- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Pipeline for Non-model Organisms for de novo Transcriptome Assembly, Annotation, and Gene Ontology Analysis Using Open Tools: Case Study with Scots Pine
Published: Vol 11, Iss 3, Feb 5, 2021 DOI: 10.21769/BioProtoc.3912 Views: 8080
Reviewed by: Antony ChettoorXinlei LiChristian Sailer
Original research article
The authors used this protocol in:

Jul 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
RNA sequencing (RNA-seq) has opened up the possibility of studying virtually any organism at the whole transcriptome level. Nevertheless, the absence of a sequenced and accurately annotated reference genome may be an obstacle for applying this technique to non-model organisms, especially for those with a complex genome. While de novo transcriptome assembly can circumvent this problem, it is often computationally demanding. Furthermore, the transcriptome annotation and Gene Ontology enrichment analysis without an automatized system is often a laborious task. Here we describe step-by-step the pipeline that was used to perform the transcriptome assembly, annotation, and Gene Ontology analysis of Scots pine (Pinus sylvestris), a gymnosperm species with complex genome. Using only free software available for the scientific community and running on a standard personal computer, the pipeline intends to facilitate transcriptomic studies for non-model species, yet being flexible to be used with any organism.
Keywords: RNA-seqBackground
Non-model organisms are valuable for environmental and evolutionary studies. However, the absence of a closely related model species to serve as reference for transcriptomic studies was a limitation until the development of the de novo assembly methods. De novo assemblers brought the non-model organisms to the omics era, but the required analyses may be computationally demanding and are often time consuming. This is especially the case for testing, evaluating, and establishing the pipeline for performing the required analyses. The costs for acquiring a computing server and/or software for automated data processing, which could speed up the process, may impose limitations to some research groups. We detail here the procedures that were employed for the transcriptome assembly, annotation, and gene ontology (GO) analysis of Scots pine (Pinus sylvestris), an organism with complex genome (Duarte et al., 2019). The pipeline was developed based on works and benchmark evaluations that describe the best practices for transcriptome studies (Conesa et al., 2016; Honaas et al., 2016; Geniza and Jaiswal, 2017). The pine transcriptome analyses were performed on a 32G of RAM 8-cores machine. The outputs of three different assemblers, namely BinPacker (Liu et al., 2016), SOAPdenovo-Trans (Xie et al., 2014), and Trinity (Grabherr et al., 2011), were evaluated individually and combined. For the pine transcriptome assembly, the best result was obtained by combining the outputs of the different assemblers, following filtering for redundancies with EvidentialGene (Gilbert, 2020). The selection and combination of assemblers, nevertheless, should be adjusted according to the input data, and the decision can be made based on the quality assessment that is described step-by-step here. InterProScan (Jones et al., 2014) performs a comprehensive signature annotation of the predicted proteins, while BLAST+ (Camacho et al., 2009) allows intra- and interspecific mapping for functional comparisons. Because of that, we combined InterProScan’s protein signatures with several BLAST+ searches for annotating the transcriptome of P. sylvestris (Duarte et al., 2019), which were integrated with Trinotate (Bryant et al., 2017). The unique GO identifiers were retrieved with a Python script that we make available, and used for GO enrichment analysis with BiNGO (Maere et al., 2005). The pipeline is based on tools that are open for the scientific community. While it is especially interesting for studies with non-model organisms, which lack closely related and well-annotated species for guiding the assembly and annotation, the pipeline is flexible to be applied to virtually any organism.
Software
Software – Linux OS:
Several programs that will be used in the protocol, such as BLAST+, Bowtie2, FASTA Splitter, and BUSCO, can easily be managed using the Bioconda channel (Grüning et al., 2018; https://bioconda.github.io/). Bioconda is a repository of packages specialized in bioinformatics, and it requires the conda package to be installed. The manager automatically installs all dependencies for a given program and makes it available in your PATH. The software support via Bioconda is continuously updated, and we suggest always to check if the required software is available through this repository. After installing conda, add the channels following the recommended order (please refer to https://bioconda.github.io/user/install.html#install-conda). Other software will need to be installed individually, and may require adjustments depending on your system. Next, we list the software that will be required for each step, which are summarized in the flowchart (Figure 1).

Figure 1. Flowchart illustrating the five steps for transcriptome assembly, annotation, and Gene Ontology analysis. The input for starting each step is represented in the boxes, and the main procedures of each step are described in black. The software that will be required for each step are listed in blue. For performing the transcriptome assembly (B), the user may use the software of choice. The three assemblers described in this pipeline were used for the Scots pine study (Duarte et al., 2019), and are marked as optional [opt].
Data pre-processing
FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) – quality control of sequencing files
FastQ Screen (https://www.bioinformatics.babraham.ac.uk/projects/fastq_screen) – screening of sequencing files
Trimmomatic (Bolger et al., 2014; available via Bioconda) – trimming of sequencing files
Trinity package (Grabherr et al., 2011) – in silico normalization
Transcriptome assembly
BinPacker (Liu et al., 2016; https://sourceforge.net/projects/transcriptomeassembly/) – assembler
EvidentialGene (Gilbert, 2020; http://arthropods.eugenes.org/EvidentialGene/) – assembly filtering
Galaxy platform (Afgan et al., 2018; usegalaxy.org) – open source online platform for data research
SOAPdenovo-Trans (Xie et al., 2014; https://github.com/aquaskyline/SOAPdenovo-Trans) – assembler
Trinity package (Grabherr et al., 2011; available in Galaxy platform) – assembler
Quality assessment
Bowtie2 (Langmead and Salzberg, 2012; available via Bioconda) – short read aligner
BLAST+ (Camacho et al., 2009; available via Bioconda) – tool for comparing sequences and searching databases
BUSCO (Simão et al., 2015; available via Bioconda) – assembly completeness assessment
Samtools (Li et al., 2009; https://github.com/samtools/samtools) – data manipulation
DETONATE (Li et al., 2014; available via Bioconda) – evaluation of de novo transcriptome assemblies
Trinity package (Grabherr et al., 2011) – estimation of full-length transcripts in the assembly
Transcriptome annotation
BLAST+ (Camacho et al., 2009; available via Bioconda) – tool for comparing sequences and searching databases
Bowtie2 (Langmead and Salzberg, 2012; available via Bioconda) – short read aligner
Corset (Davidson and Oshlack, 2014; available via Bioconda) – clustering of read counts
FASTA splitter (http://kirill-kryukov.com/study/tools/fasta-splitter/) – creation of subsets of a FASTA file
getorf (Rice et al., 2000; http://emboss.sourceforge.net/) – prediction of open reading frames
HMMER (http://hmmer.org/) – prediction of domains for annotation
InterProScan (Jones et al., 2014; https://www.ebi.ac.uk/interpro/) – protein domains identifier
RNAMMER (Lagesen et al., 2007; http://www.cbs.dtu.dk/services/RNAmmer) – ribosomal RNA annotation
SignalP (Petersen et al., 2011; https://services.healthtech.dtu.dk/) – prediction of signal peptides
TmHMM (Krogh et al., 2001; https://services.healthtech.dtu.dk/) – protein transmembrane helix prediction
TransDecoder (Haas et al., 2013; http://transdecoder.github.io) – identification of coding regions
Trinotate (Bryant et al., 2017; https://trinotate.github.io/) – suite for transcriptome annotation
Gene Ontology analysis
BiNGO (Maere et al., 2005; download available via Cytoscape platform) – Gene Ontology analysis
Cytoscape (Shannon et al., 2000; https://cytoscape.org/) – platform for performing the Gene Ontology analysis
GO retriever – provided script for retrieving transcript ID - Gene Ontology ID relationships
Procedure
The protocol requires basic bioinformatics and Linux command line skills. We provide the general instructions for using each software for the required analyses. Each software settings may be adjusted according to the user’s need, and for that, we suggest referring to the software’s manual for further information and troubleshooting. The time required for performing each step will fully depend on your computing power, and on the size of your dataset. While the assembly and quality assessment steps may require some hours, the comprehensive annotation procedure may require a few days to run. Based on the results obtained with Scots pine, an example of application is shown at the end of this protocol as supporting information.
Pre-processing the RNA-seq dataset
For starting this pipeline, it is necessary that your RNA-seq samples have been trimmed and quality checked. As a general guideline, FastQC will provide you an overview of the quality of your reads. If you observe remaining vector sequences, they can be removed with FastQ Screen providing a list of vector sequences, which can be obtained from the National Center for Biotechnology Information (NCBI, https://ftp.ncbi.nlm.nih.gov/pub/UniVec/). Trimmomatic can filter your samples based on the quality value of choice and prepares your paired reads. Trimmomatic also performs trimming of adapter sequences, which must be provided according to the method used for preparing your library. Both FastQ Screen and Trimmomatic have detailed manuals that are easy to follow.
[Tip 1] Several files will be generated for performing all steps of this protocol. Save them with descriptive names that are easy to identify, and sort them in folders accordingly.
[Tip 2] Some assemblers also accept the unpaired reads as an input. Do not discard them.
For building a common de novo transcriptome reference for a given species, it is recommended to merge all RNA-seq files (i.e., all samples and conditions) for using a unique input for the assembly. Some software may perform this step as a part of their command line. Always refer to the software's manual for specific details.
Using the Linux terminal, concatenate all trimmed fastq files (left, right, and unpaired) of all samples as three unique inputs for the assemblers (e.g., “all_left.file”, “all_right.file”, and “all_unpaired.file”). Concatenation of the data files can be done by using the “cat” function:
> cat sample_1-1.file sample_2-1.file sample_n-1.file> all_left.file
> cat sample_1-2.file sample_2-2.file sample_n-2.file > all_right.file
[Optional] Perform in silico normalization of the three concatenated files to reduce redundancy in read datasets. Trinity package provides a script for it (insilico_read_normalization.pl). If you have paired-end reads, the command will be:
> ./insilico_read_normalization.pl --seqType <string> \
--left all_left.file --right all_right.file
where seqType specifies the format of your reads (fa or fq). Several other parameters can be adjusted, as the RNA-seq read orientation (--SS_lib_type <string>; RF should be used for the dUTP method). If you intend to use Trinity and your unpaired reads, append them to the left ones (e.g., “all_left_all_unpaired.file”) using the “cat” function. Note that the backslash (\) is merely used for breaking the command into several lines for visualization purposes (it can be removed).
Transcriptome assembly (analyses performed in Linux OS)
For performing the transcriptome assembly, you may use the software of choice. Here, we provide instructions for the assemblers that were used for P. sylvestris transcriptome (Duarte et al., 2019), which therefore will be marked as optional. BinPacker and SOAPdenovo-Trans can be run locally on a standard personal computer but may require in silico normalization to reduce the input data. Because Trinity is computationally demanding, an alternative is to run it using the Galaxy platform (usegalaxy.org). Using multiple k-mer strategy can improve the assembly by balancing sensitivity and specificity (Zerbino and Birney, 2008), but it may also introduce redundancy (Xie et al., 2014). This issue will be corrected later. Trinity is optimized to run with fixed k-mer size (25 k-mers) (Grabherr et al., 2011). If RAM memory is limiting, BinPacker can be used for performing assemblies with the smaller k-mers, while SOAPdenovo-Trans is efficient for longer lengths. For Bruijn-based assemblers, the increase in k-mer size must be based on odd numbers to avoid the creation of palindromes, which may block the graph construction (Zerbino and Birney, 2008).
Perform the transcriptome assembly with different assemblers.
[Optional] BinPacker only accepts paired inputs. The parameters must be adjusted according to the data set and run using various k-mer lengths (-k option), as for instance:
> ./binpacker -s <string> -p <string> -k <int> \
-l all_left_normalized.file -r all_right_normalized.file
where -k <int> is the k-mer length of choice, -s <string> sets between fa or fq files, -p <string> sets for single-end (single, requiring -u single_reads.file for instance) or paired-end (pair, requiring -l left_reads.file -r right_reads.file). At the end, concatenate all assemblies into one file using the “cat” function.
[Optional] The easiest way to run SOAPdenovo-Trans is by setting all assembly parameters and input files in the configuration file, including the k-mer length of choice. It also allows both paired (q1=path_to_all_left_normalized.file and q2= path_to_all_right_normalized.file) and unpaired (q=path_to_ all_unpaired_normalized.file) reads to be used. SOAPdenovo-Trans has two versions: “SOAPdenovo-Trans-31mer”, which performs assemblies with k-mer sizes from 13 to 31, and “SOAPdenovo-Trans-127mer”, which accepts k-mers up to 127 (always odd values). After adjusting the configuration file, run the assemblies using different k-mers:
> ./SOAPdenovo-Trans-127mer all -s config_file -o output_prefix
Increasing k-mer value reduces the number of transcripts that are recovered, until none can be retrieved. At the end, combine all assemblies into one final file using the “cat” function.
[Optional] Trinity can be run using the Galaxy platform, either via the web servers (https://usegalaxy.org/) or using your own instance (https://galaxyproject.org/admin/get-galaxy/). Trinity can also perform the assembly combining the paired and unpaired reads, which must be appended to the left reads beforehand. Tutorials are available showing how to use the web server (https://usegalaxy.org/). Upload the quality-processed fastq files (e.g.,“all_right_normalized.file” and “all_left_all_unpaired_normalized.file”). Select Trinity in the tools list and set up the parameters according to your data. If your data have already been normalized, turn off this option and start the analysis.
[Tip 3] We suggest testing different assemblers. The quality assessment that will be described in the next section will help you to decide which assembly or assemblies will be used as your reference transcriptome.
Filter out the small length isotigs from each assembly, which might be spurious sequencing products. A routinely used threshold is 200 bp. The “Filter sequences by length” tool in Galaxy platform can perform this task. If you used other assemblers locally, upload the assemblies to Galaxy for filtering them. The tool can be found under “Genomic File Manipulation > FASTA/FASTQ > Filter sequences by length”. Set the minimal length and run. Once it is done, download all filtered assembly files.
[Recommended] If you opted for using more than one assembly file (different k-mers and/or different software), it is also possible to combine them into a single super-set. Because the combination of different assemblies creates a redundant file with transcripts of heterogeneous quality, they must be filtered. The EvidentialGene tr2aacds pipeline performs a coding potential analysis and removal of perfectly redundant transcripts rather than filtering the transcripts based solely on their length. The package provides several other scripts organized in different folders. Perform the tasks in the following order using as input your assembly files:
Regularize the transcripts ID of each assembly using the “trformat.pl” script (required for the tr2aacds pipeline, located in /scripts/rnaseq/ folder), as for instance:
> ./trformat.pl -input assembly_1.fa -output ID-assembly_1.faCombine the ID-regularized assemblies into one super-set (e.g., using the “cat” function”);
Run the tr2aacds (“tr2aacds4.pl”, which can be found in /scripts/prot/ folder) pipeline using as input the concatenated sets obtained with different assemblers and the super-set:
> ./tr2aacds4.pl -mrnaseq ID-assembly_1.fawhere -mrnaseq sets the name of the assembly to be evaluated.
The pipeline outputs the relevant transcripts as “okay” sets (“okay-main” and “okay-alts”), which may be combined and used. A folder containing discarded sequences (“drop set”) is also created.
The “assemblystats” tool may provide a summary about the general statistics of the transcriptome assembly, such as isotig length and number of assembled isotigs, called “assemblystat”. If you are running your own Galaxy instance, the tool repository may be found at https://toolshed.g2.bx.psu.edu/view/konradpaszkiewicz/assemblystats/6544228ea290. Alternatively, it may be accessed at PlantGenIE Galaxy server (http://galaxy.plantgenie.org).
Quality assessment (analyses performed in Linux OS)
For selecting the most representative assembly, it is necessary to compare their quality. In this pipeline, four criteria will be evaluated for quality assessment: (1) mapping back the reads to the transcriptome assembly; (2) completeness; (3) representation of full-length transcripts; (4) evaluation of the DETONATE scores. It is highly recommended to perform all four analyses for a complete overview of the quality of your assemblies. These four criteria must be applied for each of the assemblies to be compared.
[Recommended] For mapping back the reads to the transcriptome, a read alignment tool is necessary. For instance, Bowtie2 can be used as follows:
First, build an index for each of the transcriptome assemblies to be tested:
> bowtie2-build input_transcriptome.fasta input_transcriptome.fasta
Perform the alignment of the RNA-seq reads to each of your assemblies, as for instance:
> bowtie2 --local --no-unal -x input_transcriptome.fasta \
-q -1 all_left_normalized.file \
-2 all_right_normalized.file | samtools view -Sb - -o output.bam
The alignment statistics will be reported at the end of the run.
[Recommended] Completeness of a transcriptome can be assessed with BUSCO. Check for the newest version at https://busco.ezlab.org/. Running BUSCO is straightforward by using the following command line:
> ./busco -m transcriptome -i input_transcriptome.fasta -o output_name -l LINEAGE
where -m sets the mode for BUSCO (genome, proteins, or transcriptome), and the lineage (-l) is the dataset that will be used as reference for comparison, and must be related to the organism of your study. The lineage dataset can be downloaded from BUSCO website. Optionally, the number of CPU threads (--cpu <int>) and compression of output files (--tarzip) can be set.
[Recommended] Representation of full-length transcripts. When working with a model organism, the representation can be estimated by running a translated BLAST+ (blastx) search against the organism’s reference protein dataset. For non-model organisms, because of the absence of a specific reference for comparison, the analysis has to be done against the proteins of a closely related species (when available), or a dataset containing all known proteins (e.g., Swiss-Prot, available at https://www.uniprot.org). Next, the percentage of best aligning targets can be recovered using the script “analyze_blastPlus_topHit_coverage.pl” available in the Trinity package.
First, create a BLAST database using your reference protein set:
> makeblastdb -in ref_prot_dataset.fasta -dbtype prot
where -in sets your reference input, and -dbtype sets the type of sequence (prot, for proteins)
Run blastx against your reference:
> blastx -query input_transcriptome.fasta -db ref_prot_dataset.fasta \
-out blastx_output.outfmt6 -evalue 1e-20 -max_target_seqs 1 -outfmt 6
where -query is your transcriptome input, -db sets the reference database, -evalue sets the threshold, -max_target_seqs informs the number of sequences to be retained (1, for only the best hit), and -outfmt <list> sets the format of the output file (6 is required for using the script). Optionally, the number of CPU threads may be set (-num_threads <int>).
Run “analyze_blastPlus_topHit_coverage.pl” script on the output file:
> ./analyze_blastPlus_topHit_coverage.pl blastx_output.outfmt6 \
input_transcriptome.fasta ref_prot_dataset.fasta
The script reports for each percentage level (1st column) the number of best matching transcripts (2nd column) and the comparison to the level below (3rd column).
[Recommended] Evaluation of the DETONATE scores. RSEM-EVAL, which is a component of the DETONATE v1.11 package, performs a reference-free evaluation of the assembly quality using a probabilistic model (Li et al., 2014). DETONATE requires some settings to be adjusted prior to the quality evaluation:
First, estimate the transcript length distribution using the “rsem-eval-estimate-transcript-length-distribution” script provided in the package. If a reference dataset is not available, a closely related species can be used:
> ./rsem-eval-estimate-transcript-length-distribution \
input_reference.fasta parameter_file
where parameter_file is the output that will store the information about the estimated mean length and standard deviation of the transcripts;
Next, calculate the scores with the “rsem-eval-calculate-score” script:
> rsem-eval-calculate-score \--transcript-length-parameters parameter_file \
--paired-end all_left_normalized.file all_right_normalized.file \
input_transcriptome.fasta \
assembly_name L
where --transcript-length-parameters sets the parameter file, --paired sets the analysis for paired-end sequences, input_transcriptome.fasta should be the assembly that will be evaluated, and assembly_name is the prefix that will be used for the output files, and L is an integer for the average fragment length of your paired-end data. The program uses Bowtie2 as default. If it is not available in your PATH, use the --bowtie-path <path> option to set the destination. Several other options are available to be set if necessary. DETONATE scores will always be negative, but the higher the score values, the better the assembly.
Transcriptome annotation (analyses performed in Linux OS)
InterProScan performs the recognition of protein signatures that is useful for retrieving the possible function of the assembled transcripts. However, the analysis is time consuming. Alternatively, Trinotate can be used for a less detailed protein annotation, but provides a good way to integrate BLAST searches and to extract information from them, thus being a good option when a closely related species is well annotated. Trinotate also uses some tools for protein signature recognition, but the input file is limited by the outputs generated by Trinity, thus requiring a few extra steps when other assemblers are used. For performing a detailed protein signature recognition with InterProScan, the following steps should be performed:
First, predict the open reading frames (ORFs) of the transcripts with getorf, which is a part of EMBOSS package (http://emboss.sourceforge.net/), or Transdecoder. For instance, getorf can be used with the following command:
> getorf -sequence input_transcriptome.fasta -outseq ORF-predicted_transcriptome.fasta \-table <list> -find 0 -minsize <int>
where -minsize <int> sets the minimum nucleotide size of the ORFs (e.g., 200), -table <list> sets the genetic code to be used (0 for standard), and -find <list> sets the searching method (0 for translation of regions between STOP codons).
InterProScan analysis can be computationally demanding for large datasets. When not running on a computing server, in order to circumvent memory-related issues, an alternative is to divide the ORF-predicted transcriptome file into several parts. Next, run the analysis for each of the subsets (for instance, using a script to start sequential analyses). First, use the FASTA splitter tool for dividing the ORF-predicted transcriptome file:
> ./fasta-splitter.pl --measure count --part-size <int> ORF-predicted_transcriptome.fastawhere --measure (all, seq, or count) specifies whether all data, sequence length, or number of sequences will be used for dividing the file, --part-size <int> sets the number of fasta sequences to be present in each subset.
Next, run InterProScan analysis for each of the subsets. For best results with InterProScan, activate the databases of partner resources (e.g. Panther, SignalP, SMART and TMHMM) and make use of the lookup service which has pre-calculated results (requires internet connection and the latest version of the software). If you intend to perform Gene Ontology enrichment analysis, which will be detailed in the next section, the --goterms and --iprlookup options are required. Detailed information about InterProScan configuration can be found in InterProScan Documentation page (https://github.com/ebi-pf-team/interproscan).
[Tip 4] In the case you are using conda tool, check if your Python and Java JDK/JRE versions match InterProScan’s requirements. If not, it is possible to create new conda environments for running with specific software versions (check how to create new environments at Conda webpage; https://docs.conda.io/projects/conda/en/latest/index.html).If you divided your ORF-predicted transcriptome in several subsets, concatenate all .tsv output files using the “cat” function.
[Optional] For integrating Trinotate to the analysis when non-Trinity assemblies are included, it is necessary to provide a gene-transcript relationship of the transcriptome, which can be created with Corset (https://github.com/Oshlack/Corset/wiki):
First, create a Bowtie2 index for the transcriptome assembly file as described in Step C1a;
Next, map back each of the RNA-seq samples used for the assembly to the transcriptome with Bowtie2. If you used both paired and unpaired reads for your assembly, the command would be:
> bowtie2 --all -X <int> -x input_transcriptome.fasta \-q -1 sample_n-1.fastq -2 sample_n-2.fastq \
-U sample_n-1.unpaired.fastq,sample_n-2.unpaired.fastq | \
samtools view -Sb > sample_n.bam
where -X <int> is the estimated fragment size of your data. If only paired reads were used for the assembly, remove the -U part;
The .bam files are used as input for Corset. First, create the summaries independently for each input sample:
> ./corset -m <int> -r true-stop sample_n.bamwhere -m sets to filter out transcripts with fewer than <int> reads, and -r true-stop specifies that only the summaries must be created (outputs .corset-reads). Since we are only interested in having a rough estimation for creating a required input file for Trinotate, we can set -m 0 (consider all transcripts). Later, if performing gene expression analysis, the number of reads mapping to a transcript may be precisely considered by the tool you opt to use.
When the summaries have been created for all samples, resume Corset run defining the grouping for analysis:
> ./corset -i corset -g <list> .corset-readswhere -i corset sets the summaries previously created as input file (the default is .bam). If you have groups of samples, as for instance two replicates of both control and treatment, they can be specified with the -g option separated by commas (e.g., -g control, control, treatment, treatment). The input files have to follow the same order. If -g is not set, each sample will be considered independently. The command for a given example will be as following:
> ./corset -i corset -g control,control,treatment,treatment \control_1.bam.corset-reads control_2.bam.corset-reads \
treatment_1.bam.corset-reads treatment_2.bam.corset-reads
When the analysis is over, edit the Corset clusters file, which has the gene-transcript relationship, by switching the position of the two columns to conform to Trinotate's requirements.
[Optional] A detailed Trinotate step-by-step guide is available at http://trinotate.github.io. Trinotate relies on Swiss-Prot database for performing its annotation. An interesting feature of this tool is that it integrates BLAST searches to the analysis. When working with non-model species or samples from uncontrolled environments, BLAST searches against higher taxa can be used for identifying spurious transcripts which you might considering filtering out.
Gene Ontology analysis
Several methods and tutorials are currently available for running differential gene expression analysis (e.g., edgeR [McCarthy et al., 2012], Deseq2 [Love et al., 2014], among others), which is straightforward. Therefore, we will focus on the description of the GO enrichment analysis. Nevertheless, for this step, the annotated set of genes and the list with differentially expressed genes are both required.
BiNGO is a tool that runs in the Cytoscape platform (https://cytoscape.org/). It is available for Linux, Mac OS, and Microsoft Windows. BiNGO allows one to perform the GO enrichment analysis using a custom annotation as background. The custom file for BiNGO must be parsed as “transcript_ID = number”, where the transcript ID is associated to a unique GO ID number (one entry per line). Therefore, before performing the analysis it is necessary to retrieve the GO identifiers from the annotation file. We provide a Python script for retrieving the GO information from InterProScan (“GO_retriever.py”, available as Supplementary File S1). For each transcript ID on the 1st column, the script will return the respective GO number, if any, from the Nth column. When more than one GO term is associated to a transcript, the script splits the transcript ID into two or more lines, each one being linked to a unique GO number. The output is a .txt file. The script can also be applied for extracting the GO numbers from Trinotate output, but it would be necessary to remove the GO descriptions from the report file beforehand.
For running the script, it is necessary to edit it according to your input data:
Line 1: Substitute INPUT_FILE.tsv by the name of your input file.
[Optional] Line 2: Change the name of the output file (OUTPUT_FILE_GOs.txt).
Line 12: Based on your annotation file, set the column (N) where the GO ID numbers were reported.
Line 17: By default, the script is set to recognize the GO split character used by InterProScan (vertical bar). If you intend to use the script for Trinotate’s output, first you will need to remove the descriptions that are provided for each term (keep only GO:number), adjust the split character in the script accordingly, and change the column where the information is reported (previous step).
With the annotation file and the script in the same directory, run the script:
> python GO_retriever.py input_file
BiNGO can be installed directly through Cytoscape (on the menu bar, select Apps > App Manager ..., select BiNGO on the list, install). After installation, BiNGO will be listed in the Apps menu. With BiNGO running:
From your table with differentially expressed genes, copy the IDs of those that you want to check for overrepresentation (e.g., up-regulated or down-regulated genes).
Select "paste Genes from Text" option, and paste the IDs in the box.
"Overrepresentation" and "Visualization" are selected by default.
Select the statistical test, multiple testing correction, and significance level (e.g., Benjamini & Hochberg False Discovery Rate, P < 0.05).
Select the categories which you want to visualize. By default, only those overrepresented after the correction will be shown.
For the reference set, select the whole annotation (default).
Select the ontology file (.obo file, e.g., “go-basic.obo”), which can be downloaded from the GO Consortium website (https://geneontology.github.io/). BiNGO provides some ontology file options but they may be out of date.
Under "Select namespace", it is possible to choose which GO category will be evaluated, such as biological process, cellular component, or molecular function. For each of them, an independent run will be required.
Finally, under "Select organism/annotation", select as your background the custom annotation file created on the previous steps.
If you check the box for saving the data, you can already set the destination folder. Otherwise, you can also save the results later.
BiNGO outputs a graphical visualization of the network, representing the statistically significant enriched GO terms by colours. The nodes can be adjusted for a better visualization. The image can be exported by selecting this option under the "File" menu. At the bottom, it is possible to recover the significance levels of each enriched category for plotting a table for easier visualization.
For describing this pipeline, the datasets that were used as example are detailed in Duarte et al. (2019). In summary, RNA samples were isolated from pools of Scots pine needles belonging to three areas contaminated by radionuclides after the Chernobyl Nuclear Power Plant accident (Kulazhin, Masany, and Zabor’e), and from a reference plot. The samples were 125 bp paired-end sequenced. FastQC v0.11.5 was used for quality check the sequence data, and FastQ Screen v0.9.2 was applied for removing vector sequences from the RNA-seq reads. The reads were then trimmed with Trimmomatic v0.36, using ILLUMINACLIP:Adapters.fa:2:30:10 HEADCROP:10, MINLEN:35, LEADING:5, TRAILING:5, and SLIDINGWINDOW:4:30 parameters. Preliminary tests were performed with three different Phred qualities for trimming (5, 20, and 30), and indicated that the highest Phred cut-off was the most reliable for performing the transcriptome assembly (Duarte et al., 2019). All Linux-dependent analyses were performed on Ubuntu 16.04 or 18.04 releases, while the GO enrichment evaluation was run on Windows 10. The processed reads are available in Sequence Read Archive (BioProject PRJNA497687).
Transcriptome assembly
For pine transcriptome assembly (Duarte et al., 2019), we opted for normalizing the concatenated read files to reduce redundancy using the insilico_read_normalization.pl script available in Trinity v2.2.0 package. Next, three different assemblers were used, namely BinPacker v1.1, SOAPdenovo-Trans v1.04, and Trinity v2.2.0. Five different assemblies were generated and filtered for isotigs with minimum length of 200 bp. BinPacker was run with default parameters except for the gap length (-g) set to 500 according to the insert size of the data set (Duarte et al., 2019). The combined set comprised five different k-mers (23, 25, 27, 29, and 31), which was then filtered for redundancies using EvidentialGene pipeline (release 19-Mar-2015). For SOAPdenovo-Trans assemblies, ten different k-mer lengths were used and then combined (25, 31, 37, 43, 49, 55, 65, 75, 85, and 95). The program was run using following parameters: max_rd_len = 115, rd_len_cutoff = 115, avg_ins = 500, reverse_seq = 0, asm_flags = 3, map_len = 32, −f, -F. No data were retrieved with k-mer > 95. Using a 32G of RAM 8-cores machine, BinPacker and low-kmer SOAPdenovo-Trans assemblies required a few hours to run. The concatenated set was filtered with EvidentialGene. Trinity was run on Galaxy platform (usegalaxy.org), using the paired-end mode, with unpaired reads appended to the left reads, and default parameters except for the normalisation, which was set off. Although Trinity was run only with the optimized k-mer length, filtering with EvidentialGene was also performed for comparison. Finally, “assemblystat” tool was used for retrieving the general statistics of each assembly, which is shown in Table 1.
Table 1. General statistics for P. sylvestris assemblies comparing the output of BinPacker, SOAPdenovo-Trans, Trinity, and the super-set combining all assemblies . Adapted from Duarte et al. (2019), Table S1.
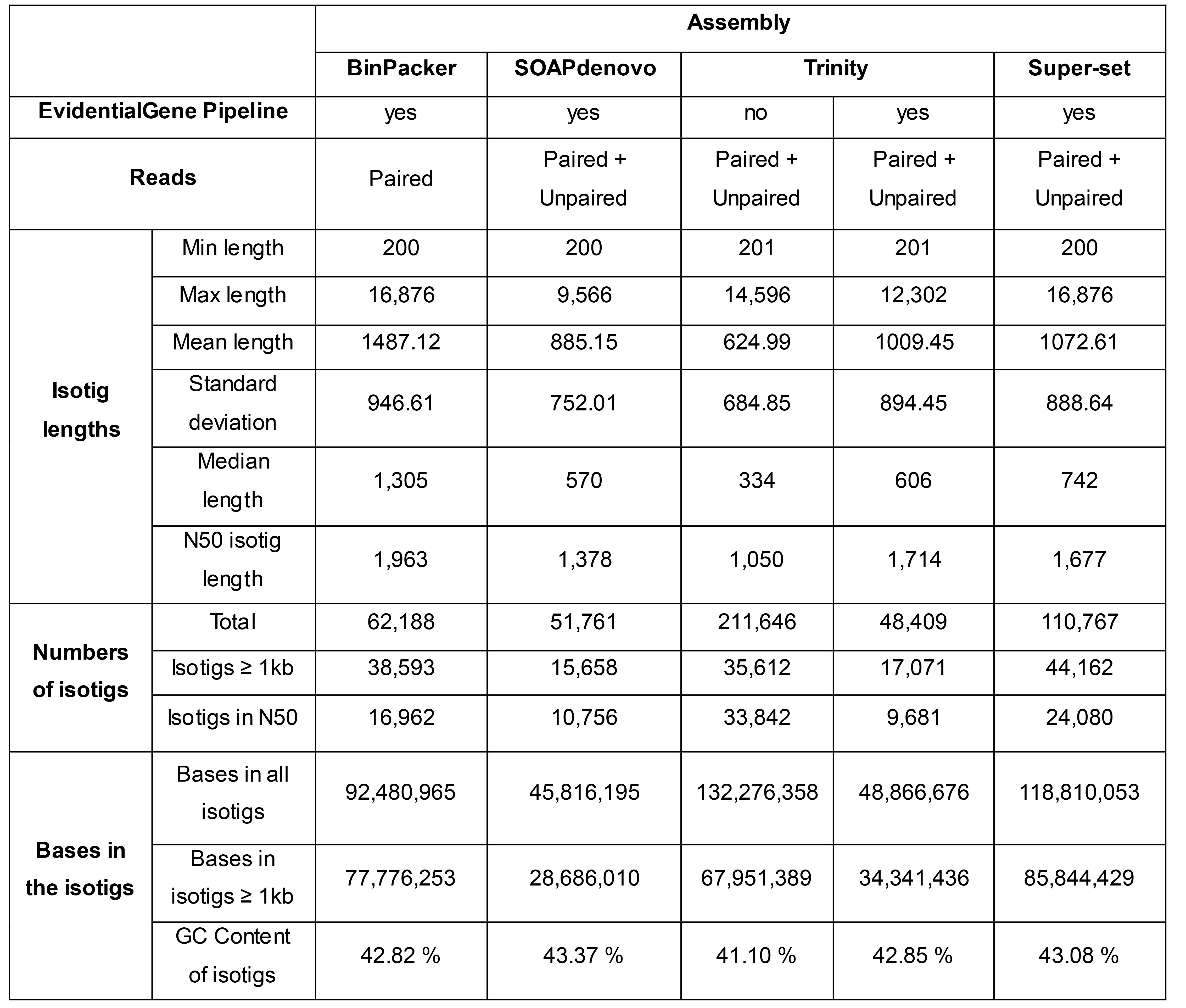
In general, SOAPdenovo-Trans performed poorly for assembling long isotigs, as shown by the max isotig length. However, considering the mean length, this assembler provided a higher score than unfiltered Trinity. Apparently, Trinity provides a highly redundant output, as indicated by the number of isotigs before and after filtering. Trinity assembly must also contain a high proportion of short isotigs, which would explain the shortest median length and N50 length retrieved among the assemblers. However, filtering Trinity’s output significantly reduced the number of isotigs in N50. Considering these general statistical scores, BinPacker, the filtered super-set, and unfiltered Trinity had the most promising initial statuses.
Quality assessment
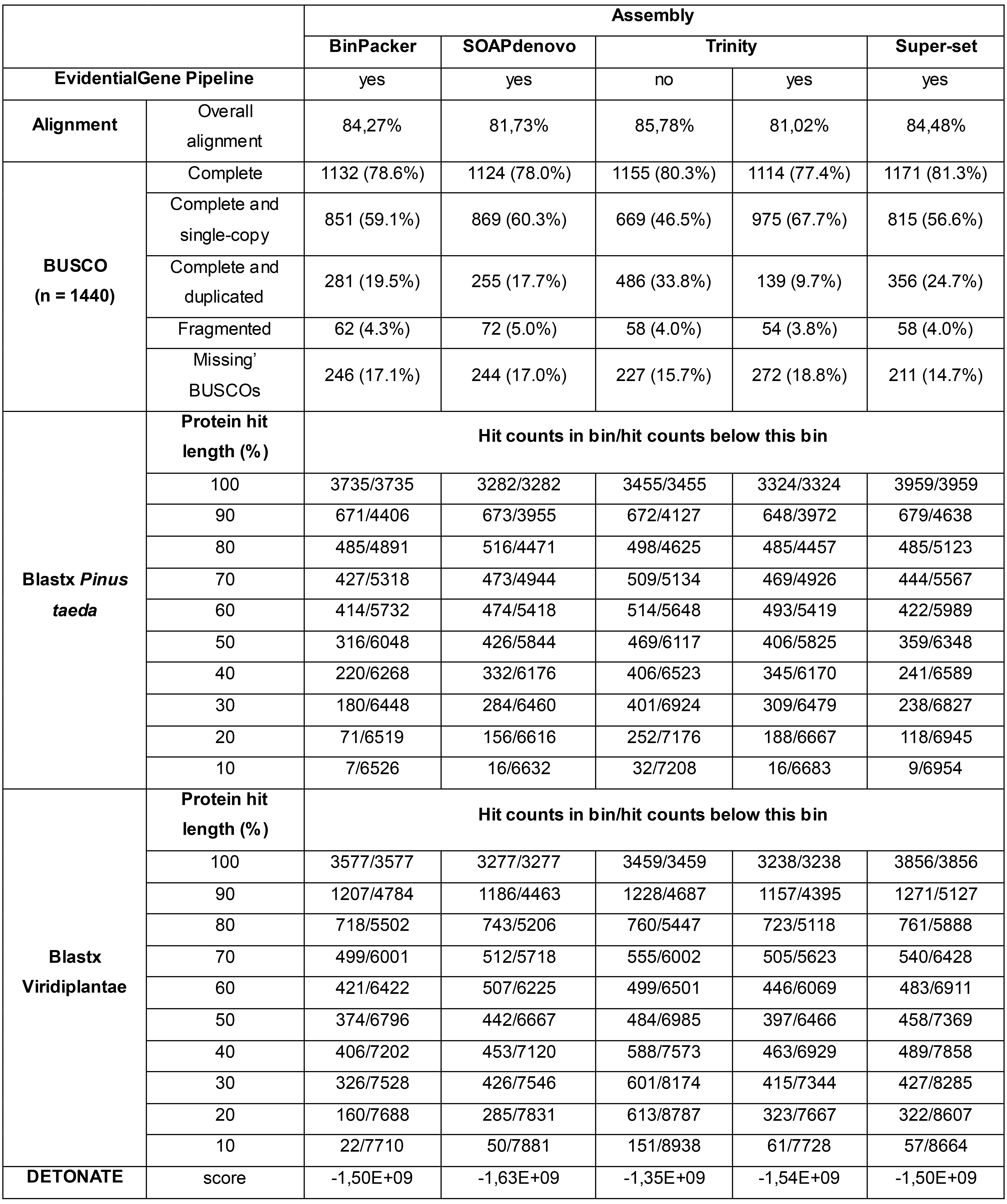
The four criteria for transcriptome assembly quality assessment described in this protocol were also applied for the P. sylvestris transcriptome study (Duarte et al., 2019). They are summarized in Table 2. The reads were mapped back to each of the five assemblies using Bowtie v2.2.9. Completeness with BUSCO v2 was assessed using the plants set (embryophyta_odb9). Translated nucleotide BLAST (blastx v2.7.1) search was performed against Pinus taeda v1.01 proteins retrieved from TreeGenes Database (https://treegenesdb.org/), and TrEMBL Viridiplantae protein sequences (release 2017_03). Finally, DETONATE v1.11 was used for scoring the quality, using P. taeda proteins nucleotides sequences for estimating the transcript length distribution. Performing all four analyses required some hours, and the results for each assembly are provided in Table 2. The filtered SOAPdenovo-Trans set had the lowest scores for all four criteria evaluated, while filtering Trinity’s output did not improve its quality scores. On the other hand, the filtered BinPacker set comprising five different k-mer lengths, the filtered super-set comprising all assemblies, and unfiltered Trinity had similar alignment rates and completeness. Although the unfiltered Trinity assembly had a slightly better DETONATE score, the super-set showed better representation of full-length transcripts and BUSCO scores (highest completeness and lowest number of missing BUSCOs). The combined super-set was selected for the evaluation of differentially expressed genes and GO enrichment analysis (Duarte et al., 2019).
Table 2. Quality assessment of P. sylvestris assemblies comparing the output of BinPacker, SOAPdenovo-Trans, Trinity, and the super-set with all assemblies. Adapted from Duarte et al. (2019), Table S1.
Transcriptome annotation and gene ontology enrichment analysis
For the pine transcriptome study (Duarte et al., 2019), InterProScan v5.26-65.0 results were combined with the summary of BLAST searches reported by Trinotate v3.1.1. For running InterProScan, the ORFs were predicted with getorf (EMBOSS v6.6.0). Then, FASTA splitter v0.2.6 was used to divide the ORF-predicted file in subsets containing 10000 transcripts. InterProScan was run with the following methods: CDD, Gene3D, Hamap, PANTHER, Pfam, PIRSF, PRINTS, ProDom, ProSitePatterns, ProSiteProfiles, SFLD, SMART, SUPERFAMILY, TIGRFAM. The comprehensive annotation with InterProScan required some days to complete. For integrating Trinotate to the analysis, the gene-transcript relationship was created using Corset v1.06. The prediction of coding regions for Trinotate was performed with TransDecoder v5.3.0 (http://transdecoder.github.io). Blastp and blastx searches were run against Swiss-Prot database (release 2017_03), reporting only the best hit (-max_target_seqs 1). Blastp and blastx analyses were also conducted for a collection of gymnosperm-specific protein sequences retrieved from the TreeGenes Database (https://treegenesdb.org/) and ConGenIE (Sundell et al., 2015) (http://congenie.org/), namely Picea abies v1.0, Picea glauca PG29V4, Pinus lambertiana v1.0, Pinus taeda v1.01, and Pseudotsuga menziesii v1.0. In addition, blastp was run against TrEMBL Viridiplantae protein sequences (release 2017_03). Only gymnosperm and/or Viridiplantae-related sequences were retained. Protein domains were identified with HMMER v2.3 (http://hmmer.org/). Prediction of signal peptides, rRNA transcripts, and transmembrane regions were performed using SignalP v4.1, RNAMMER v1.2, and TmHMM v2.0, respectively. As the final result, the combination of Trinotate and InterProScan analyses allowed 75652 out of the 98870 pine transcripts (76.5%) to be successfully annotated (Duarte et al., 2019). Using this final dataset, the transcript ID GO ID relationships were retrieved using the GO_retriever.py script provided in this protocol for generating the background file which was used for the GO enrichment analysis with BiNGO v3.03. The enrichment analysis is provided as supplementary Figures S2 and S3, and Table S3 in Duarte et al. (2019).
Supplementary material
Supplementary File S1. GO_retriever: Python script for extracting GO IDs from an annotation file.
Acknowledgments
The authors thank M. J. Neave for the Python script, and Duarte et al. (2019) for the Scots pine data. This work was supported by the Russian Science Foundation (Grant #14-14-00666) and by the Russian Foundation for Basic Research (Grant # 18-34-20012).
Competing interests
The authors declare no competing interests.
References
- Afgan, E., Baker, D., Batut, B., van den Beek, M., Bouvier, D., Cech, M., Chilton, J., Clements, D., Coraor, N. and Grüning, B. A., et al. (2018). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res 46(W1): W537-W544.
- Bolger, A. M., Lohse, M. and Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30(15): 2114-2120.
- Bryant, D. M., Johnson, K., DiTommaso, T., Tickle, T., Couger, M. B., Payzin-Dogru, D., Lee, T. J., Leigh, N. D., Kuo, T. H. and Davis, F. G., et al. (2017). A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Rep 18(3): 762-776.
- Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J. and Bealer, K.and Madden (2009). BLAST+: architecture and applications. BMC Bioinformatics 10: 421.
- Conesa, A., Madrigal, P., Tarazona, S., Gomez-Cabrero, D., Cervera, A., McPherson, A., Szcześniak, M. W., Gaffney, D. J., Elo, L. L., Zhang, X. and Mortazavi, A. (2016). A survey of best practices for RNA-seq data analysis. Genome Biol 17: 13.
- Davidson, N. M. and Oshlack, A. (2014). Corset: enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol 15(7): 410.
- Duarte, G. T., Volkova, P. Yu. and Geras’kin, S. A. (2019). The response profile to chronic radiation exposure based on the transcriptome analysis of Scots pine from Chernobyl affected zone. Environ Pollut 250: 618-626.
- Geniza, M. and Jaiswal, P. (2017). Tools for building de novo transcriptome assembly. Curr Plant Biol 11-12: 41-45.
- Gilbert, D. (2020). Longest protein, longest transcript or most expression, for accurate gene reconstruction of transcriptomes. bioRxiv. doi: https://doi.org/10.1101/829184
- Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z. and Thompson, D. A., et al. (2011). Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat Biotechnol 29(7): 644-652.
- Grüning, B., Dale, R., Sjödin A, Chapman, B. A., Rowe, J. et al. (2018). Bioconda: sustainable and comprehensive software distribution for the life sciences. Nat Methods 15(7): 475-476.
- Haas, B. J, Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D. and Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-Seq: reference generation and analysis with Trinity. Nat Protoc 8(8): 1494-1512.
- Honaas, L. A., Wafula, E. K., Wickett, N. J., Der, J. P., Zhang, Y., Edger, P. P., Altman, N. S., Pires, J. C., Leebens-Mack, J. H. and dePamphilis, C. W. (2016). Selecting superior de novo transcriptome assemblies: lessons learned by leveraging the best plant genome. PLoS One 11(1): e0146062.
- Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., McWilliam, H., Maslen, J., Mitchell, A. and Nuka, G., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30(9): 1236-1240.
- Krogh, A., Larsson, B., von Heijne, G. and Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305(3): 567-580.
- Lagesen, K., Hallin, P., Rødland, E. A., Staerfeldt, H. H., Rognes, T. and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35(9): 3100-3108.
- Langmead, B. and Salzberg, S. (2012). Fast gapped-read alignment with Bowtie. Nat Methods 9(4): 357-359.
- Li, B., Fillmore, N., Bai, Y., Collins, M., Thomson, J. A., Stewart, R. and Dewey, C. N. (2014). Evaluation of de novo transcriptome assemblies from RNA-Seq data. Genome Biol 15(12): 553.
- Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G.and Durbin, R., 1001 Genome Project Data Processing Subgroup. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16): 2078-2079.
- Liu, J., Li, G., Chang, Z., Yu, T., Liu, B., McMullen, R., Chen, P. and Huang, X. (2016). BinPacker: packing-based de novo transcriptome assembly from RNA-seq data. PLoS Comput Biol 12(2): e1004772.
- Love, M.I., Huber, W. and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. Genome Biol 15: 550.
- Maere, S., Heymans, K. and Kuiper, M. (2005). BiNGO: a Cytoscape plugin to assess overrepresentation of Gene Ontology categories in biological networks. Bioinformatics 21(16): 3448-3449.
- McCarthy, D. J., Chen, Y. and Smyth, G. K. (2012). Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 40(10): 4288-4297.
- Petersen, T. N., Brunak, S., von Heijne, G. and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8(10): 785-786.
- Rice, P., Longden, I. and Bleasby, A. (2000). EMBOSS: the european molecular biology open software suite. Trends Genet 16(6): 276-277.
- Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., Amin, N., Schwikowski, B. and Ideker, T. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13(11): 2498-2504.
- Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31(19): 3210-3212.
- Sundell, D., Mannapperuma, C., Netotea, S., Delhomme, N., Lin, Y. C., Sjödin, A., Van de Peer, Y., Jansson, S., Hvidsten, T. R. and Street, N. R. (2015). The Plant Genome Integrative Explorer Resource: PlantGenIE.org. New Phytol 208(4): 1149-1156.
- Xie, Y., Wu, G., Tang, J., Luo, R., Patterson, J., Liu, S., Huang, W., He, G., Gu, S. and Li, S. (2014). SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-seq reads. Bioinformatics 30(12): 1660-1666.
- Zerbino, D. R. and Birney, E. (2008). Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18(5): 821-829.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Duarte, G. T., Volkova, P. Y. and Geras’kin, S. A. (2021). A Pipeline for Non-model Organisms for de novo Transcriptome Assembly, Annotation, and Gene Ontology Analysis Using Open Tools: Case Study with Scots Pine. Bio-protocol 11(3): e3912. DOI: 10.21769/BioProtoc.3912.
Category
Plant Science
Plant Science
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.