- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Primer ID Next-Generation Sequencing for the Analysis of a Broad Spectrum Antiviral Induced Transition Mutations and Errors Rates in a Coronavirus Genome
Published: Vol 11, Iss 5, Mar 5, 2021 DOI: 10.21769/BioProtoc.3938 Views: 7803
Reviewed by: Alka MehraSuresh PantheeKirsten A. Copren
Original research article
The authors used this protocol in:

Apr 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Next generations sequencing (NGS) has become an important tool in biomedical research. The Primer ID approach combined with the MiSeq platform overcomes the limitation of PCR errors and reveals the true sampling depth of population sequencing, making it an ideal tool to study mutagenic effects of potential broad-spectrum antivirals on RNA viruses. In this report we describe a protocol using Primer ID sequencing to study the mutations induced by antivirals in a coronavirus genome from an in vitro cell culture model and an in vivo mouse model. Viral RNA or total lung tissue RNA is tagged with Primer ID-containing cDNA primers during the initial reverse transcription step, followed by two rounds of PCR to amplify viral sequences and incorporate sequencing adaptors. Purified and pooled libraries are sequenced using the MiSeq platform. Sequencing data are processed using the template consensus sequence (TCS) web-app. The Primer ID approach provides an accurate sequencing protocol to measure mutation error rates in viral RNA genomes and host mRNA. Sequencing results suggested that β-D-N4-hydroxycytidine (NHC) greatly increased the transition substitution rate but not the transversion substitution rate in the viral RNA genomes, and cytosine (C) to uridine (U) was found as the most frequently seen mutation.
Keywords: CoronavirusBackground
Next generation sequencing (NGS) has been extensively used in biomedical research for the last decade. When applying NGS to study intra-host viral populations for RNA viruses, modifications in library prep and sequencing protocols need to be considered. The virus titers (or viral loads) vary greatly from specimen to specimen. While conventional NGS platforms require 1-500 ng of DNA (or RNA) in a sequencing run, in most cases a clinical sample will have less than 100 fg of viral RNA, requiring that the viral RNA first be converted to cDNA using reverse transcriptase, followed by one or two rounds of PCR amplification to generate enough material for sequencing. However, the extensive cycles of PCR amplification will cause nucleotide mis-incorporation, recombination and amplification bias. Furthermore, the NGS platforms have relatively high error rates adding additional uncertainty to the resulting sequences (Liu et al., 2012).
We have developed the Primer ID NGS approach to overcome the errors and bias from the conventional NGS approaches (Jabara et al., 2011; Zhou et al., 2015). During the initial cDNA synthesis step, we use cDNA primers with an embedded 11-base degenerate block of nucleotides (the Primer ID) to tag each viral RNA template with a unique ID, a version of the approach of adding a unique molecular identifier (UMI). The Primer ID is carried on throughout the downstream amplifications and sequencing. After sequencing, all raw sequences with the same ID are collapsed to make a template consensus sequence (TCS). Each TCS is linked to an original template that was queried in the initial cDNA synthesis step, and the number of TCS shows the sequencing sampling depth of the viral population. By the construction of consensus sequence from multiple raw sequencing reads for each viral template, we can greatly reduce the PCR and sequencing errors. We have shown that the Primer ID approach can greatly reduce the sequencing error to 1 in 10,000 nucleotides (100-fold reduction from the raw sequencing reads) and also reveal the true sampling depth of the viral population, making it possible to accurately measure the substitution rates and diversity in the viral genomes in a population (Figure 1) (Zhou et al., 2015). We have further developed a multiplexing Primer ID approach, allowing the sequencing of multiple regions of the viral genome in a single cDNA synthesis/PCR (Figure 2) (Dennis et al., 2018).

Figure 1. Primer ID Approach. A. The structure of the Primer ID primer and its binding to the viral RNA template. B. An example of creating the template consensus sequence from raw sequence reads.

Figure 2. Library prep steps for the Multiplexed Primer ID sequencing. Biological sequence region in blue color, Primer ID region in yellow color, forward spacer region in orange, MiSeq barcode sequence region in purple.
This approach is ideal for studying intra-host RNA virus populations due to its ability to accurately characterize individual viral genomes. We have used this approach to determine the genetic structures of intra-host viral populations in HIV (Zhou et al., 2016; Lee et al., 2017; Dennis et al., 2018; Abrahams et al., 2019; Adewumi et al., 2020; Council et al., 2020), to study drug resistance mutations in HIV and HCV (Clutter et al., 2017; Zhou et al., 2018; Keys et al., 2015), and to detect nucleotide mutation rates in single-stranded DNA molecule after heating (Lewis et al., 2016).
In this report we describe a protocol using the Primer ID MiSeq sequencing to detect the mutagenic effect of EIDD-1931(β-D-N4-hydroxycytidine, NHC) and EIDD-2801 (an orally bioavailable prodrug of EIDD-1931 and is also recognized as MK-4482 or Molnupiravir) in the Middle East respiratory syndrome coronavirus (MERS-CoV) genome from in vitro cell culture and in vivo mouse models. In the cell culture sequencing experiment, we used a multiplexed Primer ID protocol to sequence multiple regions in the MERS-CoV ORF1b in a single library prep and sequencing reaction, while in the sequencing of total lung RNA from MERS-CoV infected mice, we used a multiplexed Primer ID protocol targeting both MERS-CoV genomes and two selected mouse mRNAs in a single library prep and sequencing reaction, in which we can directly compared the mutagenic effect induced by EIDD-2801 on MERS-CoV genomes and host mRNA. This protocol is not restricted to MERS-CoV and EIDD-1931 study only, but it can be used to detect viral RNA mutation rate in general.
Materials and Reagents
Pipette tips
Collection tubes (QIAGEN, catalog number: 19201 , stored at room temperature)
Qubit Assay Tubes (ThermoFisher Scientific, Invitrogen, catalog number: Q32856 , stored at room temperature)
SuperScript III One-Step RT-PCR System (ThermoFisher Scientific, Invitrogen, catalog number: 12574026 , stored at -20 °C)
Primer ID primers (Integrated DNA Technologies) at 100 µM in Tris-HCl buffer stored at -20 °C
SuperScript III Reverse Transcriptase (ThermoFisher Scientific, Invitrogen, catalog number: 18080085 , stored at -20 °C)
RNaseOUT Recombinant Ribonuclease Inhibitor (ThermoFisher Scientific, Invitrogen, catalog number: 10777019 , stored at -20 °C)
Ribonuclease H (ThermoFisher Scientific, Invitrogen, catalog number: 18021071 , stored at -20 °C)
KAPA2G Robust PCR kits (Hotstart PCR kits with dNTPs) (Roche, catalog number: KK5516 , stored at -20 °C)
KAPA2G HiFi HotStart (with dNTPs) (Roche, catalog number: KK2502 , stored at -20 °C)
RNAClean XP (Beckman Coulter, catalog number: A63987 , stored at 4 °C)
AMPure XP (Beckman Coulter, catalog number: A63881 , stored at 4 °C)
Ethanol 200 Proof (Decon Laboratories, catalog number: 2716 )
DNase/RNase-free water
MinElute Gel Extraction kit (QIAGEN, catalog number: 28606 , stored at 4 °C for MinElute columns, room temperature for other reagents)
Qubit dsDNA BR Assay Kit (ThermoFisher Scientific, Invitrogen, catalog number: Q32853 , stored at 4 °C for standard 1 and standard 2, room temperature for other reagents)
Experion DNA 12K analysis kit (Bio-Rad, catalog number: 7007108 , stored at room temperature for the chips, 4 °C for other reagents)
MiSeq Reagent Kit v3 (600-cycle) (Illumina, catalog number: MS-102-3003 , stored at -20 °C for box 1 and 4 °C for box 2)
Sodium hydroxide (Sigma-Aldrich, catalog number: 221465 , stored at room temperature)
Tris hydrochloride (Sigma-Aldrich, catalog number: 10812846001 , stored at room temperature)
PhiX Control V3 (Illumina, catalog number: FC-110-3001 , stored at -20 °C)
10 mM Tris-HCl with 0.1% TWEEN 20 (Teknova, catalog number: T7724 , stored at room temperature)
Equipment
Pipettes
Magnetic rack
Thermal cycler (ThermoFisher Scientific, Applied Biosystems, catalog number: 4384638 )
Centrifuge (Eppendorf, model: 5424 )
DynaMag-2 Magnet (ThermoFisher Scientific, Invitrogen, catalog number: 12321D )
Qubit 2.0 Fluorometer (ThermoFisher Scientific, Invitrogen, catalog number: Q32866 )
Experion Electrophoresis Station (Bio-Rad, catalog number: 7007010 )
MiSeq System (Illumina, catalog number: SY-410-1003 )
Software
Primer-BLAST (NCBI, https://www.ncbi.nlm.nih.gov/tools/primer-blast/) (Ye et al., 2012)
bcl2fastq pipeline (v.2.20.0) (Illumina, https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html)
TCS web app (UNC, https://tcs-dr-dept-tcs.cloudapps.unc.edu/)
TCS pipeline (v.1.3.8) (UNC, https://github.com/SwanstromLab/PID)
RUBY package viral_seq (v.1.0.8) (UNC, https://github.com/ViralSeq/viral_seq)
Procedure
We first design the primers for the multiplexed Primer ID library prep. After extraction of viral RNA from the tissue culture supernatants or total RNA from lung tissues, we construct MiSeq sequencing libraries using the multiplexed Primer ID library prep protocol from each specimen. The multiplexed Primer ID protocol allows us to make a sequencing library of multiple regions of the viral RNA in one reaction. We pool up to 24 barcoded libraries (from 24 different specimens) for each sequencing run. We use Illumina MiSeq V3 kit (600 cycle) for the sequencing. We choose paired-end sequencing to have longer read length and better error correction from overlapping regions over single-end sequencing.
Primer ID cDNA primer and PCR primer design
Design primer sequences using NCBI Primer-BLAST.
Use ORF1b region of the reference genome MERS-CoV NC_019843 as “PCR template” in the Primer-BLAST interface to design primers for MERS-CoV. Use mouse interferon-induced protein with tetratricopeptide repeats 2 (Ifit2, reference: NM_008332), interferon-induced protein with tetratricopeptide repeats 3 (Ifit3, reference: NM_010501), ISG15 ubiquitin-like modifier (Isg15, reference: NM_015783), interferon-induced protein with tetratricopeptide repeats 1 (Ifit1, reference: NM_008331) and chemokine (C-X-C motif) ligand 10 (Cxcl10, reference: NM_021274) genomes as templates to design primers for mouse mRNA.
Set PCR product size as Min: 400 bp and Max: 600 bp.
Set Primer melting temperatures as Min: 52, Opt: 55, Max: 58.
Select 3-4 pairs of primers for each template region.
Use SuperScript III one-step RT-PCR system to test Primers with best amplification efficiency for each region.
Use MERS-CoV viral RNA from tissue culture described above for MERS-CoV primer testing. Use extracted total mouse lung RNA as template for mouse mRNA primer testing. Make serial 1:10 titrations of the template RNA in DNase/RNase-free water as RT-PCR templates. Make dilutions up to 10-6.
Use following recipe to make one one-step RT-PCR mixture (total volume of 10 µl) in PCR tubes.
5 µl of 2× buffer
1.8 µl of DNase/RNase-free water
0.4 µl of Forward primer (20 µM)
0.4 µl of Reverse R (20 µM)
0.4 µl of Enzyme mix
2 µl of Template RNA
Use the following thermal cycler condition for RT-PCR.
50 °C for 30 min
94 °C for 2 min
40 cycles of:
94 °C for 15 s
55 °C for 30 s
68 °C for 30 s
68 °C for 5 min
Examine the PCR products using gel electrophoresis with 1% agarose gel.
Figure 3 shows the example gel image of amplicons from tested primers for MERS-CoV.
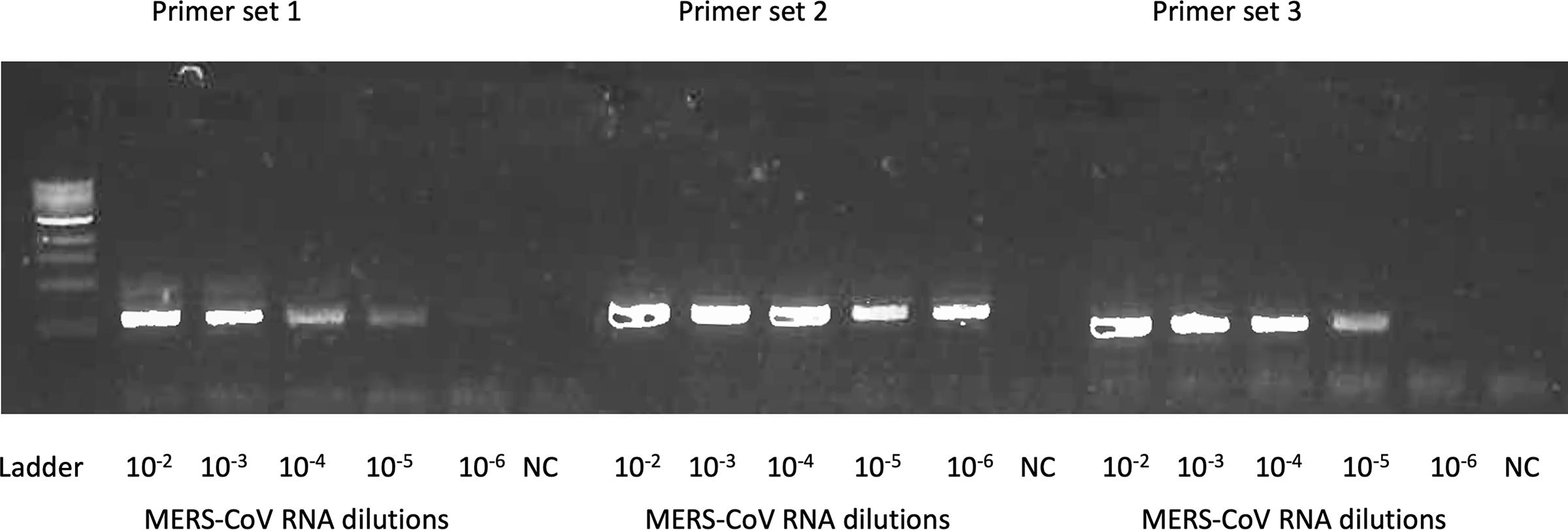
Figure 3. An example one-step RT-PCR gel image for the primer design. Three sets of primers targeting same MERS-CoV genome were tested. A serial dilution of MERS-CoV RNA up to 10-6 dilution was used to test the primers. The SSIII one-step PCR system was used for the amplification. PCR products were examined by gel electrophoresis with 1% agarose gel. Primer set 2 had the best amplification efficiency thus it was selected for downstream Primer ID primer design. NC, no-reaction control.Design the Primer ID cDNA primers and other primers for the MiSeq library prep.
Design cDNA primers by attaching the Primer ID (a degenerate block of 11-base long sequence) and adaptor sequence to the reverse primers.
Forward primer of the 1st round PCR is designed by joining a common sequence “GCCTCCCTCGCGCCATCAGAGATGTGTATAAGAGACAGNNNN” on the 5’ end with the forward primer sequencing designed at step a) on the 3’. The Ns in the joined primer sequences are random sequences to increase the sequencing quality.
Reverse primer of the 1st round PCR is designed against the adaptor sequence section of the Primer ID cDNA primer, and is universal regardless of regions (Table 1).
The 2nd round PCR primers are designed to incorporate the MiSeq sequencing adaptors with barcodes.
Order primers designed above in Steps A1a-A1d from Integrated DNA Technologies (IDT). Request hand-mixing of the precursors for the degenerate block of sequence of the cDNA primers to have an even distribution of bases at each position.
Table 1 shows the complete list of primers used in this protocol including primers for Primer ID MiSeq library prep and customized MiSeq sequencing primer. Table 2 shows the sequences of the indexed primers.
Notes:
Primer-BLAST is not necessary to design primers. Primers can be manually designed based on known reference genomes.
Hand-mixing of nucleotide precursors prior to Primer ID cDNA primer synthesis is important to ensure the degenerate sequences have an even distribution of bases (A, C, G, T) at each position of the degenerate block.
Table 1. Primers used for MiSeq library prep and sequencing. Primer sequences with gene-specific regions are highlighted in blue. They can be replaced with other gene-specific primer sequences when designing primers for other viruses.
Primer 5'-3' sequence Comment 41R_PID11* GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNNNNNNNCAGTATGACCTTCCTGTTGCTTCT cDNA primer. Targeting 20331-20350 on the reference genome. nsp10_PID11* GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNNNNNNNCAGTCCTAAAGACGACATCAGTGG cDNA primer. Targeting 13488-13507 on the reference genome. nsp12_PID11* GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNNNNNNNCAGTATAGCCAAAGACACAAACCG cDNA primer. Targeting 15983-16002 on the reference genome. nsp14_PID11* GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNNNNNNNCAGTGAACATCGACAAAGAAAGGG cDNA primer. Targeting 18715-18734 on the reference genome. ifit3_PID11* GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNNNNNNNCAGTTTCAGCCACTCCTTTATCCC cDNA primer. Targeting mice IFIT3 mRNA. isg15_PID11* GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNNNNNNNCAGTGGGGCTTTAGGCCATACTC cDNA primer. Targeting mice ISG15 mRNA. 41F_AD GCCTCCCTCGCGCCATCAGAGATGTGTATAAGAGACAGNNNNGCTACAAGTTCGTCCTTTGG 1st round PCR forward primer. Targeting 19812-19831 on the reference genome nsp10_AD GCCTCCCTCGCGCCATCAGAGATGTGTATAAGAGACAGNNNNTGCTCAGGTGCTAAGCGAAT 1st round PCR forward primer. Targeting 12983-13002 on the reference genome nsp12_AD GCCTCCCTCGCGCCATCAGAGATGTGTATAAGAGACAGNNNNATAGGCTTCGATGTTGAGGG 1st round PCR forward primer. Targeting 15388-15407 on the reference genome nsp14_AD GCCTCCCTCGCGCCATCAGAGATGTGTATAAGAGACAGNNNNATTGCAAGCTGGTTCTAACA 1st round PCR forward primer. Targeting 18260-18279 on the reference genome ifit3_AD GCCTCCCTCGCGCCATCAGAGATGTGTATAAGAGACAGNNNNCGATCCACAGTGAACAACAG 1st round PCR forward primer. Targeting mice IFIT3 mRNA. isg15_AD GCCTCCCTCGCGCCATCAGAGATGTGTATAAGAGACAGNNNNTGGGACCTAAAGGTGAAGATG 1st round PCR forward primer. Targeting mice ISG15 mRNA. Adapter R GTGACTGGAGTTCAGACGTGTGCTC 1st round PCR reverse primer Universal Adapter AATGATACGGCGACCACCGAGATCTACACGCCTCCCTCGCGCCATCAGAGATGTG 2nd round PCR forward primer with Illumina adapter sequence Indexed Adapter** CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCAGACGTGTGCTC 2nd round PCR reverse primer with Illumina adapter sequence and indices (NNNNNNN) Old Nextera GCCTCCCTCGCGCCATCAGAGATGTGTATAAGAGACAG Customized sequencing primer
* cDNA primer has a block of degenerate nucleotides (Ns) as the Primer ID.
** Ns in the Indexed Adapter primers are a set of 24 pre-designed barcodes for multiplexing samples for one MiSeq run. See the index sequences and adapter sequences in Table 2.
Table 2. Indexed adapter sequences. Note the index sequences are the reverse complement sequences of the highlighted (red) region in the adapter sequencesIndexed Adapter Index Index Sequence Sequence (5’-3’) PCR Primer, Index 1 1 ATCACGA CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 2 2 CGATGTA CAAGCAGAAGACGGCATACGAGATACATCGGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 3 3 TTAGGCA CAAGCAGAAGACGGCATACGAGATGCCTAAGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 4 4 TGACCAA CAAGCAGAAGACGGCATACGAGATTGGTCAGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 5 5 ACAGTGA CAAGCAGAAGACGGCATACGAGATCACTGTGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 6 6 GCCAATA CAAGCAGAAGACGGCATACGAGATATTGGCGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 7 7 CAGATCA CAAGCAGAAGACGGCATACGAGATGATCTGGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 8 8 ACTTGAA CAAGCAGAAGACGGCATACGAGATTCAAGTGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 9 9 GATCAGA CAAGCAGAAGACGGCATACGAGATCTGATCGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 10 10 TAGCTTA CAAGCAGAAGACGGCATACGAGATAAGCTAGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 11 11 GGCTACA CAAGCAGAAGACGGCATACGAGATGTAGCCGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 12 12 CTTGTAA CAAGCAGAAGACGGCATACGAGATTACAAGGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 13 13 TCCATAA CAAGCAGAAGACGGCATACGAGATTATGGAGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 14 14 GTACTAA CAAGCAGAAGACGGCATACGAGATTAGTACGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 15 15 ACAGTAA CAAGCAGAAGACGGCATACGAGATTACTGTGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 16 16 CTCATGA CAAGCAGAAGACGGCATACGAGATCATGAGGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 17 17 ACGATAA CAAGCAGAAGACGGCATACGAGATTATCGTGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 18 18 TGCAGAA CAAGCAGAAGACGGCATACGAGATTCTGCAGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 19 19 TTCATAA CAAGCAGAAGACGGCATACGAGATTATGAAGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 20 20 TGCTGTA CAAGCAGAAGACGGCATACGAGATACAGCAGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 21 21 TATCACA CAAGCAGAAGACGGCATACGAGATGTGATAGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 22 22 TGGATAA CAAGCAGAAGACGGCATACGAGATTATCCAGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 23 23 CGCATTA CAAGCAGAAGACGGCATACGAGATAATGCGGTGACTGGAGTTCAGACGTGTGCTC PCR Primer, Index 24 24 GCCTTAA CAAGCAGAAGACGGCATACGAGATTAAGGCGTGACTGGAGTTCAGACGTGTGCTC
Primer ID MiSeq library prep
cDNA synthesis
Use viral RNA extracted from cell culture supernatants from infected cells or total lung tissue RNA from infected mice as amplification templates. cDNA synthesis step should be performed in BSL2+ facility until after the Ribonuclease H step.
Completely thaw, vortex and spin down 5× buffer, dNTPs and DTT.
Make cDNA primer working mix from the 100 µM primer stock. The final concentration for each primer is 10 µM in the primer mix.
Determine regions for sequencing.
Add 10 µl of each cDNA primer (no more than 5 different primers to multiplex) to an Eppendorf microcentrifuge tube, and add DNase/RNase-free water to a total volume of 100 µl.
Increase the amount of primers and DNase/RNase-free water proportionally if larger volume of primer mix is needed.
Pipette the following components into a 0.5 ml RNase-free PCR tube.
2 µl of dNTP mix (10 mM each)
1 µl of Primer ID cDNA primer mix (10 µM each)
23 µl of RNA template (vRNA or total lung RNA)
Heat the PCR tube on a thermal cycler at 65°C for 5 min, then cool down to 4 °C for 2 min with lid closed (lid temperature at 105 °C).
Add the following components to the tube. If doing the cDNA synthesis in batch, make a master mix and add 14 µl of the mixture to each tube.
8 µl of 5× buffer
2 µl of DTT (100 mM)
2 µl of RNaseOUT (40 U/µl)
2 µl of SSIII reverse transcriptase (200 U/µl)
Mix well by pipetting up and down for a few times. Transfer the tube(s) to a thermal cycler. Use the following thermal cycler program for the cDNA synthesis.
55 °C for 1 h
50 °C for 1 h
70 °C for 15 min
To each tube, add 0.5 μl (1U) Ribonuclease H, incubate at 37 °C for 20 min. After this step, the cDNA can be handled on a regular workbench (BSL1).
Purify cDNA with RNAClean XP.
Resuspend the beads by vortexing and take an aliquot out. Keep in room temperature for at least 30 min before use.
Resuspend the beads thoroughly by vortexing. Add 28 µl of RNAClean XP beads to 1.5 ml tubes. Transfer all of the cDNA reaction (40.5 µl) to each tubes with beads.
Mix the RNAClean XP and sample thoroughly by pipette mixing 15 times. Vortexing is not recommended. Let the tube incubate at room temperature for 20 min. Spin down briefly if samples touches the upper part of the tube.
Place the tube onto the magnetic tube rack for 5 min to separate the beads from solution.
Slowly aspirate the cleared solution from the tube and discard. This step should be performed while the tube is situated on the rack. Do not disturb the magnetic beads, which have formed a spot on the side of the tube.
Dispense 400 μl of 70% ethanol into the tube and incubate for 30 s at room temperature. Aspirate out the ethanol and discard. Repeat for a total of three washes. It is important to perform these steps with the tube situated on the magnetic rack. Do not disturb the separated magnetic beads. Be sure to remove all of the ethanol from the bottom of the well as it may contain residual contaminants.
Let the reaction tube air-dry up to 10 min on the rack with the cap open. The tube(s) should air-dry until the last visible traces of ethanol evaporate. However, over drying the sample may result in a lower recovery.
Remove tube from magnetic rack and resuspend beads in 24 µl DNase/RNase-free water by pipetting up and down. Place tube back on rack and leave for 3 min.
Pipette the eluent (23.5 µl) from the tube while it is situated on the magnetic tube rack.
First round PCR amplification with KAPA2G Robust PCR kit.
In the first round PCR, we amplify all the cDNA we make from the previous step using a mixture of forward primers targeting multiple regions of the genome.
Make forward primer working mix from the 100 µM primer stock.
Determine regions of primer binding based on the initial choice of placement of the cDNA primers.
Add 10 µl of each forward primer (no more than 5 primers) to an Eppendorf microcentrifuge tube, and add DNase/RNase-free water to a total volume of 100 µl.
Increase the amount of primers and DNase/RNase-free water proportionally if larger volume of primer mix is needed.
Completely thaw, vortex and spin down reagents (except for enzyme) before use.
Use the following recipe to make one first round PCR mix (total volume of 50 µl).
10 µl of 5× Buffer A
10 µl of Enhancer
1 µl of dNTPs
2.5 µl of Forward primer mix
2.5 µl of adapter R primer
0.5 µl of KAPA Robust polymerase
23.5 µl of template cDNA
Mix well by pipetting up and down for a few times. Transfer the tube(s) to a thermal cycler. Use the following thermal cycler program for the first round PCR amplification.
95 °C for 1 min
25 cycles of:
95 °C for 15 s
58 °C for 60 s
72 °C for 30 s
72 °C for 3 min
Purify PCR products using AMPure XP cleanup beads.
Vortex the 1 ml aliquot of AMPure XP cleanup beads and remove the needed volume. Keep in room temperature for at least 30 min before use.
Transfer the first round of PCR reactions into 1.5 ml DNase-free tubes.
Resuspend the beads. Add 35 µl AMPure XP beads to each PCR reaction.
Mix the AMPure XP and sample thoroughly by vortexing. Let the tube incubate at room temperature for 5 min before proceeding to the next step (incubate off the rack).
Place the tube onto the magnetic tube rack for 5 min to separate the beads from solution.
Slowly aspirate the cleared solution from the tube and discard. This step should be performed while the tube is situated on the rack. Do not disturb the magnetic beads, which have formed a spot on the side of the tube.
Dispense 400 μl of 70% ethanol into the tube and incubate for 30 s at room temperature. Aspirate out the ethanol and discard. Repeat for a total of two washes. It is important to perform these steps with the tube situated on the rack. Do not disturb the separated magnetic beads. Be sure to remove all of the ethanol from the bottom of the well as it may contain residual contaminants.
Let the reaction tube air-dry up to 10 min on the rack with the cap open. The tube(s) should air-dry until the last visible traces of ethanol evaporate. Over drying the sample may result in a lower recovery.
Remove tube from rack and resuspend beads in 50 µl DNase-free water by pipetting up and down. Place tube back on rack and leave for 3 min.
Pipette the 45 µl eluent from the tube while it is situated on the magnetic tube rack. Transfer the eluent to a new 1.5 ml microcentrifuge tube.
Second round of PCR amplification with KAPA2G HiFi PCR kits.
Complete thaw, vortex and spin down reagents (except for enzyme) before use.
Use the following recipe to make one second round PCR mix (total volume of 25 µl).
10 µl of 5× KAPA HiFi Buffer
1 µl of dNTPs
1 µl of Universal Adapter
1 µl of indexed Adapter
0.5 µl of KAPA HiFi polymerase
2 µl of purified DNA from the first round PCR.
Mix well by pipetting up and down for a few times. Transfer the tube(s) to a thermal cycler. Use the following thermal cycler program for the cDNA synthesis.
95 °C for 2 min
35 cycles of:
98 °C for 20 s
63 °C for 15 s
72 °C for 30 s
72 °C for 3 min
Gel purification of PCR products using QIAGEN MinElute Gel extraction kit.
Before gel extraction, run 2 µl PCR products on 1% agarose gel to check if the library has the right size.
Make 1.2% (w/v) agarose gel with ethidium bromide (final concentration of 0.2-0.5 µg/ml).
Load all the second round PCR products on the 1.2% agarose gel. Adjust the voltage based on the size of the gel. The ideal electric field strength is 4 V/cm. Run the gel for 30 min.
Excise target DNA fragments of the right size (500 bp to 1 kb) on a non-UV blue light transilluminator.
Weigh the gel; add 3 volumes of Buffer QG to 1 volume of gel.
Incubate at 50 °C for 10 min to completely dissolve the gel fragment. Vortex every 2-3 min to help dissolve.
Check the color of the gel solution (should be yellow, otherwise add 10 µl 3 M sodium acetate of pH 5.0).
Apply the sample to the MinElute column and centrifuge at maximum speed for 1 min.
Transfer the column to a new collection tube, discard the filtrate, add 500 µl buffer QG and centrifuge at maximum speed for 1 min.
Transfer the column to a new collection tube, discard the filtrate, add 0.75 ml buffer PE, incubate for 5 min at room temperature, centrifuge at maximum speed for 1 min.
Transfer the column to a new collection tube, discard the filtrate, centrifuge at maximum speed for additional 3 min.
Put the column in a new 1.5 ml tube, add 10 µl buffer EB. Stand for 4 min, centrifuge at maximum speed for 2 min.
Quantification, pooling and QA/QC.
Quantify each gel-purified library using Qubit dsDNA BR Assay kit, following the manufacturer’s protocol. Use 1 µl of the library for each quantification.
Pool the quantified individual libraries in equal amounts. We suggest pooling a total number of 20-24 libraries, each with 100 to 150 ng DNA per library. Use the maximum available volume of libraries if the calculated volume exceeds the actual volume.
Purify the pool of libraries with AMPure XP beads following the protocol above in the First Round PCR section. The volume of AMPure beads to use equals the volume of pooled libraries multiplied by 0.7.
Quantify the purified pooled libraries using Qubit dsDNA BR Assay kit, following the manufacturer’s protocol.
Run the pooled libraries with the Experion Automated Electrophoresis System with DNA 12k analysis kit following the manufacturer’s protocol. An example of gel image of Experion is shown in Figure 4.
Check the size of DNA peaks on the Experion gel image. Make sure there is no visible peak around 100 bp (primer dimer). If it is present, purify the pooled libraries with AMPure XP beads again and repeat the quantification and Experion electrophoresis.
Calculate the average DNA size (bp) of the library pool based on the Experion molarity (nM) and Experion concentration (ng/µl) using the following equation. Do NOT use the Qubit concentration to calculate the average DNA size.

It is important to set up the cDNA reaction in a separate room from where the PCR amplification takes place to avoid potential contamination from PCR products.
Viral RNA in this protocol that have very little quantity may not be measurable. qRT-PCR is usually recommended to confirm the copy numbers of viral RNA before library prep. We recommend at least 1,000 copies of input viral RNA for each library prep. However, this protocol may still work with less than 1,000 copies of input viral RNA, with compromised sampling depth.
Primer mix is only needed when multiplexed regions are sequenced. It is possible to use one set of cDNA/forward primers at 10 µM concentration when sequencing only one region.
Multiplexed regions normally should not overlap as this may inhibit the efficiency of priming and/or amplification.
Include one negative control and possibly one positive control for each batch of library prep.
It is recommended to aliquot the RNAclean XP beads and the AMPure XP beads into 1 ml aliquots. It is very important to warm the beads at room temperature for 30 min before use.
The ratio of the volume of beads to cDNA/DNA is 0.7. It is optimized to remove long Primer ID cDNA primers or PCR primers.
We recommend a second round of cDNA purification for some clinical specimens, especially plasma samples, to remove inhibitors of the downstream amplification. After the cDNA is eluted in water (in this case, use 40 µl water), add 28 µl of new RNAclean XP beads and purify the cDNA for a second time.
Residual beads in the cDNA or PCR products eluent won’t affect the downstream amplification.
First and second round PCR amplifications use two different KAPA PCR kits. The enzyme and buffer tubes look similar. It is very important to use the right buffer with the right enzyme for PCR 1 and PCR 2.
Save the first round PCR product in the 1.5 ml tube in -20 °C, in case it is needed to repeat PCR2.
Make sure to excise the fragment of the right size during gel extraction. Occasionally larger fragments (1.5-3 kb) can be present on a gel, which should not be extracted.
Library concentration normally should be at least 20 ng/µl. Discard libraries with concentration less than 10 ng/µl as they are less likely to have high quality sequences.
Qubit 2.0 fluorometer has been discontinued. Qubit 4.0 fluorometer is available at the time this protocol is written.
Qubit dsDNA HS Assay kit is not recommended to quantify the libraries. The HS kits have an upper detection limit of 100 ng/µl. In some cases the library concentration can be higher than 100 ng/µl.
The Bio-Rad Experion system is used in this protocol but it is close to obsolete. A better substitution is Agilent TapeStation System (Agilent, catalog number: G2991AA). The TapeStation system can directly measure the average DNA size of the library pool.
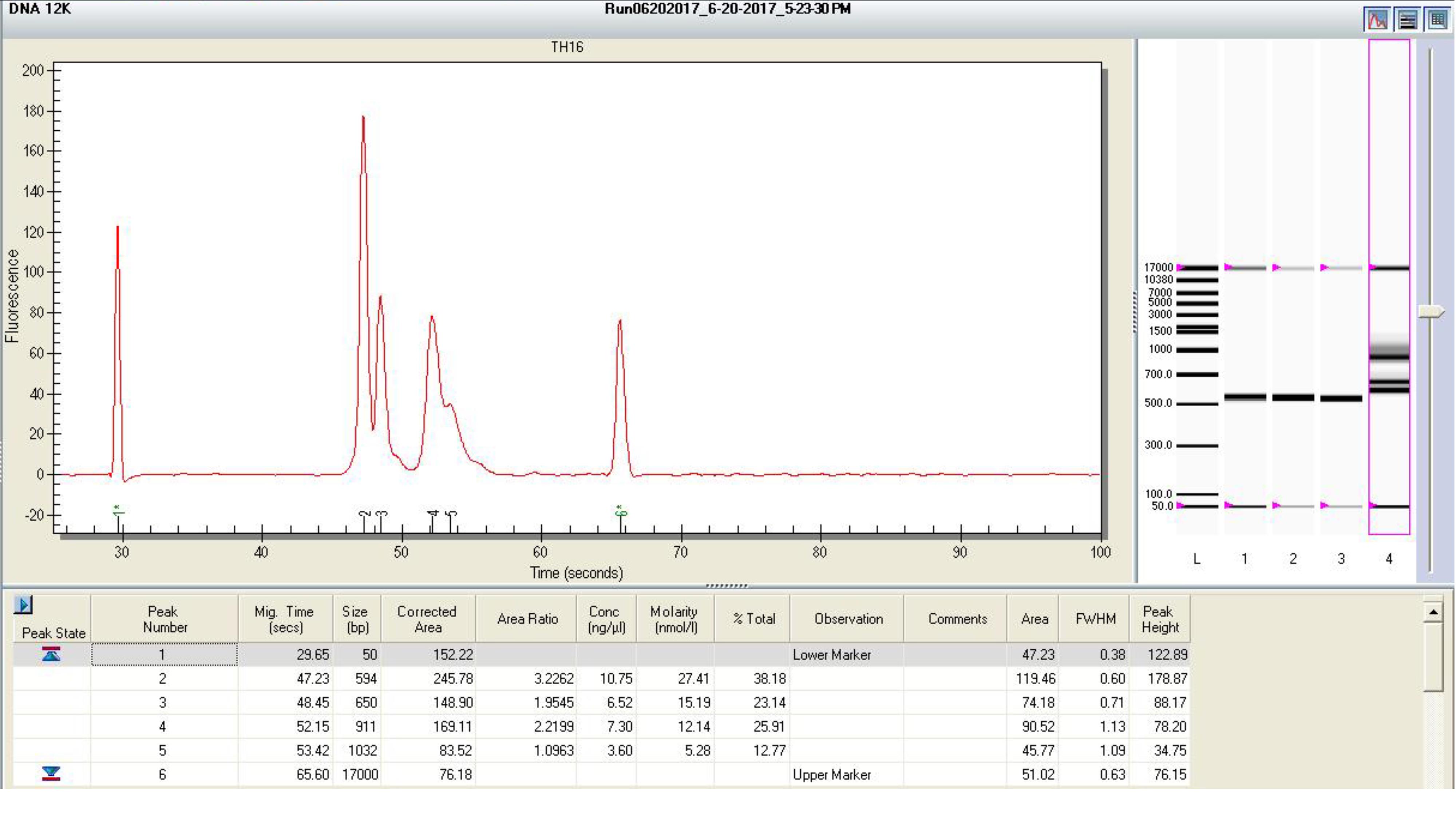
Figure 4. An example of Experion gel imageMiSeq sequencing run
The MiSeq is an integrated instrument that performs clonal amplification, DNA sequencing based on sequencing-by-synthesis, and data analysis with base calling, alignment, variant calling, and reporting in a single run. In our protocol, we use the MiSeq reagent kit V3 (600 cycle) for sequencing. Figure 5 shows the MiSeq reagent cartridge and content by well numbers. This step is modified based on the MiSeq V3 protocol.
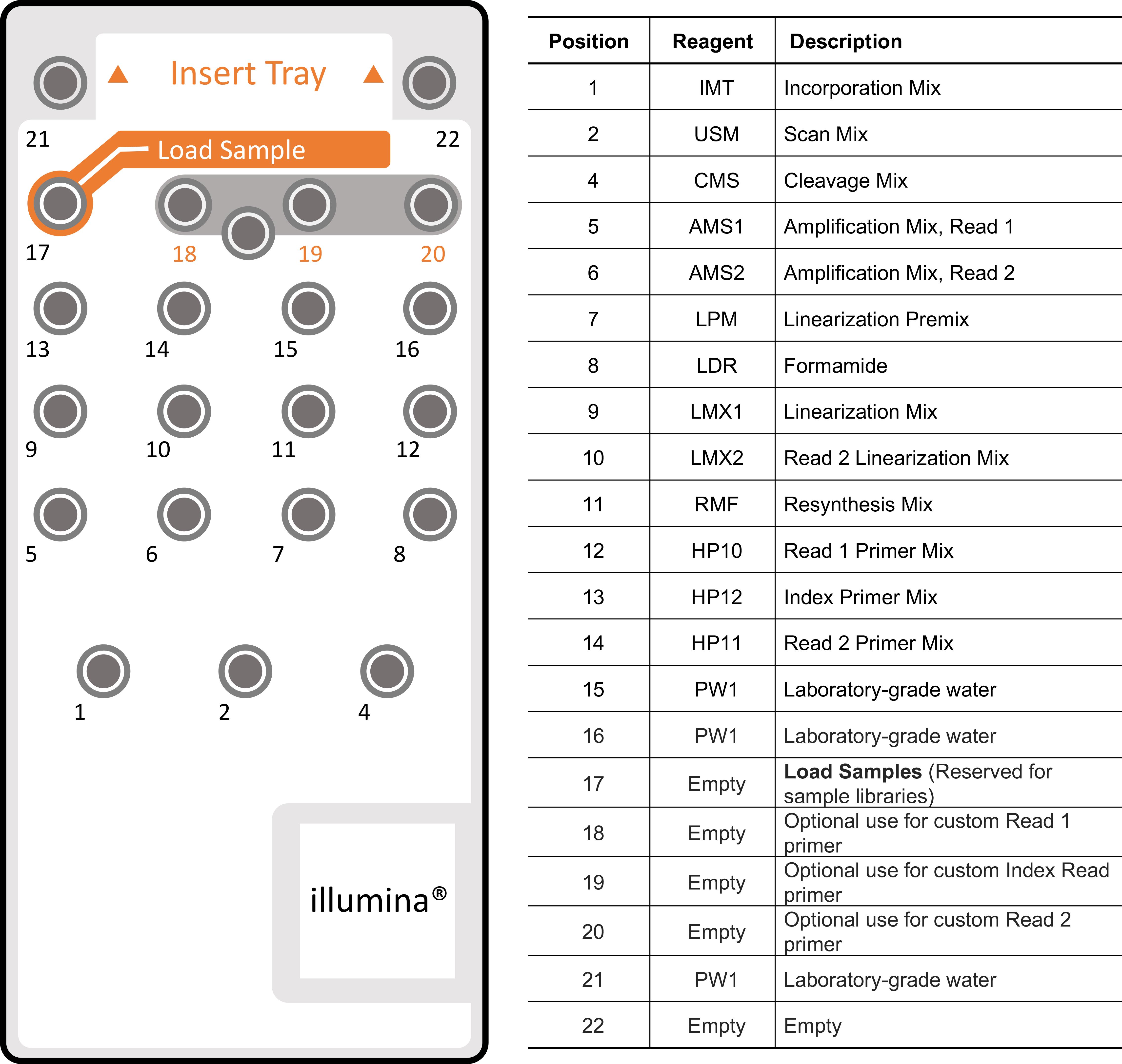
Figure 5. MiSeq Reagent Cartridge and content by well numbersPrepare the reagent cartridge.
Remove the reagent cartridge from -20 °C storage.
Place the reagent cartridge in a water bath containing only enough room temperature, deionized water to submerge the base of the reagent cartridge. Do not allow the water to exceed the maximum water line printed on the reagent cartridge.
Allow the reagent cartridge to thaw in the room temperature water bath for approximately 60 min or until completely thawed.
Remove the cartridge from the water bath and gently tap it on the bench to dislodge water from the base of the cartridge. Dry the base of the cartridge, if necessary.
Invert the reagent cartridge ten times to mix the thawed reagents, and visually inspect that all positions are thawed.
Visually inspect the reagent marked IMT (incorporation mix) to make sure that it is fully mixed and free of precipitates.
Gently tap the cartridge on the bench to dislodge water from the base of the cartridge and reduce air bubbles in the reagents.
Note: The MiSeq sipper tubes go to the bottom of each plate well to aspirate the reagents so it is important that there are no air bubbles stuck at the bottom of the reagent wells.
Place the reagent cartridge on ice or set aside at 2 °C to 8 °C until you are ready to load the sample onto the reagent cartridge and set up the run.
Prepare the libraries.
Denature DNA
Prepare a fresh dilution of 0.2 N NaOH on the day of sample loading. This is essential in order to completely denature samples for cluster generation on the MiSeq. Always start with a concentrated stock of NaOH that is 10 N or higher, or you may get a decrease in cluster density.
Calculate the molarity of the pooled library based on the Qubit concentration and average DNA size, dilute the pooled library to 4 nM.
Combine the following volumes of sample DNA and 0.2 N NaOH in a microcentrifuge tube:
4 nM sample DNA (5 µl)
0.2 N NaOH (5 µl)
Vortex briefly to mix the sample solution, and then briefly centrifuge the sample solution.
Incubate for five minutes at room temperature to denature the DNA into single strands.
Add the following volume of pre-chilled HT1 to the tube containing denatured DNA to result in a 20 pM denatured library:
Denatured DNA (10 μl)
Pre-chilled HT1 (990 μl) (Shipped frozen in the MiSeq reagent cartridge box)
Place the denatured DNA on ice until you are ready to proceed to final dilution.
Dilute Denatured DNA
Dilute the denatured DNA to 8 pM concentration using the following volumes:
20 pM DNA (400 µl)
Pre-chilled HT1 (600 µl)
Vortex briefly to mix the DNA solution.
Pulse centrifuge the DNA solution.
Place the denatured and diluted DNA on ice until you are ready to load your samples onto the MiSeq reagent cartridge directly or after adding the PhiX control.
Denature and Dilute PhiX control
PhiX is used as an internal sequencing control provided by Illumina. Use the following instructions to denature and dilute the 10 nM PhiX library to 8 pM. This should result in a cluster density of 750-850 K/mm2.
Combine the following volumes to dilute the PhiX library to 2 nM:
10 nM PhiX library (2 μl)
10 mM Tris-HCl, pH 8.5 with 0.1% Tween 20 (8 μl)
Combine the following volumes of 2 nM PhiX library and 0.2 N NaOH in a microcentrifuge tube to result in a 1 nM PhiX library:
2 nM PhiX library (10 μl)
0.2 N NaOH (10 μl)
Vortex briefly to mix the 1 nM PhiX library solution.
Briefly centrifuge the template solution.
Incubate for five minutes at room temperature to denature the PhiX library into single strands.
Add the following volume of pre-chilled HT1 to the tube containing denatured PhiX library to result in a 20 pM PhiX library.
Denatured PhiX library (20 μl)
Pre-chilled HT1 (980 μl)
Dilute the denatured 20 pM PhiX library to 8 pM as follows:
20 pM denatured PhiX library (400 μl)
Pre-chilled HT1 (600 μl)
Mix Sample Library and PhiX Control
Combine the following volumes of denatured PhiX control library and your denatured sample library to result in a 15% volume ratio:
8 pM PhiX control library (105 μl)
Denatured sample library (595 μl)
Set the combined sample library and PhiX control aside on ice until you are ready to load it onto the MiSeq reagent cartridge.
Load Sample Libraries onto Cartridge
Use a separate, clean, and empty 1 ml pipette tip to pierce the foil seal over the reservoir labeled Load Samples, position 17 on the reagent cartridge.
Pipette 700 μl of your sample libraries into the Load Samples reservoir.
Load Old Nextera Primer onto Cartridge.
Old Nextera Primer is a custom Read 1 primer that gets spiked into position 12 on the reagent cartridge. Old Nextera Primer sequence can be found in Table 1.
In a new microcentrifuge tube, add 3.8 µl of 100 µM Old Nextera Primer, and place on ice.
Use a separate, clean, and empty 1 ml pipette tip to pierce the foil seal over the reservoir position 12 on the reagent cartridge.
Pipette approximately 500 μl of the Read 1 Primer Mix out of the cartridge using a 1 ml serological pipette.
Add the Read 1 Primer Mix to the tube with the Old Nextera Primer.
Vortex briefly to mix the primer solution, and then briefly centrifuge the solution.
Pipette the entire primer solution back into position 12 on the reagent cartridge using a 1 ml pipette tip.
Note: Do not pierce any other reagent positions, they will be pierced when the cartridge is loaded on the instrument.
Proceed directly to the run setup steps using the MiSeq Control Software (MCS) interface. Index sequences required for MCS can be found in Table 2.
Notes:
In the Illumina Experiment Manager (the software where you create the sample sheet) there is an option to select “Custom Primer for Read 1”. Since we are spiking in the custom primer into position #12, we do NOT select that option.
Double check the primer sequence is correct and it is diluted to 100 µM. It is important that the custom primer gets mixed well with the other primers in position #12. We recommend pipetting 3.8 µl of primer into a new Eppendorf tube. Then remove the contents of position #12 and add them to the Eppendorf tube. Vortex to mix well. Then pipette the entire contents back into position #12 of the reagent cartridge. Tap the cartridge to make sure the liquid falls to the bottom of the tube.
Do NOT add Old Nextera Primer into Positions 18 to 20 (custom sequencing primers).
It is very important that the stock solution of NaOH is 10 N or higher. Some people try to use either 5 N or 2 N stock. For some reason these lower concentration stocks don’t work as well for denaturation and the 0.2 N dilution has to be made on the day of loading samples on the MiSeq.
The denatured 20 pM PhiX library can be stored up to three weeks if stored at -15 °C to -25 °C. After three weeks, cluster numbers tend to decrease.
Notes:
Data analysis
Use the Illumina bcl2fastq pipeline (v.2.20.0) for initial data processing. This pipeline will de-multiplex sequences based on their barcodes. In this experiment design, each barcoded library is a library constructed from an RNA specimen, either from the treatment group or from the control group. In this step, only the library barcodes are de-multiplexed. The region and Primer ID de-multiplexing will be done in the following steps.
Use TCS pipeline (v.1.3.8) to de-multiplexed each target region and create template consensus sequences. There are two ways to process it.
(Recommended) Use TCS web app (https://tcs-dr-dept-tcs.cloudapps.unc.edu/) to process the raw MiSeq sequences automatically. The web app asks for the information of each sequenced regions including a name, the cDNA primer sequence and the first round PCR forward primer sequence. Choose direct upload, UNC Longleaf sequence repo or Dropbox to upload the raw MiSeq sequences. The processed sequences will be returned to the email address provided by the user. The process time is from 20 min to 2 h, based on the size of files for each batch submitted.
Use the TCS pipeline script (https://github.com/SwanstromLab/PID/blob/master/TCS.rb) to process the MiSeq raw sequence data manually, following the instructions in the script. It is recommended to use the file-sorting and logging script in the TCS pipeline ‘log_mutli.rb’ (https://github.com/SwanstromLab/PID/blob/master/log_multi.rb) to sort the files after running the TCS pipeline. The script can be run on the Linux/MacOS platform using Terminal and Ruby without additional support, and in Windows platform with terminal and Ruby installed.
Use the ‘end_join.rb’ script (https://github.com/SwanstromLab/PID/blob/master/end_join.rb) in the TCS pipeline to join the paired-end sequences.
The number of TCS is an important parameter that reveals the sampling depth of sequencing as the number of initial templates actually queried.
Use the RUBY package ‘viral_seq’ (1.0.8) to calculate the mutation rates of each specimen. An executable script can be found in the TCS pipeline (https://github.com/SwanstromLab/PID/blob/master/mut_table.rb). This script compares each individual sequence with a sample consensus sequence and generates a table listing frequencies of each type of substitutions.
Notes:
The newer version of the TCS pipeline (version ≥ 2.0.0) is included in the RUBY package ‘viral_seq’ (version ≥ 1.0.8) as an executable command line tool. It has several major updates including the end-joining functions along with the TCS creation, thus it is recommended for future use.
The authors dedicated to improve the bioinformatics analysis pipelines. Users are welcome to contact the authors for any related questions.
Acknowledgments
We would like to acknowledge the following funding sources: National Institutes of Allergy and Infectious Disease R01 AI132178 awarded to T.P.S. and R.S.B,. and NIAID 5R01 AI140970 awarded to R.S.. We thank the UNC High Throughput Sequencing Facility for help developing this protocol. This protocol was used in our previously published work (Sheahan et al., 2020).
Competing interests
The University of North Carolina is pursuing IP protection for Primer ID and R.S. has received nominal royalties.
References
- Abrahams, M. R., Joseph, S. B., Garrett, N., Tyers, L., Moeser, M., Archin, N., Council, O. D., Matten, D., Zhou, S., Doolabh, D., Anthony, C., Goonetilleke, N., Karim, S. A., Margolis, D. M., Pond, S. K., Williamson, C., and Swanstrom, R. (2019). The replication-competent HIV-1 latent reservoir is primarily established near the time of therapy initiation. Sci Transl Med 11(513): eaaw5589.
- Adewumi, O. M., Dukhovlinova, E., Shehu, N. Y., Zhou, S., Council, O. D., Akanbi, M. O., Taiwo, B., Ogunniyi, A., Robertson, K., Kanyama, C., Hosseinipour, M. C. and Swanstrom, R. (2020). HIV-1 Central Nervous System Compartmentalization and Cytokine Interplay in Non-Subtype B HIV-1 Infections in Nigeria and Malawi. AIDS Res Hum Retroviruses 36(6): 490-500.
- Clutter, D. S., Zhou, S., Varghese, V., Rhee, S. Y., Pinsky, B. A., Jeffrey Fessel, W., Klein, D. B., Spielvogel, E., Holmes, S. P., Hurley, L. B., Silverberg, M. J., Swanstrom, R. and Shafer, R. W. (2017). Prevalence of Drug-Resistant Minority Variants in Untreated HIV-1-Infected Individuals With and Those Without Transmitted Drug Resistance Detected by Sanger Sequencing. J Infect Dis 216(3): 387-391.
- Council, O. D., Zhou, S., McCann, C. D., Hoffman, I., Tegha, G., Kamwendo, D., Matoga, M., Kosakovsky Pond, S. L., Cohen, M. S. and Swanstrom, R. (2020). Deep Sequencing Reveals Compartmentalized HIV-1 in the Semen of Men with and without Sexually Transmitted Infection-Associated Urethritis. J Virol 94(12): e00151-20.
- Dennis, A. M., Zhou, S., Sellers, C. J., Learner, E., Potempa, M., Cohen, M. S., Miller, W. C., Eron, J. J., and Swanstrom, R. (2018). Using Primer-ID Deep Sequencing to Detect Recent Human Immunodeficiency Virus Type 1 Infection.J Infect Dis 218(11): 1777-82.
- Jabara, C. B., Jones, C. D., Roach, J., Anderson, J. A. and Swanstrom, R. (2011). Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc Natl Acad Sci U S A 108(50): 20166-20171.
- Keys, J. R., Zhou, S., Anderson, J. A., Eron, J. J., Jr., Rackoff, L. A., Jabara, C. and Swanstrom, R. (2015). Primer ID Informs Next-Generation Sequencing Platforms and Reveals Preexisting Drug Resistance Mutations in the HIV-1 Reverse Transcriptase Coding Domain. AIDS Res Hum Retroviruses 31(6): 658-668.
- Lee, S. K., Zhou, S., Baldoni, P. L., Spielvogel, E., Archin, N. M., Hudgens, M. G., Margolis, D. M. and Swanstrom, R. (2017). Quantification of the Latent HIV-1 Reservoir Using Ultra Deep Sequencing and Primer ID in a Viral Outgrowth Assay. J Acquir Immune Defic Syndr 74(2): 221-228.
- Lewis, C. A., Jr., Crayle, J., Zhou, S., Swanstrom, R. and Wolfenden, R. (2016). Cytosine deamination and the precipitous decline of spontaneous mutation during Earth's history. Proc Natl Acad Sci U S A 113(29): 8194-8199.
- Liu, L., Li, Y., Li, S., Hu, N., He, Y., Pong, R., Lin, D., Lu, L. and Law, M. (2012). Comparison of next-generation sequencing systems. J Biomed Biotechnol 2012: 251364.
- Sheahan, T. P., Sims, A. C., Zhou, S., Graham, R. L., Pruijssers, A. J., Agostini, M. L., Leist, S. R., Schafer, A., Dinnon, K. H., 3rd, Stevens, L. J., Chappell, J. D., Lu, X., Hughes, T. M., George, A. S., Hill, C. S., Montgomery, S. A., Brown, A. J., Bluemling, G. R., Natchus, M. G., Saindane, M., Kolykhalov, A. A., Painter, G., Harcourt, J., Tamin, A., Thornburg, N. J., Swanstrom, R., Denison, M. R., and Baric, R. S. (2020). An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci Transl Med 12(541): eabb5883.
- Ye, J., Coulouris, G., Zaretskaya, I., Cutcutache, I., Rozen, S. and Madden, T. L. (2012). Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13: 134.
- Zhou, S., Bednar, M. M., Sturdevant, C. B., Hauser, B. M. and Swanstrom, R. (2016). Deep Sequencing of the HIV-1 env Gene Reveals Discrete X4 Lineages and Linkage Disequilibrium between X4 and R5 Viruses in the V1/V2 and V3 Variable Regions.J Virol 90(16): 7142-7158.
- Zhou, S., Jones, C., Mieczkowski, P. and Swanstrom, R. (2015). Primer ID Validates Template Sampling Depth and Greatly Reduces the Error Rate of Next-Generation Sequencing of HIV-1 Genomic RNA Populations. J Virol 89(16): 8540-8555.
- Zhou, S., Williford, S. E., McGivern, D. R., Burch, C. L., Hu, F., Benzine, T., Ingravallo, P., Asante-Appiah, E., Howe, A. Y. M., Swanstrom, R. and Lemon, S. M. (2018). Evolutionary pathways to NS5A inhibitor resistance in genotype 1 hepatitis C virus. Antiviral Res 158: 45-51.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Zhou, S., Hill, C. S., Clark, M. U., Sheahan, T. P., Baric, R. and Swanstrom, R. (2021). Primer ID Next-Generation Sequencing for the Analysis of a Broad Spectrum Antiviral Induced Transition Mutations and Errors Rates in a Coronavirus Genome . Bio-protocol 11(5): e3938. DOI: 10.21769/BioProtoc.3938.
- Sheahan, T. P., Sims, A. C., Zhou, S., Graham, R. L., Pruijssers, A. J., Agostini, M. L., Leist, S. R., Schafer, A., Dinnon, K. H., 3rd, Stevens, L. J., Chappell, J. D., Lu, X., Hughes, T. M., George, A. S., Hill, C. S., Montgomery, S. A., Brown, A. J., Bluemling, G. R., Natchus, M. G., Saindane, M., Kolykhalov, A. A., Painter, G., Harcourt, J., Tamin, A., Thornburg, N. J., Swanstrom, R., Denison, M. R., and Baric, R. S. (2020). An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci Transl Med 12(541): eabb5883.
Category
Microbiology > Antimicrobial assay > Antiviral assay
Molecular Biology > RNA
Systems Biology > Transcriptomics
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.