- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Simultaneous Imaging of Single Protein Size, Charge, and Binding Using A Protein Oscillation Approach
Published: Vol 11, Iss 5, Mar 5, 2021 DOI: 10.21769/BioProtoc.3934 Views: 5378
Reviewed by: Zinan ZhouAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Electrophoresis and Western blot are important tools in protein research for detection and identification of proteins. These traditional techniques separate the proteins based on size and charge differences and identify the proteins by antibody binding. Over the past decade, the emergence of single-molecule techniques has shown great potential in improving the resolution of the traditional protein analysis methods to the single-molecule level. However, such single-molecule techniques measure either size or charge, and it is challenging to measure both at the same time. Recently, we have developed a single-molecule approach to address this problem. We tether the single proteins to a surface with a polymer linker and drive them into oscillation with an electric field. By tracking the electromechanical response of the proteins to the field using an optical imaging method, the size and charge can be obtained simultaneously. Binding of antibodies or ions to the tethered protein also changes the size and charge, which allows us to probe the interactions. This protocol includes fabrication of protein oscillators, configuration of the optical detection system, and analysis of the oscillation signal for quantification of protein size and charge. We wish this protocol will enable researchers to perform comprehensive single-protein analysis on a single platform.
Keywords: Single-molecule imagingBackground
Proteins play essential roles in many biological processes and serve as drug targets and biomarkers. Analysis of proteins rely on technologies including electrophoresis, Western Blot, and mass spectrometry, which separate and identify proteins based on the size and charge of protein molecules. Although powerful in detecting proteins, these technologies are not sensitive to single molecules, which is required for elucidating molecular heterogeneity and precision diagnosis. Recently, several single-molecule technologies have been developed to measure the size or charge of single biomolecules. Examples include interferometric scattering microscopy (iSCAT) (Young et al., 2018) and plasmonic scattering microscopy (PSM) (Zhang et al., 2020), which quantify the size by measuring the scattered light from the molecules. Single-molecule electrometry (Ruggeri et al., 2017) and anti-Brownian electrokinetic (ABEL) trap (Wang et al., 2012) measure the charge of single molecules by monitoring single molecule motion in a potential trap. However, simultaneously measuring the size and charge on a single platform remains challenging.
Recently, we have developed a single-molecule technique to solve this problem (Ma et al., 2020). We tether proteins to an indium tin oxide (ITO) surface by polyethylene glycol (PEG) linkers and drive them into oscillation in vertical direction by applying an alternating electric field (Figure 1A). To track the protein oscillation, we place the ITO on an inverted optical microscope and generate evanescent field on the ITO surface. The oscillating proteins scatter the evanescent field, which is collected by the microscope and detected by a CMOS camera (Figure 1B). Using this method, the oscillation can be tracked with nanometer precision. By analyzing the oscillation and electromechanical response of the proteins to the field, the size and charge of individual proteins can be determined. The current protocol describes the assembly of protein oscillators and the optical detection system. Applications including protein size, charge, and binding measurements will also be presented. This protocol is also applicable to other single-molecule detection platforms, such as PSM and iSCAT, to expand the capability in simultaneous charge measurements.
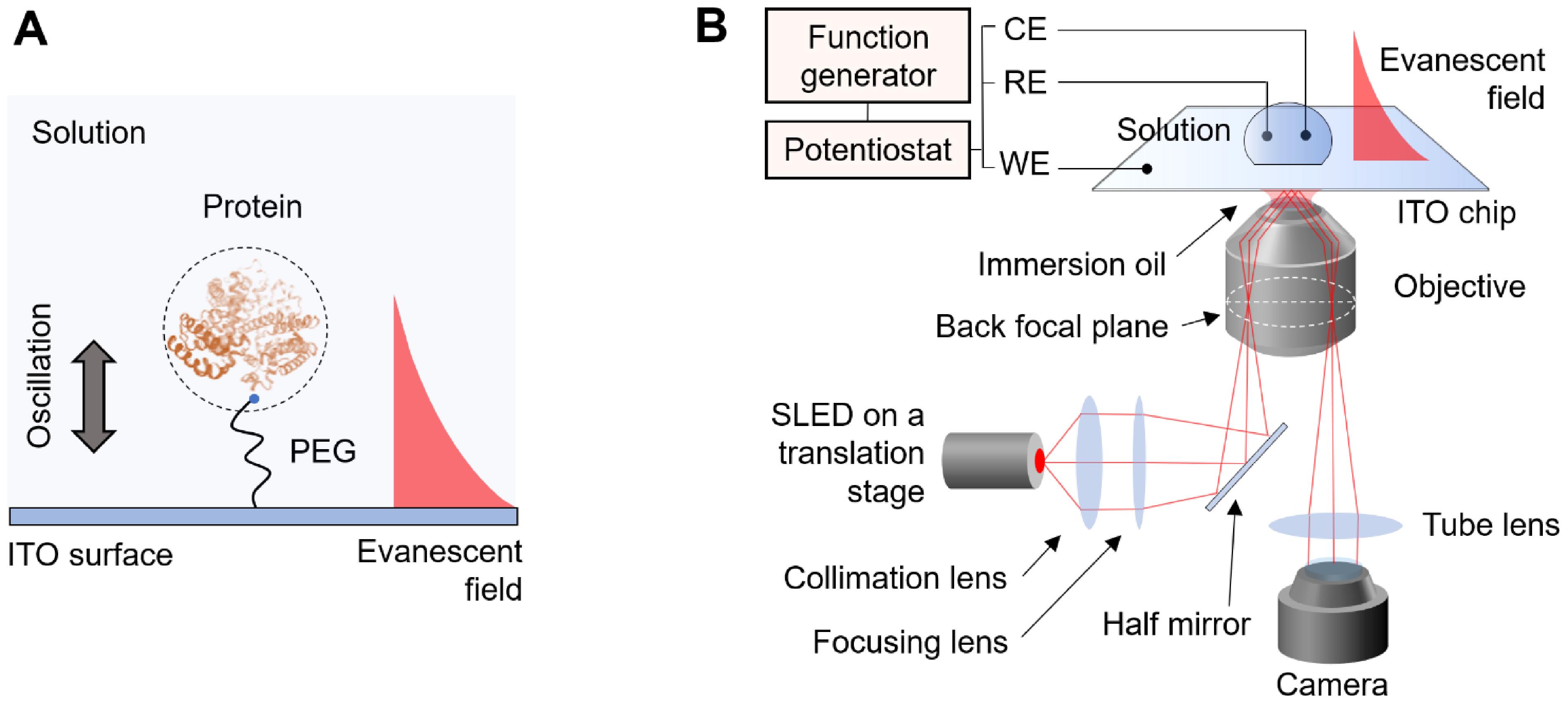
Figure 1. Detection principle and schematic of the detection system. A. The protein is tethered to the ITO surface by a PEG tether. An electric field is applied to the surface which drives the protein into oscillation. An evanescent field is generated on the surface to probe the oscillation of the protein. B. The detection system is based on an inverted microscope. A p-polarized incident light is directed onto the ITO surface via a high-numeric aperture objective with an incident angle slightly lower than the total internal reflection angle. The electric field is applied by a three-electrode electrochemical system, where the WE, RE, and CE represent working electrode, reference electrode, and counter electrode, respectively.
Materials and Reagents
ITO coated cover slips, 22 × 22 mm, thickness #1, 70-100 Ohms resistivity (SPI Supplies, catalog number: 06470-AB )
Silver wire (Alfa Aesar, catalog number: 11468-G9 )
Platinum wire (Alfa Aesar, catalog number: 10958-CB )
Reusable silicone well (SARSTEDT, flexiPERM®, catalog number: 94.6032.039 )
Streptavidin (VWR, catalog number: 97062-810 )
(3-glycidyloxypropyl) trimethoxylsilane (Sigma-Aldrich, catalog number: 440167 )
Bovine serum albumin (Sigma-Aldrich, catalog number: 0 5470 )
Goat IgG (MW = 150 kDa) (Abcam, catalog number: ab76907 )
Rabbit anti-goat IgG (MW = 150 kDa) (Invitrogen, catalog number: A16138 )
Biotin-PEG10k-NHS (Nanocs, catalog number: PG2-BNNS-10k )
Polystyrene (PS) nanoparticles, diameters of 50, 100, 150 and 200 nm (Bangs Laboratories, catalog numbers: PS2002 , PS2004 , PS2006 , and PS2008 )
(Optional) Sodium azide purification kit (Nanopartz, catalog number: PPZ-KIT-10-MAG )
1× phosphate buffered saline (Corning, catalog number: 21-040-CV )
Immersion oil type A (Cargille, catalog number: 16482 )
Isopropanol (Sigma-Aldrich, catalog number: 109827 )
Acetone (VWR, BDH®, BDH2002 )
Ethanol (Koptec’s Pure Ethanol 200 Proof, Decon Labs, catalog number: V1001 )
Ammonium Hydroxide (Mallinckrodt Chemicals, catalog number: 3256 )
Hydrogen peroxide (VWR, catalog number: BDH7814-3 )
H2O2/NH3·H2O/H2O (1:3:5) mixture (see Recipes)
0.1 mg/ml BSA blocking solution (see Recipes)
100 times diluted PBS buffer (see Recipes)
Equipment
Inverted optical microscope (Olympus, model: IX-81 )
60× NA 1.49 oil immersion TIRF objective (Olympus, model: APON60XOTIRF )
Antivibration optical table (Newport, model: RS2000 )
XYZ translation stage (Thorlabs, model: PT3 )
Superluminescent light emitting diode (SLED) (Superlum, model: SLD-260-HP-TOW-PD-670 )
SLED current driver (Superlum, model: PILOT4-AC )
CMOS camera (Hamamatsu, model: ORCA-Flash 4.0 )
Function generator (Agilent, model: 33521A )
Potentiostat (Pine Instrument Company, model: AFCBP1 )
USB data acquisition card (National Instruments, model: NI USB-6251 )
Vacuum pump (TOPSFLO, model: TM30A-B6-P9504 (V6004 ))
Plastic tubing, 0.02” ID × 0.06” OD (Saint-Gobain, TYGON®, model: AAD04103 )
Syringe with Luer-LokTM tip, 20 ml (Fisher Scientific, catalog number: 22-124-967 )
Perfusion manifold, 4 to 1 ports (Warner Instruments, model: MM-4 )
Syringe 1-way stopcock (B. Braun Medical Inc., catalog number: 455980 )
Dispensing needles (Weller, catalog number: KDS2312P )
Cover glass staining jars (Fisher Scientific, catalog number: 02-912-636 )
Ultrasonic cleaner (Fisher Scientific, model: FS20D )
Tweezers (Electron Microscopy Sciences, catalog number: 72750-D )
Computer (Processor: Intel Xeon E-2274G; RAM: 64 GB; Hard drive: Samsung SSD 970 PRO 1TB)
Software
HCImage Live (Hamamatsu)
MATLAB (MathWorks)
Fiji (https://imagej.net/Fiji)
Origin 2019 (OriginLab)
Office 365 (Microsoft)
Procedure
Cleaning and silanization of ITO surface
Put the ITO chips in a staining jar and sonicate the chips sequentially in acetone, ethanol, and DI water each for 20 min to remove surface contaminates.
Incubate the clean ITO chips in another jar filled with a mixture of H2O2/NH3·H2O/H2O (1:3:5) at room temperature for 1 h to increase the surface density of OH.
Rinse the ITO chips with DI water and dry with N2.
Fill a pre-dried staining jar with 1% (3-glycidyloxypropyl) trimethoxylsilane in isopropanol, place the dried ITO chips in the jar and incubate at room temperature overnight to functionalize the chips with terminal epoxy groups (Figures 2A and 2B).
Rinse the ITO chips with isopropanol and water, each for 3 times, and dry with N2. Mount a silicone well on the ITO side of the chip before fabricating protein oscillators.
Preparation of protein-PEG complex
Dilute the protein with PBS to reach a final concentration of 100 nM with a volume of 100 μl.
Note: The protein sample should not contain any NHS-reactive additives such as BSA and azide. If azide-free protein sample is unavailable, sodium azide purification kits can be used to remove the azide (example: Nanopartz, catalog number: PPZ-KIT-10-MAG ).
Prepare 10 nM NHS-PEG10k-biotin in 100 μl PBS.
Note: NHS degrades fast in water, always use fresh solution for the reaction.
Mix the 100 μl protein solution and the 100 μl NHS-PEG10k-biotin solution and incubate the mixture at 4 °C overnight (Figures 2D-2E).
Note: The 10/1 molar ratio of protein/PEG prevents multiple PEG molecules linking to the same protein molecule.
Assembly of protein oscillators
Add 200 μl 0.1 mg/ml streptavidin (dissolved in PBS) to the ITO chip in the sample well and incubate for 2 h to allow immobilization of streptavidin (Figure 2C).
Wash the chip with PBS for 3 times.
Add 0.1 mg/ml BSA solution to ITO and incubate for 20 min to block non-specific binding sites.
Wash the chip with PBS for 3 times.
Add 200 μl protein-PEG complex solution to the chip and incubate for 2 h to allow the terminal biotin in the PEG couple to the streptavidin on the ITO surface (Figure 2F).
Slowly remove 100 μl solution from the well, and then gently add 400 μl of 100 times diluted PBS to the well. Do not generate fast flow in the solution because it may break the tethered protein.
Slowly remove 400 μl solution from the well. Repeat Steps C6-C7 for ~10 times to reduce the amount of free protein molecules and unbound protein-PEG complexes remaining in the solution.
Note: The surface should always be covered with at least 100 μl solution, drying out the surface will lead to irreversible adhesion of the tethered proteins to the surface. Also, avoid generating air bubbles on the surface when pipetting.
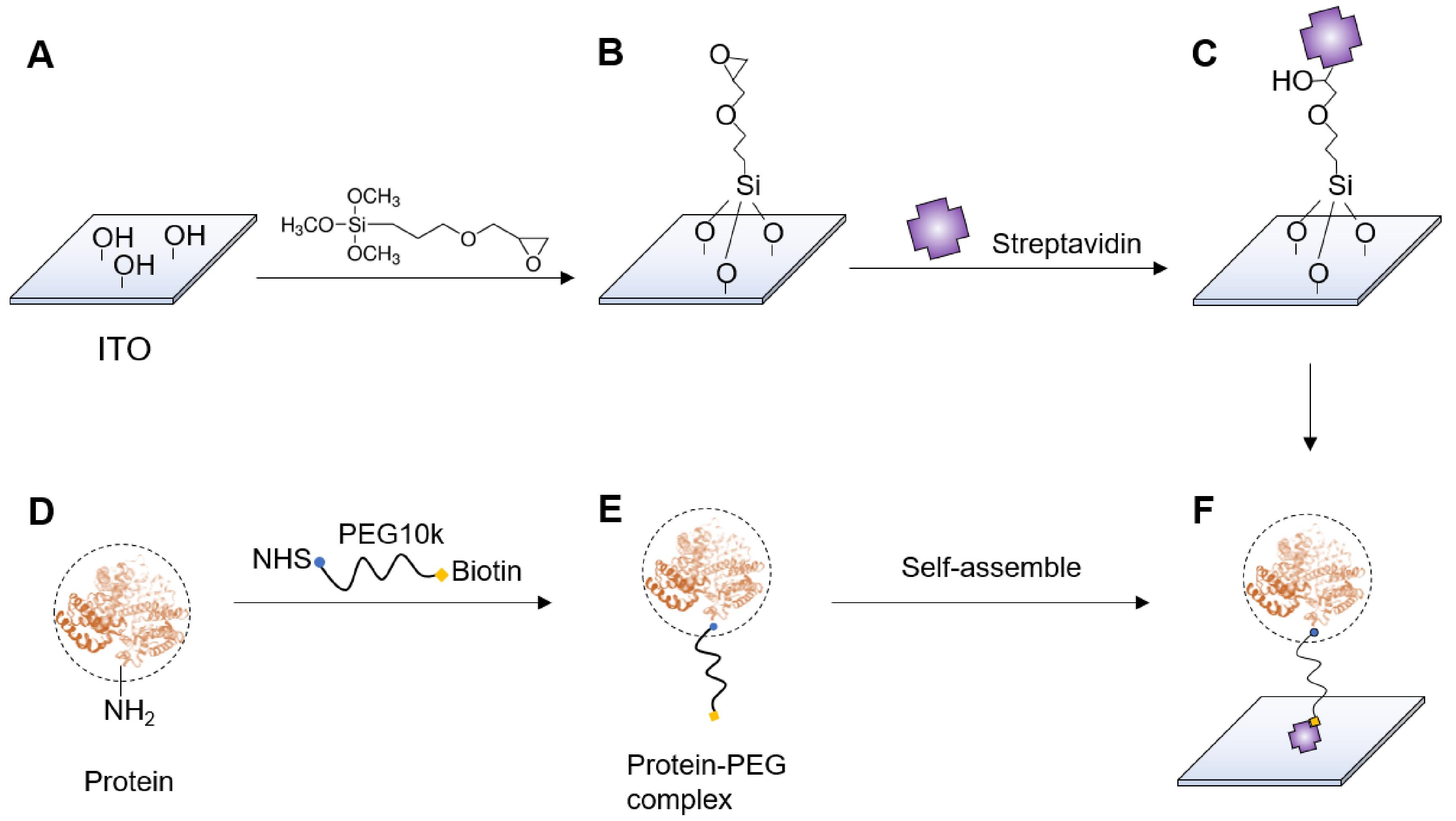
Figure 2. Fabrication of protein oscillators. A-C. Immobilization of streptavidin on ITO surface. D-F. Assembly of protein-PEG complex and protein oscillator.
Measuring the electromechanical response of protein oscillators
Mount the ITO chip on the microscope with half drop of immersion oil placed between the objective and the chip. Set the camera exposure time to 1 ms. Set the camera frame rate to 800 fps at 2048 × 256 pixels. Turn off the camera cooling fan to reduce mechanical noise.
Park the incident angle above the total internal reflection angle, where all the incident light is reflected. Increase the incident power by tuning the SLED current until the camera is saturated (maximum image intensity ~65,535).
Slowly lower the incident angle until it is slightly below the total internal reflection angle, and half of the light is reflected to the camera (image intensity ~30,000).
Set up the electrochemical system and gravity sample delivery system as shown in Figures 3A and 3B.
Double-check if the electrodes are correctly connected. Short circuit or open circuit can damage the ITO surface.
Find a large impurity on the surface and roughly adjust the focus until the pattern of the impurity turns to V-shape (Figure 3C) from parabolic shape (Figure 3D). Move the stage and find a clean region with no impurities (Figure 4A).

Figure 3. Experimental setup for measuring protein oscillation. A. A picture showing the whole setup. B. A zoom-in of the sample stage (the red square in A). The ITO chip is placed on the objective. A self-adhesion silicone well is sticked to the chip for holding solution. The ITO surface, an Ag/AgCl wire, and a Pt coil serve as the working electrode (WE), reference electrode (RE), and counter electrode (CE), respectively. The sample inlet directs the sample to the surface within the field of view to minimize diffusion. The outlet tubing (connected to a vacuum pump) is fixed near the upper edge of the well and continuously removing excess solution from the well. C. An image showing a large impurity that is properly focused (inverted V shape). D. An image showing an impurity that is out of focus (parabolic patterns).Apply a sinusoidal potential of 6 V at 80 Hz to the ITO surface, and record the images for 1 s (Video 1).
 Video 1. An image sequence recorded at 800 fps for 1 s. Any applied potential between 0 V to 10 V should generate only minor changes to the raw images that are not observable without image processing. The top video shows the raw image sequence recorded by the camera, and the bottom video shows differential images obtained by subtracting the first frame from each frame in the raw image sequence (the background is removed). The applied potential in the video is 8 V at 80 Hz. The sensor surface is modified with IgG oscillators.
Video 1. An image sequence recorded at 800 fps for 1 s. Any applied potential between 0 V to 10 V should generate only minor changes to the raw images that are not observable without image processing. The top video shows the raw image sequence recorded by the camera, and the bottom video shows differential images obtained by subtracting the first frame from each frame in the raw image sequence (the background is removed). The applied potential in the video is 8 V at 80 Hz. The sensor surface is modified with IgG oscillators.Extract the oscillation signal at 80 Hz using a custom MATLAB fast Fourier transform (FFT) filter. The program can be found in the supporting information in Ma et al. (2020) .
Examine the FFT image to see if there are sufficient amount of protein molecules oscillating in the field of view and whether they are correctly focused. If there are not enough proteins, move the stage to another region. If the focus is incorrect, slightly tune the focus knob and repeat Steps D7-D9.
Apply sinusoidal potentials from 0 V to 10 V with 1 V or 2V interval at 80 Hz. Record 1 s video at 800 fps for each potential. If needed, record the applied potential and current simultaneously using data acquisition card.
Measuring binding induced size and charge changes using Anti-IgG and IgG oscillators as an example
Add 20 ml of 100 times diluted PBS buffer and 2 ml of 130 nM anti-IgG in 100 times diluted PBS buffer to two different syringes in the sample delivery system (Figure 3A).
Turn on the buffer channel to flow buffer over the ITO surface modified with IgG oscillators (the flow rate is about 300 μl/min if the valve is fully turned on).
Apply a potential with 9 V amplitude at 80 Hz to the ITO (which fully stretches the PEG). Start recording images and if needed, use data acquisition card to record the potential and current.
After flowing the buffer for 20 s to establish a baseline, turn off the buffer syringe and turn on the anti-IgG channel to start the association process. Flow anti-IgG for 30 s.
Switch back to the buffer channel and flow buffer for 30 s for dissociation measurement.
For charge detection, use a lower potential to oscillate the protein (~5 V) so that the PEG is not completely stretched. Do not apply high potential to fully stretch the PEG because charge change can be either positive or negative, which will either increase or decrease the oscillation amplitude.
Size calibration using PS nanoparticles
Dilute each PS nanoparticle stock solution (50, 100, 150, 200 nanometers) with PBS for 1000 times.
Add 300 μl PBS to the sample well mounted on a bare ITO surface, and then inject 50 μl of diluted 50 nm nanoparticle solution to allow nanoparticles binding to the surface. Upon injection, start recording images at 400 fps (512 × 2048 pixels) for 1 min.
Repeat Step F2 to measure the other sizes. Use a new ITO chip for each measurement.
Note that the optimal nanoparticle concentration should lead to ~5 binding events per image. Particle concentration may vary, adjust the concentration if it is not appropriate.
Data analysis
Constructing a size calibration curve
Use Fiji software to obtain a differential image sequence showing nanoparticle binding by subtracting the first frame from each frame in the sequence (remove the background) (Video 2).
Select 10 × 10 pixels regions of interest (ROIs) on the center of the particle images (red squares in Video 2) and calculate the mean intensity (I) within each ROI.
Video 2. Nanoparticle of different sizes binding to the surface. Each video is cropped from the original video showing a single particle (50, 100, 150, and 200 nm) binding to the surface. For better visualization, background noise is reduced by averaging every 11 neighboring frames using the moving average function in Fiji software (Select “Plugins>Stowers>Jay_Unruh>Detrend Tools>stack divide moving average jru v2”).Measure ~100 individual nanoparticles for each size (D).
Plot log(I) vs. log(D) and find the slope of the curve, which should be around 2.
Single protein analysis – temporal fast Fourier transform
Perform fast Fourier transform (FFT) on the image sequence recorded in Procedure D (Figure 4A) using a custom MATLAB program (the same program mentioned in Procedure D8).
Use the 81st image in the FFT image sequence, which shows the signal at 80 Hz, for single protein analysis (Figure 4B).
Single protein analysis – background removal
Perform k-space FFT to the temporal FFT image obtained above and convert the image to k-space (Figure 4C) using Fiji (Select “Process>FFT>FFT”).
Apply a band-block filter (Figure 4D) which filters out the low-frequency background patterns while keeping the signal. Use the rectangle selection tool to select the low-frequency region, and then use “Edit>Clear” to remove the selected region.
Use “Process>FFT>Inverse FFT” to transform the image back to time domain (Figure 4E).
Oscillation amplitude vs. applied potential
Select ROIs (10 × 10 pixels) on the bright spots which are tethered proteins (Figure 4F). Measure the mean intensity of each ROI and subtract background intensity (the mean intensity of the whole image).
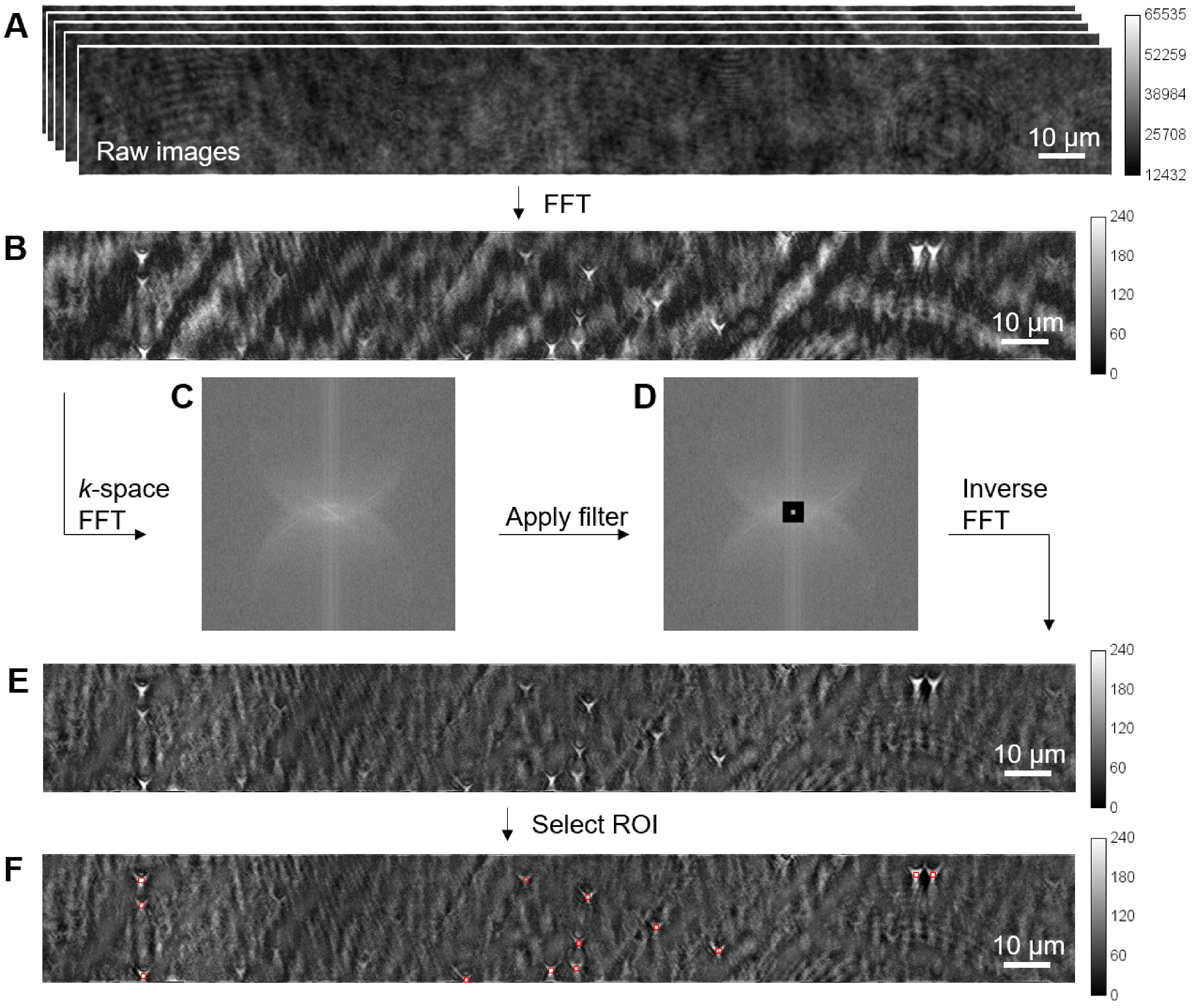
Figure 4. Image processing. A. Raw image sequence obtained in Procedure D. The calibration bar shows the image intensity in gray scale. B. Temporal FFT image of IgG molecules obtained by performing FFT on the image sequence. Applied potential: 8 V at 80 Hz; FFT integration time: 1 s. C. k-space FFT is performed on the temporal FFT image. D. A band-block filter (black region) is applied to partially remove the background. E. Image after background removal. F. ROIs of 10 × 10 pixels are selected for single protein size and charge analysis.Measure the mean intensity (with background subtraction) of each tethered protein at different applied potentials.
Plot intensity (y-axis) vs. applied potentials (x-axis). The intensity increases linearly with applied potential at first and reaches a plateau at high potentials (often > 6 V) because the tether is fully stretched (Figure 5).
Convert the intensity to oscillation amplitude in nanometers using the equation:
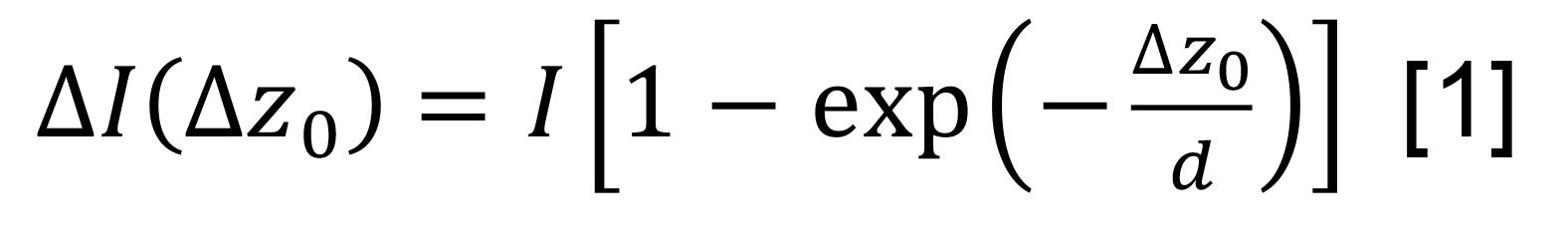
where ∆z0 is the oscillation amplitude, I is the evanescent wave scattering intensity of a protein, ∆I(∆z0 ) is the protein oscillation intensity at z0, and d is the decay constant of evanescent field. I is determined from the oscillation plateau regime when ∆z0 is 63 nm (the length of the PEG) and ∆I(∆z0=63 nm) is the maximum FFT image intensity of the protein.
Single protein size analysis
Find the ∆I(∆z0=63 nm) value for each individual protein using the intensity vs. applied potential plots (Figure 5). Determine I for each protein using equation [1].
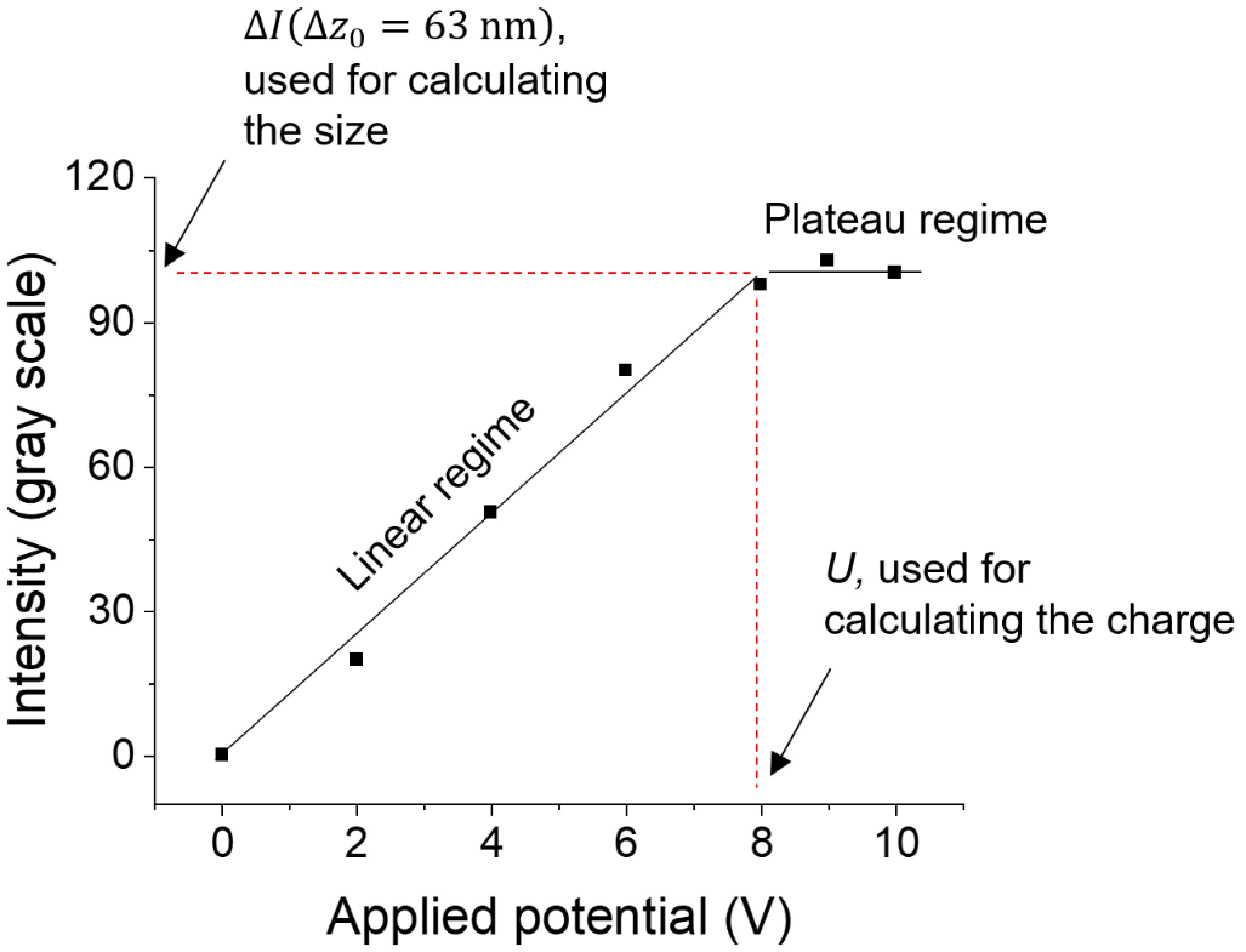
Figure 5. Calculating the size and charge. The plot shows the oscillation response (intensity) vs. the applied potential of a single IgG molecule. At low potentials, the oscillation increases with the potential in a roughly linear manner, because the PEG is stretched by the electric field. At high potentials, the PEG is fully stretched, and the oscillation (intensity) reaches the maximum value. The protein size is determined by the intensity at the plateau (∆I(∆z0=63 nm)), and the charge is determined by the potential (U) or field when the PEG is fully stretched. The positions for ∆I(∆z0=63 nm) and U are marked on the plot.Convert I to diameter Dapp using the size calibration curve. Here, Dapp is the effective diameter of the protein-PEG complex.
Calculate the diameter of protein using
 , where DPEG is the diameter of PEG coil measured by dynamic light scattering. DPEG is ~4.5 nm for PEG10k in 100 times diluted PBS according to our dynamic light scattering measurement.
, where DPEG is the diameter of PEG coil measured by dynamic light scattering. DPEG is ~4.5 nm for PEG10k in 100 times diluted PBS according to our dynamic light scattering measurement.
Single protein charge analysis
Find the potential U on the intensity vs. applied potential plot where the oscillation amplitude reaches the plateau (Figure 5).
Convert the potential U to electric field E0 using the equation
E0 = 3.53×106 U/m. Note: This relationship is determined on our system, and we recommend the readers to calibrate it before experiment in consideration of system differences. Please refer to Ma et al. (2020) for details.
Determine the charge q using
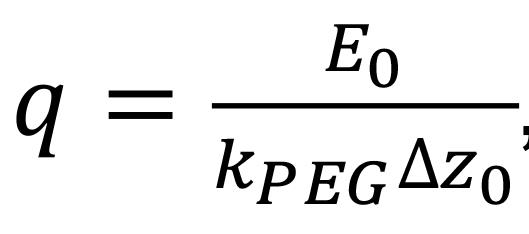 , where kPEG is the entropic spring constant of the PEG tether. For PEG10k, kPEG~3.62×10-4 N/m.
, where kPEG is the entropic spring constant of the PEG tether. For PEG10k, kPEG~3.62×10-4 N/m.Determine the mobility μ of each protein by μ=q/(3πηD), where η is the solution viscosity.
Recipes
H2O2/NH3·H2O/H2O (1:3:5) mixture
To make 45 ml:
5 ml H2O2
15 ml NH3·H2O
25 ml DI water
0.1 mg/ml BSA blocking solution
Add 1 mg of BSA to 10 ml PBS buffer (pH 7.4)
100 times diluted PBS buffer
To make 40 ml:
400 μl PBS
39.6 ml H2O
Acknowledgments
We thank financial support from National Institute of Health (R44GM126720). This protocol is based on our work published in Nature Communication ( Ma et al., 2020 ).
Competing interests
A US patent application (16/584,120) has been filed by Arizona Board of Regents on behalf of Arizona State University based on the method described in this protocol and published on 03/26/2020.
References
- Ma, G., Wan, Z., Yang, Y., Zhang, P., Wang, S. and Tao, N. (2020). Optical imaging of single-protein size, charge, mobility, and binding. Nat Commun 11(1): 4768.
- Ruggeri, F., Zosel, F., Mutter, N., Rozycka, M., Wojtas, M., Ozyhar, A., Schuler, B. and Krishnan, M. (2017). Single-molecule electrometry. Nat Nanotechnol 12(5): 488-495.
- Wang, Q., Goldsmith, R. H., Jiang, Y., Bockenhauer, S. D. and Moerner, W. (2012). Probing single biomolecules in solution using the anti-Brownian electrokinetic (ABEL) trap. Accounts Chem Res 45(11): 1955-1964.
- Young, G., Hundt, N., Cole, D., Fineberg, A., Andrecka, J., Tyler, A., Olerinyova, A., Ansari, A., Marklund, E. G., Collier, M. P., Chandler, S. A., Tkachenko, O., Allen, J., Crispin, M., Billington, N., Takagi, Y., Sellers, J. R., Eichmann, C., Selenko, P., Frey, L., Riek, R., Galpin, M. R., Struwe, W. B., Benesch, J. L. P. and Kukura, P. (2018). Quantitative mass imaging of single biological macromolecules. Science 360(6387): 423-427.
- Zhang, P., Ma, G., Dong, W., Wan, Z., Wang, S. and Tao, N. (2020). Plasmonic scattering imaging of single proteins and binding kinetics. Nat Methods 17(10): 1010-1017.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Ma, G., Wan, Z. and Wang, S. (2021). Simultaneous Imaging of Single Protein Size, Charge, and Binding Using A Protein Oscillation Approach. Bio-protocol 11(5): e3934. DOI: 10.21769/BioProtoc.3934.
Category
Biochemistry > Protein > Imaging
Biophysics > Single-molecule technique
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.