competing interests
The authors report no conflict of interest.
Ethics
Animal experiments reported in this protocol have been approved by the ethics commission of the Ministry of Energy Change, Agriculture, Environment and Digitalization (MELUR) of the State of Schleswig-Holstein (Az 4_2019-10-01_Oster; 2019-2021).
References
- Biffi, E., Regalia, G., Menegon, A., Ferrigno, G. and Pedrocchi, A. (2013). The Influence of Neuronal Density and Maturation on Network Activity of Hippocampal Cell Cultures: A Methodological Study. PLOS ONE 8(12): e83889.
- Behringer, R., Gertsenstein, M., Nagy K.V. and Nagy A. (2016). Selecting Female Mice in Estrus and Checking Plugs. Cold Spring Harb Protoc. 2016(8). doi: 10.1101/pdb.prot092387.
- Brown, S. A., Fleury-Olela, F., Nagoshi, E., Hauser, C., Juge, C., Meier, C. A., Chicheportiche, R., Dayer, J.-M., Albrecht, U. and Schibler, U. (2005). The Period Length of Fibroblast Circadian Gene Expression Varies Widely among Human Individuals. PLOS Biol 3(10): e338.
- Cedernaes, J., Huang, W., Ramsey, K. M., Waldeck, N., Cheng, L., Marcheva, B., Omura, C., Kobayashi, Y., Peek, C. B. and Levine, D. C., et al. (2019). Transcriptional Basis for Rhythmic Control of Hunger and Metabolism within the AgRP Neuron. Cell Metab 29(5): 1078-1091.e5.
- Fang, M., Kang, H.-G., Park, Y., Estrella, B. and Zarbl, H. (2017). In Vitro Bioluminescence Assay to Characterize Circadian Rhythm in Mammary Epithelial Cells. J Vis Exp 127: e55832.
- Fick, L. J., Fick, G. H. and Belsham, D. D. (2010). Rhythmic clock and neuropeptide gene expression in hypothalamic mHypoE-44 neurons. Mol Cell Endocrinol 323(2): 298-306.
- Fick, L. J., Fick, G. H. and Belsham, D. D. (2011). Palmitate alters the rhythmic expression of molecular clock genes and orexigenic neuropeptide Y mRNA levels within immortalized, hypothalamic neurons. Biochem Biophys Res Commun 413(3): 414-419.
- Gordon, J., Amini, S. and White, M. K. (2013). General overview of neuronal cell culture. Methods Mol Biol 1078: 1-8.
- Hughes, M. E., Hogenesch, J. B. and Kornacker, K. (2010). JTK_CYCLE: An efficient non-parametric algorithm for detecting rhythmic components in genome-scale datasets. J Biol Rhythms 25(5): 372-380.
- Ko, C. H. and Takahashi, J. S. (2006). Molecular components of the mammalian circadian clock. Human Molecular Genetics 15(suppl_2): R271-R277.
- Kos, A., Wanke, K. A., Gioio, A., Martens, G. J., Kaplan, B. B. And Aschrafi, A. (2016). Monitoring mRNA Translation in Neuronal Processes Using Fluorescent Non-Canonical Amino Acid Tagging. J Histochem Cytochem 64(5):323-333.
- Landgraf, D., Tsang, A. H., Leliavski, A., Koch, C. E., Barclay, J. L., Drucker, D. J. and Oster, H. (2015). Oxyntomodulin regulates resetting of the liver circadian clock by food. eLife 4: e06253.
- Pan, C., Kumar, C., Bohl, S., Klingmueller, U. and Mann, M. (2009). Comparative proteomic phenotyping of cell lines and primary cells to assess preservation of cell type specific functions. Mol Cell Proteomics 8(3): 443-450.
- Parsons, R., Parsons, R., Garner, N., Oster, H. and Rawashdeh, O. (2020). CircaCompare: A method to estimate and statistically support differences in mesor, amplitude and phase, between circadian rhythms. Bioinformatics 36(4): 1208-1212.
- Ramanathan, C., Khan, S. K., Kathale, N. D., Xu, H. and Liu, A. C. (2012). Monitoring Cell-autonomous Circadian Clock Rhythms of Gene Expression Using Luciferase Bioluminescence Reporters. J Vis Exp 67: e4234.
- Sharma, V. K. (2003). Adaptive Significance of Circadian Clocks. Chronobiol Int 20(6): 901-919.
- Tsang, A. H., Koch, C. E., Kiehn, J.-T., Schmidt, C. X. and Oster, H. (2020). An adipokine feedback regulating diurnal food intake rhythms in mice. eLife 9: e55388.
- Verma, A., Verma, M. and Singh, A. (2020). Animal tissue culture principles and applications. Animal Biotechnology 269-293.
- Yu, W. and Hardin, P. E. (2007). Use of firefly luciferase activity assays to monitor circadian molecular rhythms in vivo and in vitro. Methods Mol Biol 362: 465-480.
Article Information
Copyright
![]() Schmidt et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Schmidt et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Schmidt, C. X., Tsang, A. H. and Oster, H. (2021). Generation of Mouse Primary Hypothalamic Neuronal Cultures for Circadian Bioluminescence Assays . Bio-protocol 11(5): e3944. DOI: 10.21769/BioProtoc.3944.
- Tsang, A. H., Koch, C. E., Kiehn, J.-T., Schmidt, C. X. and Oster, H. (2020). An adipokine feedback regulating diurnal food intake rhythms in mice. eLife 9: e55388.
Category
Neuroscience > Cellular mechanisms > Cell isolation and culture
Cell Biology > Cell isolation and culture
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.