- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Plant ARGONAUTE Protein Immunopurification for Pathogen Cross Kingdom Small RNA Analysis
(*contributed equally to this work) Published: Vol 11, Iss 3, Feb 5, 2021 DOI: 10.21769/BioProtoc.3911 Views: 6400
Reviewed by: GANG YUAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2020
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

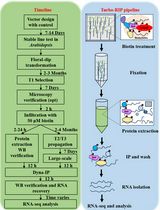
Abstract
Over the last decade, it has been noticed that microbial pathogens and pests deliver small RNA (sRNA) effectors into their host plants to manipulate plant physiology and immunity for infection, known as cross kingdom RNA interference. In this process, fungal and oomycete parasite sRNAs hijack the plant ARGONAUTE (AGO)/RNA-induced silencing complex to post-transcriptionally silence host target genes. We hereby describe the methodological details of how we recovered cross kingdom sRNA effectors of the oomycete pathogen Hyaloperonospora arabidopsidis during infection of its host plant Arabidopsis thaliana. This Bio-protocol contains two parts: first, a detailed description on the procedure of plant AGO/sRNA co-immunopurification and sRNA recovery for Illumina high throughput sequencing analysis. Second, we explain how to perform bioinformatics analysis of sRNA sequence reads using a Galaxy server. In principle, this protocol is suitable to investigate AGO-bound sRNAs from diverse host plants and plant-interacting (micro)organisms.
Keywords: Cross kingdom RNA interferenceBackground
Small RNAs (sRNAs) can serve as pathogen effectors that hijack the plant ARGONAUTE (AGO)/RNA-induced silencing complex (RISC) and silence host mRNAs for infection, a virulence mechanism termed cross kingdom RNA interference (Weiberg et al., 2015; Zeng et al., 2019). Profiling the repertoire of sRNAs bound to the plant AGO during infection is the method of choice, to gain a global overview on plant-invasive pathogen sRNAs that might function through the host AGO/RISC. Antibody-based, co-immunopurification (co-IP) of plant AGO/sRNAs, the functional components of a RISC, coupled to sRNA high throughput sequencing is the gold standard to quantify silencing sRNAs in plants (Mi et al., 2008; Montgomery et al., 2008; Carbonell et al., 2012). Such approaches have led to the discovery of specifications for the binding of sRNAs to distinct members of the plant AGO protein family (Mi et al., 2008; Montgomery et al., 2008) and revealed characteristic changes of AGO-bound sRNA profiles according to plant environmental and stress responses (Zhang et al., 2011). In this context, protocols have been published describing how to co-immunopurify plant AGO/sRNAs in order to study AGO-bound, endogenous plant sRNAs under various conditions (Qi and Mi, 2009; Zhao et al., 2012; Carbonell, 2017).
In this bio-protocol, we provide a detailed description of A. thaliana AGO1/sRNAs co-IP isolated from H. arabidopsidis-infected seedlings and the recovery of both plant and pathogen AGO1-bound sRNAs for high throughput sequencing analysis. By this method, we discovered several novel pathogen sRNA effectors as well as plant silencing sRNAs that were responsive to H. arabidopsidis infection (Dunker et al., 2020). Applying this protocol allowed us to investigate sRNAs bound to other members of the plant AGO family, as well. For instance, we successfully co-immunopurified A. thaliana AGO2/sRNAs using a proAGO2:HA-AGO2 transgenic A. thaliana line (Montgomery et al., 2008) in combination with commercial anti-Human influenza hemagglutinin (HA) antibody, and could identify several AGO2-bound H. arabidopsidis sRNAs (Dunker et al., 2020). Although experimentally validated in the A. thaliana system, we propose this protocol being suitable for AGO/sRNAs co-IP and analysis of silencing sRNAs in various plant species and plant-interacting (micro)organisms, given a suitable antibody for AGO co-IP is available and host and microbe genome sequences are known.
Materials and Reagents
Materials
Blotting paper (Ahlstrom Munksjö, catalog number: BF4 )
DNA LoBind® 1.5 ml reaction tubes (Eppendorf, catalog number: 0030108051 )
Falcon tubes 50 ml and 15 ml (Greiner Bio-One, catalog numbers: 227261 and 188271 )
Glass pipettes (10 ml)
Miracloth (Merck Millipore, catalog number: 475855 )
Propagation soil substrate (Stender, catalog number: A210 )
Reaction tubes 1.5 and 2 ml (Sarstedt, catalog numbers: 72.690 and 72.691 )
14-day-old Arabidopsis thaliana seedlings (ecotype Col-0)
Hyaloperonospora arabidopsidis spores (strain Noco2)
Reagents
Note: If this protocol refers to water, it always implies de-ionized, ultrapure water.
Liquid nitrogen
Acrylamide/bis-acrylamide solution (Rotiphorese Gel A, Carl Roth, catalog number: 3037 )
Anti-AGO1 polyclonal antibody (Agrisera, catalog number: AS09 527 )
Ammonium persulfate (APS) p.a. (Carl Roth, catalog number: 9592 )
Bromophenol blue Dye (Carl Roth, catalog number: A512 )
cOmplete® protease inhibitor cocktail (Roche, ordered via Sigma-Aldrich, catalog number: 04693116001 )
Diethyl pyrocarbonate (DEPC, Carl Roth, catalog number: K028 )
1,4-Dithiothreitol p.a. (DTT, Carl Roth, catalog number: 6908 )
Desoxyribonucleotide mix (dNTP, 10 mM each nucleotide type) for molecular biology (New England Biolabs, catalog number: N0447 )
96% Ethanol Ph. Eur. (VWR chemicals, catalog number: 20905.296 )
Ethylenediamine tetraacetic acid (EDTA) disodium salt dehydrate (Gerbu Biotechnik Gmbh, catalog number: 1034 )
Glacial acetic acid (Carl Roth, catalog number: 3738 )
Glycerol Ph. Eur. (Carl Roth, catalog number: 6967 )
Glycine p.a. (PanReac/AppliChem, catalog number: 131340 )
Glycogen RNA grade (Thermo Fisher Scientific, catalog number: R0551 )
GoTaq® G2 DNA Polymerase (Promega, catalog number: M7841 )
IRDye® 800CW Goat anti-Rabbit IgG Secondary Antibody (LI-COR, catalog number: 926-32211 )
Magnesium chloride (25 mM; for molecular biology) (New England Biolabs, catalog number: B9021 )
NEBNext® Multiplex Small RNA Library Prep Set for Illumina (New England Biolabs, catalog number: E7300 )
Nonidet P-40 (NP-40, no longer available, the replacement product is IGEPAL CA-630 Sigma-Aldrich, catalog number: I8896 )
10 bp O'RangeRuler DNA ladder (Thermo Fisher Scientific, catalog number: SM1313 )
Potassium chloride (KCl) molecular biology grade (Merck Millipore, catalog number: 529552 )
Potassium dihydrogen phosphate (KH2PO4) p.a. (Carl Roth, catalog number 3904 )
Protein A agarose (Roche, ordered via Sigma-Aldrich, catalog number: PROTAA-RO )
Proteinase K (Thermo Fisher Scientific, catalog number: EO0491 )
RiboLock® RNase inhibitor (Thermo Fisher Scientific, catalog number: EO0381 )
ROTI C/I (Chlorofom/Isoamyl alcohol mixture, Carl Roth, catalog number: X984 )
ROTI-Phenol (Carl Roth, catalog number: 0038 )
ROTI-Phenol/Chloroform/Isoamyl alcohol (Carl Roth, catalog number: A156 )
Sodium chloride (NaCl) p.a. (Carl Roth, catalog number: 3957 )
Sodium dodecyl sulfate (SDS) ultrapure (Carl Roth, catalog number: 2326 )
SuperScript® III (Thermo Fisher Scientific, catalog number: 18080093 )
Disodium hydrogen phosphate (Na2HPO4) p.a. (Carl Roth, T876 )
Tetramethylethylenediamine (TEMED) for electrophoresis (Carl Roth, catalog number: 2367 )
1 M Tris-HCl pH 6.8 stock solution (made from Tris ultrapure, PanReac/AppliChem, catalog number: A1086 , pH adjusted with HCl, Carl Roth, catalog number: X896 )
1 M Tris-HCl pH 7.5 stock solution (made from Tris ultrapure, PanReac/AppliChem, catalog number: A1086 , pH adjusted with HCl, Carl Roth, catalog number: X896 )
1 M Tris-HCl pH 8.0 stock solution (made from Tris ultrapure, PanReac/AppliChem, catalog number: A1086 , pH adjusted with HCl, Carl Roth, catalog number: X896 )
1.5 M Tris-HCl pH 8.8 stock solution (made from Tris ultrapure, PanReac/AppliChem, catalog number: A1086 , pH adjusted with HCl, Carl Roth, catalog number: X896 )
Tris ultrapure (PanReac/AppliChem, catalog number: A1086 )
Triton X-100 (Carl Roth, catalog number: 6683 )
Tween 20 (Sigma-Aldrich, catalog number: P9416 )
5× Protein SDS loading buffer (see Recipes)
10× Protein SDS running buffer (see Recipes)
10× Protein transfer buffer (see Recipes)
10× PBS pH 7.4 (see Recipes)
50× TAE buffer (see Recipes)
DEPC-treated water (see Recipes)
IP extraction buffer (see Recipes)
IP washing buffer (see Recipes)
RNA release buffer (see Recipes)
6% 0.5× TAE gel (see Recipes)
10% 0.5× TAE gel (see Recipes)
8% SDS resolution gel (see Recipes)
SDS stacking gel (see Recipes)
Equipment
Falcon cooling centrifuge with 15 ml and 50 ml adapters (Eppendorf, model: 5810R, catalog number: 5811000325 )
Funnel (e.g., Plastic funnel, Carl Roth, catalog number: 2041 )
Growth chamber
Hemocytometer (Neubauer counting chamber, Carl Roth, catalog number: PC73.1 )
Mortar and pestle
PAGE electrophoresis system (Mighty small II system, Hoefer Inc., catalog number: SE250 )
PCR cycler (FlexCycler, Analytik Jena, succession product is Biometra TOne, Analytik Jena)
Standard pipettes of 100-1000 µl, 20-200 µl, 2-20 µl and 1-10 µl (Gilson, catalog numbers: F123602 , F123601 , F123600 , F144802 )
Pipette controller (Integra Biosciences, ordered via VWR, catalog number: 612-0927 )
Rolling shaker (TRM 50, IDL GmbH, catalog number: 5200330100 )
Rotator (AG, FINEPCR, no order number found)
Small scissors
Spatula
Spray unit (Carl Roth, catalog number: YC44.1 )
Tabletop centrifuge for micro tubes (Eppendorf, model: 5424R, catalog number: 5404000410)
Table top mixer (Scientific Industries, model: Vortex Genie 2, catalog number: SI-0236 )
Thermo shaker with 1.5 ml reaction tube adapter (Eppendorf, model: Thermomixer C, catalog number: 5382000015 , can be also used as heat block)
Wet blot tank system (Mighty small transfer tank, Hoefer Inc., catalog number: TE22 )
Software
Software, bioinformatics tools, and databases:
Galaxy server (release 19.01)
Illumina Demultiplex (Galaxy Version 1.0.0) (alternative tool for demultiplexing: bcl2fastqc
https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html)
Clip adaptor sequence (Galaxy Version 1.0.0) (alternative tool for adapter trimming: Trimmomatic [Bolger et al., 2014])
Filter FASTQ reads by quality score and length (Galaxy Version 1.0.0)
FastQC Read Quality reports (Galaxy Version 0.72)
Map with Bowtie for Illumina (Galaxy Version 1.1.0)
SAM to FASTQ creates a FASTQ file (Galaxy Version 1.56.1)
Filter with SortMeRNA of ribosomal RNAs in metatranscriptomic data (Galaxy Version 2.1b.6)
Collapse FASTA sequences (Galaxy Version 1.0.0)
TAPIR: target prediction for plant microRNAs (Bonnet et al., 2010)
A. thaliana TAIR10.0 genome sequence, cDNA sequences
H. arabidopsidis Noks1 (PRJNA298674), Noks1 is a single spore isolate from a Noco2 sample (Bailey et al., 2011)
Procedure
Note: Figure 1 provides an overview scheme of the protocol wet-lab part for your consideration. Plant AGO/sRNA co-immunopurification does not require in vivo cross-linking. Before starting, we suggest carefully read the entire protocol. During the procedure, work as quickly as possible, on ice and in a 4 °C cold room, when possible, in order to prevent RNA or protein degradation.
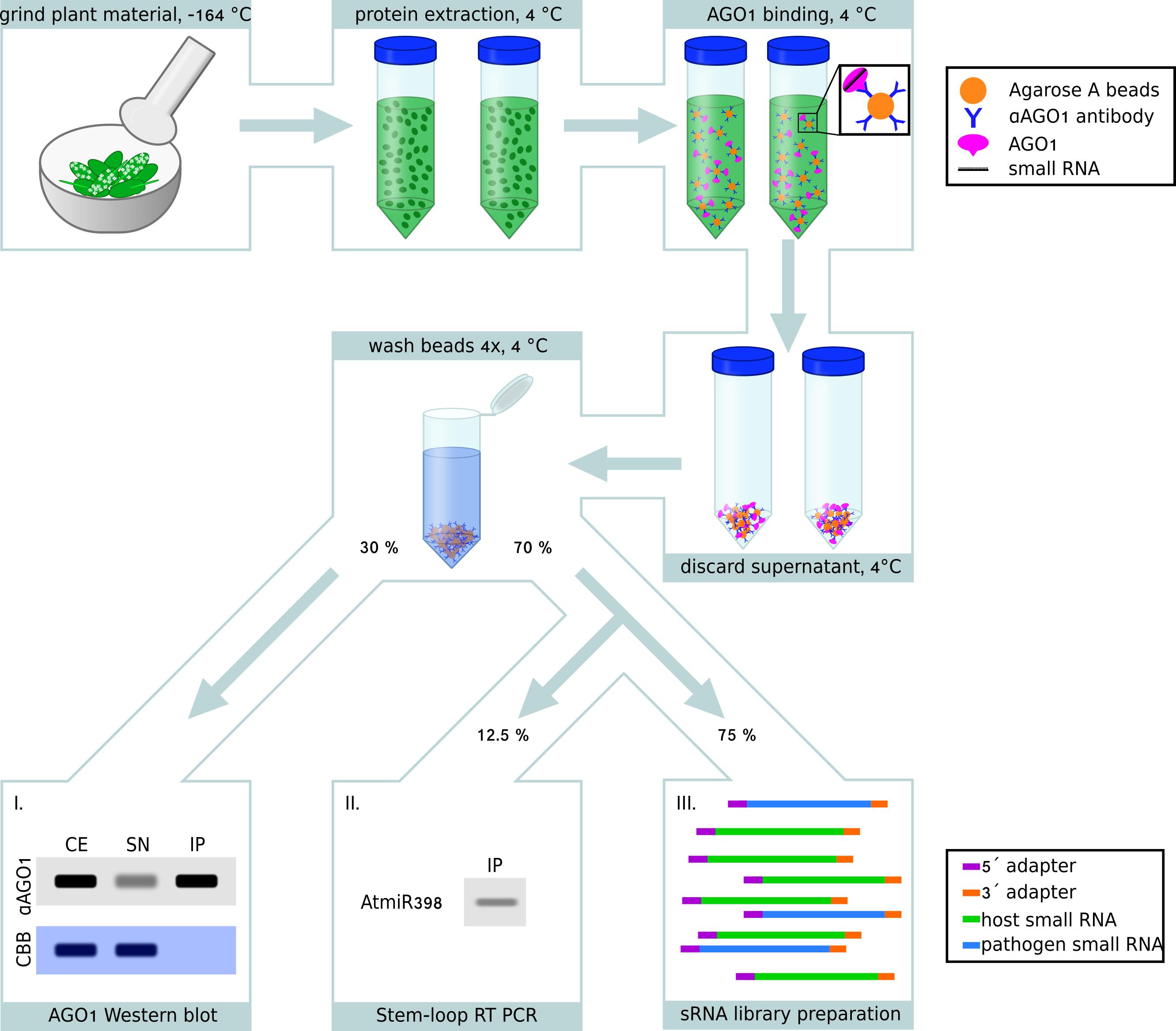
Figure 1. Schematic workflow of plant AGO co-IP for sRNA analysis. A. thaliana seedling leaves infected with H. arabidopsidis were ground under liquid nitrogen with pestle and mortar. After adding the IP extraction buffer to the ground leaf material, A. thaliana AGO1 proteins were immunopurified (IP) using an AGO1-specific antibody and Protein A agarose beads. After washing the beads, IP samples were used for Western blot analysis and small RNA extraction in a sample volume ratio of 30%/70%, respectively. As a quality control for successful IP, AGO1 was tested in the crude extract (CE), in the supernatant (SN), and in the IP fractions by the Western blot analysis, using total protein reference stained by Coomassie Brilliant Blue (CBB) as a loading control (I.), refer to Figure 2 for a Western blot example. As a quality control for successful co-IP and sRNA extraction, an aliquot (12.5%) of the extracted RNA was used for stem-loop RT PCR amplifying the A. thaliana miR398 (AtmiR398) known to bind AGO1 (II.), refer to Figure 3 for a stem-loop RT PCR example. Upon positive results of protein and RNA quality control experiments, the sRNA library was prepared for Illumina-based sequencing (III.).
A. thaliana seedling inoculation with H. arabidopsidis
Note: We normally perform dual co-IP experiment of a mock-treated and a pathogen-inoculated sample. However, if more samples are to be prepared, we suggest to process them sequentially in order to prevent protein or RNA degradation. The following inoculation procedure was adapted from Asai et al. (2015).
Use 14-day-old A. thaliana Col-0 seedlings grown under long-day condition (16 h light/8 h dark) at 22 °C and 60% relative humidity in propagation soil substrate. Prepare 10 squared pots (7 × 8 cm) of seedlings per AGO co-IP experiment to obtain enough leaf material.
Collect conidiospores of H. arabidopsidis strain Noco2 from A. thaliana Col-0 infected seedlings at 7 days post inoculation using small scissors. Pool collected leaves in a 50 ml Falcon tube avoiding any soil particles.
Harvested approximately 2 g of infected fresh leaf material and add 10 ml water to wash off the conidiospores from infected leaves by vigorously shaking the Falcon tube. Filter the conidiospore suspension through a Miracloth and estimate conidiospore concentration by counting conidiospores with a hemocytometer using a light microscope. Adjust the conidiospore suspension to a concentration of 2.5 × 104 spores/ml.
Evenly spray 10 ml of 2.5 × 104 spores/ml suspension (or water as mock treatment) on top of A. thaliana seedlings using a spray unit.
Cover the inoculated or mock-treated seedlings with a transparent lid and seal it, for instance with adhesive tape, to keep high humidity for infection. Incubate the seedlings under long-day conditions (16 h light/8 h dark) at 18 °C for 4-7 days.
Plant AGO/sRNA co-immunopurification
Harvest 5 grams of fresh leaf material at a given time point (in this protocol, we refer to 4 and 7 days post inoculation) with a small pair of scissors and directly transfer leaf material into a pre-cooled mortar containing liquid nitrogen. Grind the leaves into a fine powder with a pre-cooled pestle. Avoid carrying over any soil particles.
Once the entire leaf material is powdered, pre-cool a 50 ml Falcon tube and a spatula tip using liquid nitrogen. Transfer the powdered leaf material from the mortar into the Falcon tube using the pre-cooled spatula. Optionally, use a pre-cooled funnel for the transfer. Double-check that all liquid nitrogen has been evaporated before closing the Falcon tube with the plastic lid to avoid explosion of the tube. Place the closed Falcon tube back into liquid nitrogen.
Place the Falcon tube from the liquid nitrogen into an ice box and add immediately 20 ml of immunopurification (IP) extraction buffer per 5 grams leaf fresh weight. Due to low temperature of the sample, the IP extraction buffer might freeze at this step. Close the Falcon tube again and thoroughly mix the leaf material in the buffer by placing it on a rolling shaker in a 4 °C cold room to let it thaw. Thawing might take up to 50 min.
Double-check that thawing is complete, before proceeding.
From this step onwards, always keep the sample placed on ice. Spin-down the leaf debris at 3,200 × g for 15 min using a pre-cooled (4 °C) centrifuge.
In a 4 °C cold room, filter the supernatant through a two-layered Miracloth into a new Falcon tube with the help of a glass pipette to remove the cell debris. At this point, split a sample into two aliquots of 10 ml each. Set aside a 200 µl aliquot from this filtered crude extract (CE = input sample) for Western blot analysis. Mix the Western blot sample with 50 µl 5× protein SDS loading buffer and boil it for 5 min at 95 °C in a thermo shaker. Store the Western blot sample at -20 °C until further use.
To continue with AGO co-immunopurification, add 5 µg anti-AGO1 antibody per 5 g original leaf tissue weight as well as 200 µl of protein A agarose beads to the crude extract. Incubate the sample on a rotation wheel for 2 h in a 4 °C cold room.
Note: The Protein A agarose beads in the Materials list are provided being pre-equilibrated and ready to use by the manufacturers. Read carefully the manufacturers’ manual on how to use this product.
Spin-down the sample in a pre-cooled centrifuge at 200 × g and 4 °C for 30 s. Take a 200 µl aliquot from the supernatant (SN = unbound fraction) for Western blot analysis. Mix the Western blot sample with 50 µl 5× protein SDS loading buffer and boil it for 5 min at 95 °C. Store the Western blot sample at -20 °C until further use.
To continue with AGO co-IP, discard the rest of the supernatant. Add 1 ml of ice-cold IP wash buffer to the pelleted beads of the sample, carefully resuspend the beads by pipetting up and down and unify the two sample aliquots into a single 2 ml micro tube.
Spin down the sample in a pre-cooled centrifuge at 200 × g and 4 °C for 30 s. Remove the supernatant and wash the pelleted beads with 1 ml freshly prepared, ice-cold IP wash buffer by pipetting up and down.
Repeat the washing step of the pelleted beads 3 more times.
Resuspend the pelleted beads in 1 ml wash buffer and transfer 300 µl (30% of the sample volume) into a new micro tube for Western blot analysis (IP = immunopurified fraction), keeping the remaining 700 µl (70% of the sample volume) for RNA extraction. Spin-down both sample aliquots and discard the supernatant.
Add 50 µl of 1× protein SDS loading buffer (prepared from 5× protein SDS loading buffer by diluting with IP wash buffer) to the pelleted beads of the Western blot sample. Boil the beads with the loading buffer for 5 min at 95 °C. Store the Western blot sample at -20 °C until further use.
sRNA recovery from co-immunopurified AGO1 bound to Protein-A agarose beads
Note: Adequate personal protection is mandatory during this part of the protocol since toxic chemicals such as Proteinase K, SDS, phenol, and chloroform are used. Dispose all toxic chemicals according to local legislation.
Add 300 µl of IP wash buffer and resuspend the pelleted beads by pipetting up and down. Add 150 µl of RNA release buffer. Incubate the sample in a thermo shaker at 300 rpm and 65 °C for 15 min.
Add 450 µl water-saturated phenol and mix the samples using a vortexer for 2 min.
Separate the phenol-water phases by centrifugation at 10,000 × g at room temperature for 8 min, and transfer the upper aqueous phase including the RNA into a new micro tube.
Add 450 µl of Phenol/Chloroform/Isoamylalcohol (PCI) mixture (25:24:1) to the RNA sample, invert the sample 10 times and separate the PCI-water phases by centrifugation at 10,000 × g and room temperature for 8 min, and transfer the upper aqueous phase containing the RNA into a new micro tube.
Repeat the Step C4 two additional times, using Chloroform/Isoamylalcohol mixture (24:1) instead of the PCI. Take great care to avoid carry-over of any traces of the organic phase before starting RNA precipitation. Optionally from this step on, DNA/RNA LoBind® plastic ware can be used to reduce RNA loss.
Precipitate the RNA of the sample by adding in the given order 0.1× volume 3 M sodium acetate, 2.5× volume 96% ethanol, and 20 µg Glycogen (RNA grade). Upon mixing, place the sample at -20 °C for a minimum of 1 h for RNA precipitation.
Note: This is a safe stopping point. RNA samples can be stored in 80% ethanol at -20 °C.
Pellet the RNA by centrifugation at 20,000 × g and 4 °C for 30 min, and wash the RNA pellet with 500 µl 80% ethanol diluted in DEPC-treated water.
Pellet the RNA by centrifugation at 20,000 × g and 4 °C for 20 min, remove all liquids and air-dry the RNA pellet until ethanol is completely evaporated.
Note: Optionally, the RNA pellet can be dried faster in an open-cap micro tube at 37 °C. However, avoid “over-drying” the RNA pellet.
Resuspend the RNA pellet in 8 µl DEPC-treated water. Completely resolve the RNA pellet by incubating the sample for 5 min at 65 °C.
Store the remaining RNA for the library preparation at -80 °C for up to 3 months.
Note: Optionally, we recommend to perform stem-loop reverse transcription PCR to detect A. thaliana microRNA(s) of choice in the AGO co-IP sample as a quality control on the successful recovery of sRNAs before starting the sRNA library cloning (see Figure 3).
Western blot analysis for AGO1 co-immunopurification quality control
Note: The following steps guide through standard Western blot to analyze the three sample types collected throughout the AGO co-immunopurification procedure (crude extract, supernatant, immunopurification). This analysis is essential to confirm efficient AGO purification. In this protocol, Western blots results were visualized on an LI-COR Odyssey detection system. However, any standard protocol for protein identification by Western blot can be used as well.
Assemble a protein gel electrophoresis chamber and fill it with protein running buffer. Load 20 µl per sample of the crude extract from Step B6 (CE), the supernatant (SN) after agarose bead collection from Step B8, and of the AGO co-IP (IP) from Step B13 on an 8% polyacrylamide gel. Load 3 µl of a pre-stained protein size marker on the same gel.
Note: For standard protein SDS polyacrylamide gel electrophoresis (PAGE), we used a Rotiphorese acrylamide/bis-acrylamide solution, APS, and TEMED to prepare a discontinuous gel consisting of a 2 ml collection gel at pH 6.8 and a 7 ml separation gel at pH 8.8.
Initiate the SDS-PAGE run for 30 min at ~10 V/cm, then increase the voltage to ~17.5 V/cm and let the PAGE run until the 50 kilo-Dalton (kDa) band of the protein size marker reaches the edge of the gel (electrophoresis run takes approximately 90 min).
Disassemble the PAGE gel from the electrophoresis chamber and measure the gel size dimensions. Prepare two blotting papers and a PVDF blotting membrane of the measured gel size. Equilibrate the blotting papers and two sponges in the protein transfer buffer for 5 min. Activate the blotting membrane by submerging in 96% ethanol for 1 min and quickly wash-off the ethanol with water. Equilibrate the blotting membrane in the protein transfer buffer until use.
Note: Use a PVDF membrane that is compatible with the Odyssey detection method.
Assemble the blotting sandwich in the following order: i) cathode, ii) sponge, iii) blotting paper, iv) polyacrylamide gel, v) PVDF membrane, vi) blotting paper, vii) sponge, viii) anode. Fill the complete blotting tank with protein transfer buffer. Set the amperage to 1 mA cm-2 of blotting membrane surface and perform blotting of the proteins overnight in a 4 °C cold room.
On the following day, increase the amperage to 2 mA cm-2 for 30 min. This step might increase the focus of protein bands.
Disassemble the blotting sandwich and roll the membrane with the blotted site to the inside to fit in a 50 ml Falcon tube. Add 10 ml of 5% (v/v) skim fat milk in 1× PBS. Block the membrane for 1 h in a 4 °C cold room on a rolling shaker.
Discard the blocking solution and quickly wash the blotting membrane with 1× PBS. Add 4 ml of primary antibody solution (anti AtAGO1 1 µg/µl diluted 1:4,000 in 1% milk in a 0.1% PBST buffer) and incubate on a rolling shaker overnight in a 4 °C cold room.
Wash the blotting membrane 4 times each for 5 min on a rolling shaker in a 4 °C cold room with 10 ml of 0.2% PBST buffer.
Remove the PBST washing buffer and add 5 ml of secondary antibody solution (anti-rabbit IRdye800 (1 µg/µl) diluted 1:3,000 in 1% milk in a 0.1% PBST with 0.02% SDS). Incubate on a rolling shaker for 1-2 h at room temperature.
Wash the blot 4 times each for 10 min on a rolling shaker in a 4 °C cold room with 10 ml of 0.2% PBST.
Take the membrane out of the Falcon tube and rinse the blotting membrane for 1 min with water and let it dry between two blotting papers. Scan the membrane using an LI-COR Odyssey Scanning device. The band of AGO1 appears at ~130 kDa.
Note: A representative example of a Western blot analysis is displayed in Figure 2. Samples with low AGO signal intensity might still be valid for sRNA library cloning and high throughput sequencing.
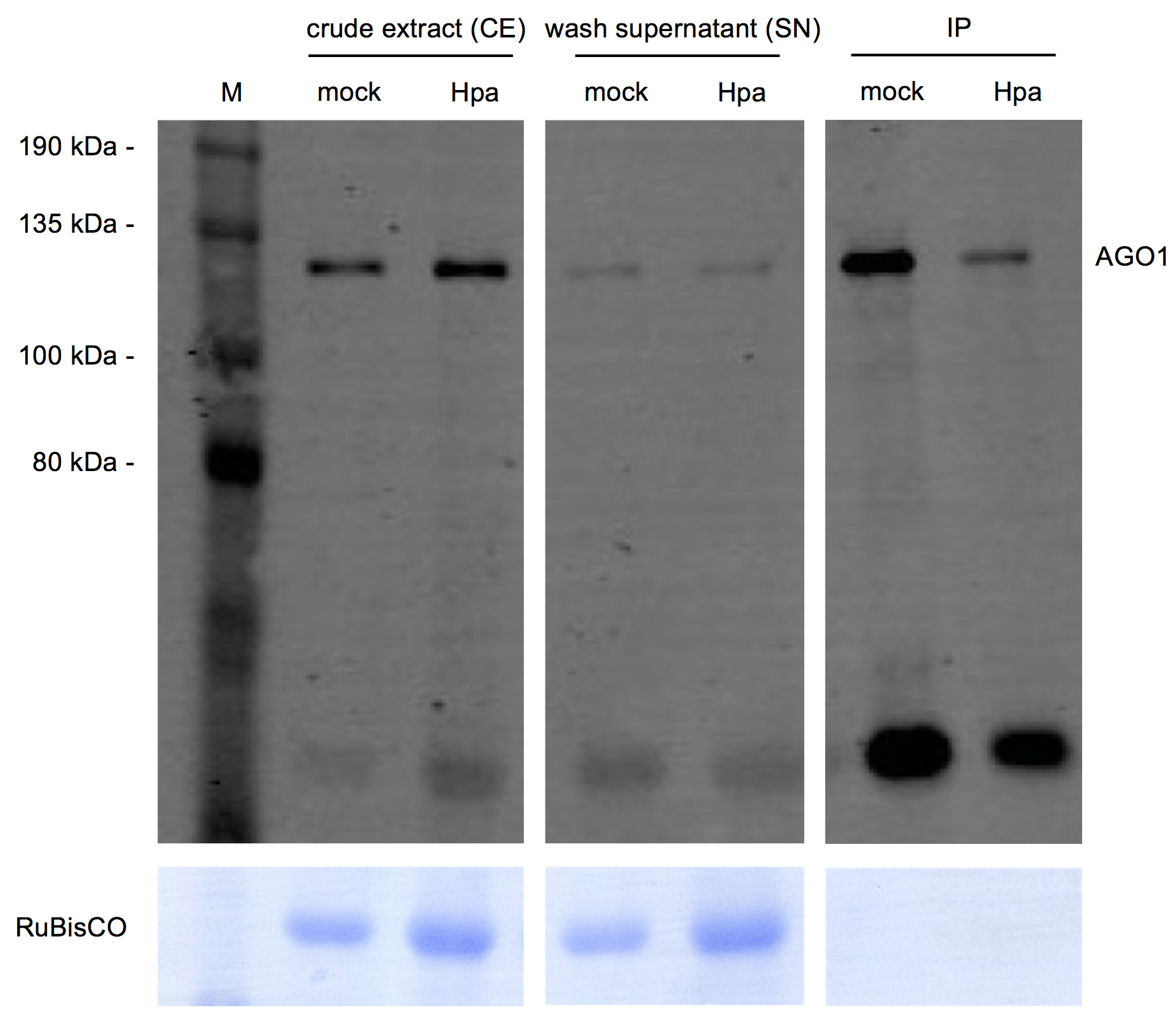
Figure 2. Quality control of AGO1 immunopurification by Western blotting. Three sample fractions of the AGO1 co-IP experiment were analyzed: crude extract (CE), supernatant (SN), and the IP fraction. These three fractions were analyzed in an A. thaliana mock-treated and in an H. arabidopsidis-infected (Hpa) sample. The top figure shows the detection of A. thaliana AGO1 using an AGO1-specific antibody at the expected size of ~130 kDa. Note that AGO1 signals were stronger in IP factions than in SN. The bottom figure displays the ribulose-1,5-bisphosphate carboxylase/-oxygenase (RuBisCO) signal from a Coomassie Brilliant Blue total protein staining of the Western blot membrane. Note that RuBisCO signals disappeared in the IP fraction. The broad range pre-stained protein marker was used as a protein size marker (M).
A. thaliana miRNA stem-loop reverse transcription (RT) PCR for AGO/sRNA co-immunopurification quality control
Note: Before proceeding with library preparation, it is recommended to validate successful RNA co-immunopurification by stem-loop RT PCR. We use the Arabidopsis miR398 known to bind AGO1. The protocol directly follows the stem-loop RT PCR protocol as previously described (Varkonyi-Gasic et al., 2007).
Use 1 µl of your eluted RNA (12.5% sample volume) for this quality control assay and pipet it into a PCR micro tube. Add the following components to your RNA sample:
1 µl of AtmiR398-specific stem-loop RT primer (1 µM), for the sequence design of stem-loop RT primers, refer to Varkonyi-Gasic et al. (2007)
0.5 µl of dNTP mix (10 mM each)
8 µl DEPC-treated water
Heat the sample to 65 °C for 5 min in a PCR cycler. Cool it down on ice for 1 min.
Add the following components per sample:
4 µl 5× first strand SuperScript III reaction buffer
2 µl DTT (0.1M)
4 µl MgCl2 (25 mM)
0.25 µl SuperScript III® reverse transcriptase
0.25 µl RiboLock
Run the following thermo cycler protocol:
step 1 16 °C 30 min
step 2 30 °C 30 s
step 3 42 °C 20 s
step 4 50 °C 1 s; back to step 2 (60x)
step 5 85 °C 10 min
step 6 End
Note: The final end-point PCR can be performed with any standard Taq-Polymerase. Below, a protocol using the GoTaq® Polymerase is described.
Amplify the reverse transcribed miRNA by end-point PCR. Dilute your stem-loop RT product 1:10 with water as a PCR template.
Pipet the following components into a PCR micro tube:
3 µl 5× Green GoTaq® reaction buffer
0.3 µl dNTP mix (10 mM each)
0.5 µl miR398 forward primer (10 µM), for the sequence design of sRNA forward primer, see Varkonyi-Gasic et al. (2007).
0.5 µl universal stem-loop reverse primer (10 µM), primer sequence according to Varkonyi-Gasic et al. (2007).
9.5 µl water
0.2 µl GoTaq® G2 Polymerase
1 µl stem-loop RT product (1:10 diluted in water)
Perform the following thermo cycler protocol:
step 1 94 °C 2 min
step 2 94 °C 30 s
step 3 60 °C 30 s
step 4 72 °C 20 s; back to step 2 (36×)
step 5 72 °C 2 min
step 6 End
Prepare a 10% 0.5× TAE polyacrylamide gel. Assemble a gel electrophoresis running system. Fill it with 0.5× TAE running buffer.
Load 7 µl of each sample in a pocket of the polyacrylamide gel. Pipet 2 µl of 10 bp O'RangeRuler as size marker one the same gel.
Run the gel at ~18 V/cm for 90 min to separate PCR products.
Dissemble the running chamber and stain the gel with ethidium bromide or any other substitutive DNA dye (e.g., SYBR Gold) for 3 min. If the AGO/sRNA co-immunopurification was successful, you should obtain a PCR band at the size of 69 bp. As an example, a stem-loop RT PCR result of AtmiR398 from an AGO1 co-IP sample is shown in Figure 3.
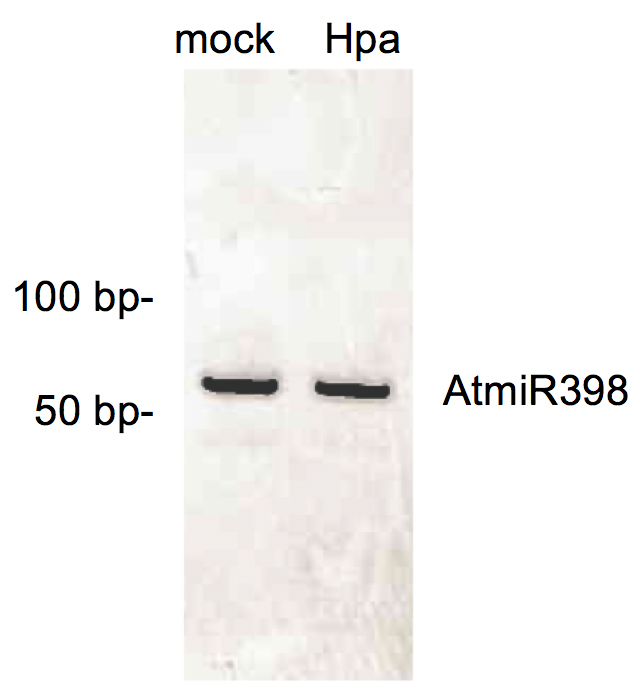
Figure 3. Quality control of sRNAs extracted from AGO1 co-IP samples. For both mock-treated and H. arabidopsidis-infected (Hpa) AGO1-co-IP RNA samples, stem-loop RT PCR was performed to detect the AGO1-bound A. thaliana AtmiR398. A PCR band of the expected size of 69 base pairs (bp) was visible for both samples. A 10 bp O'RangeRuler DNA ruler was used as a size marker. Depicted samples represent A. thaliana leaf materials harvested at 4 days post treatment.
sRNA library cloning for Illumina next generation sequencing
Note: We used the NEBNext® Multiplex Small RNA Library Prep Set for Illumina to perform library preparation that is suitable for non-modified as well as modified sRNAs, such as 3’ terminal 2’-O-methylation. The protocol is a reprint of the original manufacturer’s protocol with slight modifications we applied for AGO co-IP sRNA library cloning. In case library kit protocols are updated by the manufacturer, we recommend to consider the updated instructions.
Dilute the required amount of 3’ SR adaptor (supplied by NEB kit) 1:2 in nuclease-free water. Mix the following per sample in a 1.5 ml LoBind® reaction tube:
6 µl RNA from the AGO co-IP (75% of the eluted RNA sample volume)
1 µl diluted 3’ SR adaptor
Incubate the tube for 2 min at 70 °C in a thermo block.
Add the following:
10 µl 3’ Ligation reaction buffer (2×) (supplied by NEB kit)
3 µl 3’ Ligation enzyme mix (supplied by NEB kit)
Incubate the sample reaction for 18 h at 16 °C.
Note: This prolonged reaction time is recommended to increase the ligation efficiency of methylated sRNAs.
Dilute the SR RT primer (supplied by NEB kit) 1:2 in nuclease-free water.
Add the following components to the 3’-adaptor/RNA ligation mix (Step F4):
4.5 µl nuclease-free water
1 µl diluted SR RT primer
Incubate the mixture for 5 min at 75 °C. Transfer the tube to 37 °C for 15 min, then to 25 °C for 15 min.
In the meantime, resuspend the lyophilized 5’ SR adaptor (supplied by NEB kit) in 120 µl of nuclease-free water. Store 20 µl aliquots of the adaptor solution at -80 °C.
Mix the required amount of 5’ SR adaptor (supplied by NEB kit) in a 1:1:1 ratio with nuclease-free water and 10 mM ATP in a 200 µl PCR tube. Store remaining adaptor solution at -80 °C.
Incubate the 5’ SR adaptor mix (Step F9) in a thermo cycler for 2 min at 70 °C, and place the mix on ice immediately. Use the denatured adaptor mix for the following ligation reaction within 30 min.
Add the following components to the reaction tube from Step F7:
1 µl 5’ SR adaptor mix (Step F10)
1 µl 10× 5’ ligation reaction buffer (supplied by NEB kit)
2.5 µl 5’ ligation enzyme mix (supplied by NEB kit)
Incubate reaction mix for 1 h at 25 °C in a heat block.
After this incubation step, add the following components to the reaction and mix well:
8 µl first strand synthesis reaction buffer (supplied by NEB kit)
1 µl murine RNase inhibitor (supplied by NEB kit)
1 µl ProtoScript II reverse transcriptase (supplied by NEB kit)
Incubate for 60 min at 50 °C. Stop RT reaction at 70 °C for 15 min.
Add the following components to the cDNA and mix well:
50 µl LongAmp Taq 2× master mix
2.5 µl SR primer for Illumina
2.5 µl Index primer (use a distinct Index primer for each treatment)
Note: As PCR performs better in small reaction volumes, we run 20 µl reaction volumes. After the PCR run was completed, samples were pooled before further procedure.
Amplify the library using the following PCR protocol:
step 1 94 °C 30 s
step 2 94 °C 15 s
step 3 62 °C 30 s
step 4 70 °C 15 s; back to step 2 (18-22×)
step 5 70 °C 5 min
step 6 End
Note: The expected size of a PCR product representing cloned 21 nucleotides sRNAs is ~140 base pairs. Nevertheless, the PCR product sometimes appears smeary at this point.
Purification of the sRNA library for Illumina sequencing
Add 0.1× volume 3 M sodium acetate in DEPC-treated water to the PCR reaction in a 1.5 ml LoBind® reaction tube followed by 2.5× volume of ethanol (96%) and 20 µg glycogen. After mixing the solution, incubate the sample for a minimum of 1 hour at -20 °C.
Prepare a 6% 0.5× TAE polyacrylamide gel.
Pellet the sRNA library DNA by centrifugation at 20,000 × g and 4 °C for 30 min. Remove the supernatant and add 500 µl of 80% ethanol diluted in DEPC-treated water.
Repeat pelleting the sRNA library DNA by centrifugation at 20,000 × g and 4 °C for 20 min, remove all liquid from the sample and air-dry the pellet until the ethanol completely evaporated.
Note: Optionally, the DNA pellet can be dried faster in an open-cap micro tube at 37 °C, however, avoid “over-drying” the DNA pellet.
Resuspend the DNA pellet in 25 µl DEPC-treated water. Incubate the sample for 5 min at 65 °C, if you encounter problems with bringing DNA pellet in solution.
Mix the eluted library DNA sample with 5 µl of 6× gel loading dye (supplied by NEB kit). Mix the 6× gel loading dye well prior usage.
Assemble a gel electrophoresis running system. Fill the electrophoresis tank with 0.5× TAE running buffer.
Load 5 µl of the Quick-load pBR322 DNA-MspI digest size marker (supplied by NEB kit) on the same polyacrylamide gel.
Use two gel slots per sample by loading 15 µl per well to avoid overloading of the gel lane.
Perform gel electrophoresis run with 15 ~V/cm for 60 min; after this running time, the bromophenol blue dye of the loading buffer typically reaches the bottom edge of the gel.
Dissemble the gel cast and stain the polyacrylamide gel with ethidium bromide or any other substitutive DNA dye (e.g., SYBR Gold) for 3 min. Under a UV documentation station, cut out the DNA band of the gel at the size of 140-150 base pairs. Avoid any DNA band at ~120 base pairs, as this size usually represents adaptor dimers.
Place the gel piece into a new 1.5 ml DNA LoBind® reaction tube. Use a blue pipette tip to crush the gel piece. Add 250 µl of 1× gel elution buffer (supplied by NEB kit).
Elute the sRNA library DNA from the crushed gel pieces by rotating the sample for 2 h at room temperature or overnight in a 4 °C cold room.
Spin the sample at 10,000 × g and room temperature for 10 min, and transfer the supernatant into a new 1.5 ml DNA LoBind® reaction tube. Try to collect all liquids from the reaction tube.
Repeat spinning the sample at 10,000 × g at room temperature for 10 min, and transfer the supernatant into a new 1.5 ml DNA LoBind® reaction tube. At this step, avoid transferring any remaining gel pieces.
Add 0.1× volume 3 M sodium acetate in DEPC-treated water, 2.5× volume of 96% ethanol, and 20 µg of glycogen. For DNA precipitation, mix the solution and incubate the sample for a minimum of 30 min at -80 °C.
Prepare a 6% 0.5× TAE polyacrylamide gel.
Pellet library DNA by centrifugation at 20,000 × g and 4 °C for 30 min. Discard the supernatant and add 500 µl of 80% ethanol diluted in DEPC-treated water.
Pellet the DNA by centrifugation at 20,000 × g and 4 °C for 20 min, remove all liquids and air-dry the DNA pellet until ethanol is completely evaporated.
Note: Optionally, the DNA pellet can be dried faster in an open-cap micro tube at 37 °C, however, avoid “over-drying” the DNA pellet.
Resuspend the DNA pellet in 25 µl DEPC-treated water. Incubate the DNA pellet for 5 min at 65 °C, if encounter problems with bringing the pellet in solution.
Repeat the gel clean-up of library DNA, following the Steps G6-G18.
Store purified library DNA in 80% ethanol at -20 °C, until sequencing.
Note: Storage of library DNA is valid for up to 12 months.
To proceed with sequencing, precipitate the library DNA by centrifugation at 20,000 × g and 4 °C for 20 min, remove all liquids and air-dry the pellet until ethanol is completely evaporated.
Note: In this last step, do not use DEPC-treated water for DNA pellet resuspension as it can interfere with Illumina sequencing; instead use nuclease free water (supplied by the NEB kit).
Resuspend the sample in nuclease free water for Illumina HiSeq run.
Note: For sRNA library sequencing, we used an Illumina HiSeq1500 platform in single-end mode with 50 base read length. To obtain sufficient read numbers of pathogen sRNAs collected from infected plant tissue, we recommend to sequence minimum at a depth of 50 million reads per library.
sRNA Illumina sequencing analysis
Note: An overview workflow of the bioinformatics part of this Bio-protocol is shown in Figure 4. sRNA sequencing data were analyzed using a Galaxy Server (Giardine et al., 2005). All tools of the analysis pipeline are also freely available as stand-alone versions, except the Illumina demultiplexing package and the Clip adapter script. As an adequate substitution of these steps, the Illumina bcl2fastqc tool and Trimmomatic can be used, respectively. In the following, a step-by-step description of the bioinformatics pipeline is given for the recovery and identification of H. arabidopsidis sRNA sequences bound to A. thaliana AGO1 during infection, including a target prediction of A. thaliana mRNAs using identified H. arabidopsidis AGO1-bound sRNA sequences. As indicated in Figure 4, a similar analysis can be run to predict plant mRNA targets of endogenous A. thaliana small RNAs.
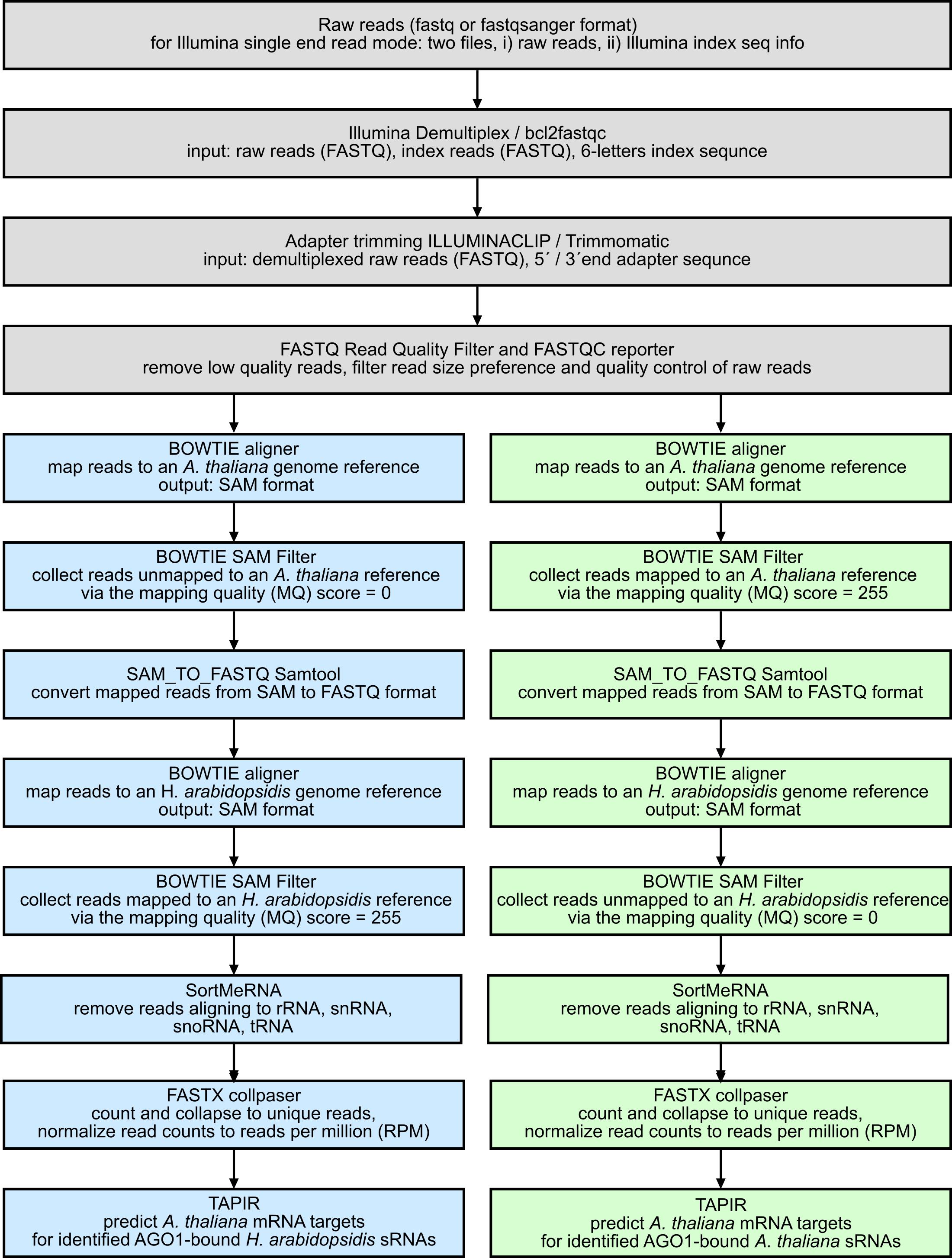
Figure 4. Bioinformatics workflow to analyze AGO1 co-IP sRNA NGS data. After standard processing of sRNA raw reads, sequences can be grouped into pathogen-derived sRNAs (H. arabidopsidis) and host plant-derived (A. thaliana) sRNAs for further analysis.If applicable; demultiplex sRNA libraries using the Illumina Demultiplex or the bcl2fastqc tool by giving the library sequence indices.
Remove 3’-end adapter sequences from sRNA reads using Clip adapter or the ILLUMINACLIP function of Trimmomatic (Bolger et al., 2014).
Remove low quality reads by setting a minimum Phred quality score of 30.0 and a size range from 19-30 nucleotides using the FASTQ Filter tool (Blankenberg et al., 2010). The FASTQ_filter command is also available as part of the USEARCH package (https://drive5.com/usearch/features.html).
Note: Optionally, useful information on sRNA sequencing quality can be found by consulting the FastQC reporter tool at any step of the bioinformatics analysis.
To remove A. thaliana sRNA reads from the dataset, align the raw reads to the A. thaliana reference genome (e.g., TAIR10 for ecotype Col-0) using the Bowtie aligner (Langmead et al., 2009). We recommend allowing one mismatch (-v 1) to map reads to the A. thaliana reference genome. By this step, a mapping quality score (MQ) is attributed to individual reads with a score of 255 (aligned) or 0 (unaligned) given in a SAM format output file.
Collect reads with MQ = 0, as these reads do not align to the A. thaliana reference genome using the Filter_tool (Galaxy Version 1.1.0).
Convert the SAM file (unaligned to A. thaliana) into a FASTQ file format using the SAM_to_FASTQ tool with SAMtools (Li et al., 2009).
Use the Bowtie aligner to align reads from Step H6 to an H. arabidopsidis Noks1 reference genome (PRJNA298674), allowing zero mismatch (-v 0).
Collect reads with MQ = 255, as these read aligned to the H. arabidopsidis reference genome, using the Filter_tool (Galaxy Version 1.1.0).
Note: We do not recommend to use the Bowtie2 short-read aligner, as this version does not allow binary mapping scores (aligned or unaligned), rather attributes low MQ values to reads with multiple alignment events. Many pathogen sRNAs that we found being loaded into plant AGOs are derived from repetitive DNA, thus such sequences could be easily lost through Bowtie2 quality aware mapping.
As a quality control for the successful A. thaliana AGO1-co-IP sRNA sequencing, display read counts aligning to the A. thaliana reference genome (go back to Step H5 and collect reads with MQ = 255), by read length and 5’-prime nucleotide distribution. A. thaliana AGO1 preferentially binds 21 nt sRNAs with 5’ prime uracil. This analysis can be run by the FastQC tool, as shown in Figure 5.
Optionally, remove reads from your dataset (Step H8) that align to ribosomal RNA (rRNA), transfer RNA (tRNA), or small nuclear/nucleolar RNA (snRNA, snoRNA) sequences using the SortMeRNA tool (Kopylova et al., 2012). In particular, rRNA-derived sRNA reads often occur in high abundance in sRNA library sequencing data, if no riboRNA-depletion was performed prior to RNA cloning.
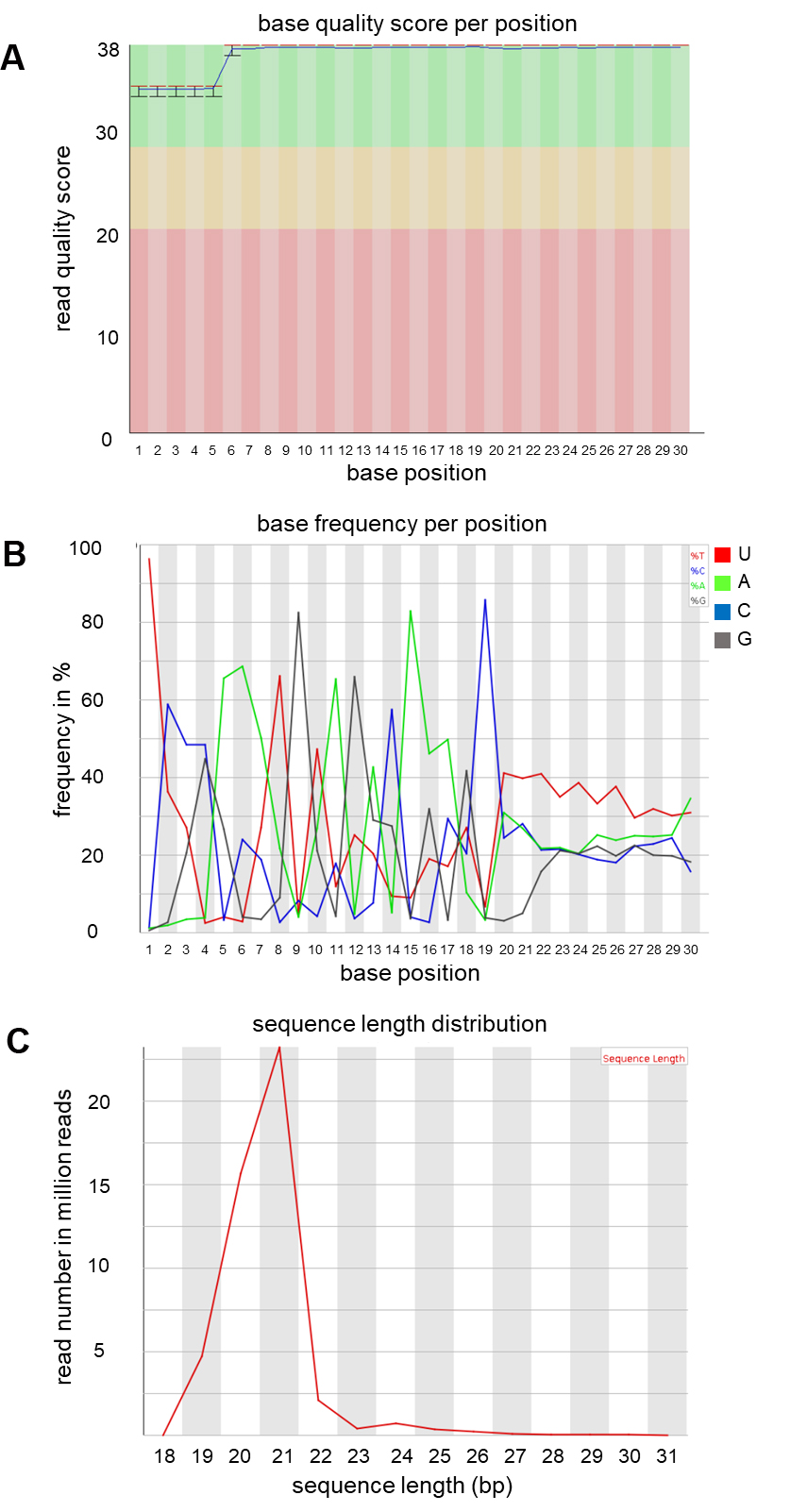
Figure 5. FastQC report of AGO1 co-IP sRNA NGS data. As plant AGOs are typically associated to distinct sRNA classes, AGO1 co-IP sRNA NGS data reflect some unique features that can be used as a quality control in such experiments. High quality (> 30 Phred score) sRNA reads of the size range of 19-30 bases were collected (A). AGO1-associated sRNAs showed preference to 5’ prime Uracil U – in sRNA sequencing data Thymine T (B), and to a size of 21 nucleotides (C). Graphs shown here represent an AGO1 co-IP sample collected from H. arabidopsidis-infected A. thaliana at 4 days post inoculation. The total read number of this sRNA library was 51,089,216.Count and unify sRNA reads to unique sequence tags using a collapse tool, e.g., the FASTX-toolkit (http://hannonlab.cshl.edu/fastx_toolkit/index.html).
Transform read numbers of unique sRNA sequences into reads per million (rpm) by normalizing to the total library read numbers (referring to the organism of interest, here H. arabidopsidis).
Note: Before starting mRNA target prediction, it can be useful to apply an abundancy filter of small RNA reads.
For predicting mRNA targets of candidate sRNAs, several target prediction tools were implemented. We have used the TAPIR tool (Bonnet et al., 2010) to predict A. thaliana mRNA targets of H. arabidopsidis sRNA candidates using an A. thaliana cDNA dataset. For target prediction, a maximal score of 4.5 penalizing mismatches in the mRNA/sRNA base pairing, and a maximal free energy ratio of 0.7 were set as thresholds.
Recipes
Buffers
5× Protein SDS loading buffer
225 mM Tris-HCl (pH 6.8)
5% (w/v) SDS
50% (v/v) glycerol
0.05% (w/v) bromophenol blue dye
455 mM DTT (added directly before use)
10× Protein SDS running buffer
30 g Tris ultrapure
144 g glycine
10 g SDS
Fill up to 1 L with ultrapure water and stir until salts are dissolved
The pH does not need to be adjusted
10× Protein transfer buffer
30 g Tris ultrapure
144 g glycine
Fill up to 900 ml with ultrapure water and stir until salts are dissolved
Adjust the pH to 8.3 with HCl
Fill up to 1 L with ultrapure water
10× PBS pH 7.4
80 g NaCl
2 g KCl
14.4 g Na2HPO4
2.4 g KH2PO4
Fill up to 900 ml with ultrapure water
Adjust the pH to 7.4 with HCl
Fill up to 1 l with ultrapure water
For PBST 0.1% and 0.2% add the respective amount of Tween 20
50× TAE buffer
242 g Tris ultrapure
18.6 g EDTA
Dissolve in 950 ml of water
Adjust the pH to 8.0 with glacial acetic acid
Fill up to 1 L with water
DEPC-treated water
Add 1 ml DEPC to 1 L of ultrapure water
Let it stir overnight
Autoclave for 20 min at 121 °C
IP extraction buffer
20 mM Tris-HCl (pH 7.5)
300 mM NaCl
5 mM MgCl2
0.5% (v/v) NP-40
5 mM DTT (add directly before use)
1 tablet protease inhibitor/50 ml sample volume (add directly before use)
5 µl RNase inhibitor (40 U)/50 ml sample volume (add directly before use)
Make up to 50 ml with DEPC-treated water
IP washing buffer
20 mM Tris-HCl (pH 7.5)
300 mM NaCl
5 mM MgCl2
0.5% (v/v) Triton X-100
5 mM DTT (add directly before use)
1 tablet protease inhibitor/50 ml sample volume (add directly before use),
Fill up to 50 ml with DEPC-treated water
RNA release buffer
100 mM Tris-HCl (prepare from 1 M Tris-HCl pH 7.5)
10 mM EDTA (prepare from 0.5 M EDTA pH 8.0)
300 mM NaCl
2% SDS
1 µg/µl Proteinase K (add directly before use).
Polyacrylamide gel recipes (for 10 ml gel volume)
Nucleic acid polyacrylamide gel recipes:
6% 0.5× TAE gel
7.8 ml H2O
2.0 ml 30% acrylamide/bis-acrylamide solution
0.1 ml 50× TAE buffer
0.1 ml 10% APS
0.008 ml TEMED
10% 0.5× TAE gel
6.5 ml H2O
3.3 ml 30% acrylamide/bis-acrylamide solution
0.1 ml 10% APS
0.1 ml 50× TAE buffer
0.004 ml TEMED
8% SDS resolution gel
4.6 ml H2O
2.7 ml 30% acrylamide/bis-acrylamide solution
2.5 ml 1.5 M Tris-HCl (pH 8.8)
0.1 ml 10% SDS
0.1 ml 10% APS
0.006 ml TEMED
SDS stacking gel
2.1 ml H2O
0.5 ml 30% acrylamide/bis-acrylamide solution
0.38 ml 1.0 M Tris-HCl (pH 6.8)
0.03 ml 10% SDS
0.03 ml 10% APS
0.003 ml TEMED
Acknowledgments
This protocol has been developed to identify H. arabidopsidis small RNAs that bind to the A. thaliana AGO/RISC during plant infection (Dunker et al., 2020). The authors wish to thank New England Biolabs® for their permission to include the library preparation kit protocol within this protocol. This work was supported by a grant from the Deutsche Forschungsgemeinschaft DFG (WE 5707/1-1) to AW. The funders had no role in the study design, data collection, interpretation, or the decision to publish.
Competing interests
The authors declare no competing interests.
References
- Asai, S., Shirasu, K. and Jones, J. D. (2015). Hyaloperonospora arabidopsidis (Downy Mildew) infection assay in Arabidopsis. Bio-protocol 5(20): e1627.
- Bailey, K., Cevik, V., Holton, N., Byrne-Richardson, J., Sohn, K. H., Coates, M., Woods-Tor, A., Aksoy, H. M., Hughes, L., Baxter, L., Jones, J. D., Beynon, J., Holub, E. B. and Tor, M. (2011). Molecular cloning of ATR5Emoy2 from Hyaloperonospora arabidopsidis, an avirulence determinant that triggers RPP5-mediated defense in Arabidopsis. Mol Plant Microbe Interact 24(7): 827-838.
- Blankenberg, D., Gordon, A., Von Kuster, G., Coraor, N., Taylor, J., Nekrutenko, A. and Galaxy, T. (2010). Manipulation of FASTQ data with Galaxy. Bioinformatics 26(14): 1783-1785.
- Bolger, A. M., Lohse, M. and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15): 2114-2120.
- Bonnet, E., He, Y., Billiau, K. and Van de Peer, Y. (2010). TAPIR, a web server for the prediction of plant microRNA targets, including target mimics. Bioinformatics 26(12): 1566-1568.
- Carbonell, A. (2017). Immunoprecipitation and high-throughput sequencing of ARGONAUTE-bound target RNAs from plants. Methods Mol Biol 1640: 93-112.
- Carbonell, A., Fahlgren, N., Garcia-Ruiz, H., Gilbert, K. B., Montgomery, T. A., Nguyen, T., Cuperus, J. T. and Carrington, J. C. (2012). Functional analysis of three Arabidopsis ARGONAUTES using slicer-defective mutants. Plant Cell 24(9): 3613-3629.
- Dunker, F., Trutzenberg, A., Rothenpieler, J. S., Kuhn, S., Pröls, R., Schreiber, T., Tissier, A., Kemen, A., Kemen, E., Huckelhoven, R. and Weiberg, A. (2020). Oomycete small RNAs bind to the plant RNA-induced silencing complex for virulence. Elife 9: e56096.
- Giardine, B., Riemer, C., Hardison, R. C., Burhans, R., Elnitski, L., Shah, P., Zhang, Y., Blankenberg, D., Albert, I., Taylor, J., Miller, W., Kent, W. J. and Nekrutenko, A. (2005). Galaxy: a platform for interactive large-scale genome analysis. Genome Res 15(10): 1451-1455.
- Kopylova, E., Noé, L. and Touzet, H. (2012). SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28(24): 3211-3217.
- Langmead, B., Trapnell, C., Pop, M. and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology 10(3): R25.
- Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16): 2078-2079.
- Mi, S., Cai, T., Hu, Y., Chen, Y., Hodges, E., Ni, F., Wu, L., Li, S., Zhou, H., Long, C., Chen, S., Hannon, G. J. and Qi, Y. (2008). Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5' terminal nucleotide. Cell 133(1): 116-127.
- Montgomery, T. A., Howell, M. D., Cuperus, J. T., Li, D., Hansen, J. E., Alexander, A. L., Chapman, E. J., Fahlgren, N., Allen, E. and Carrington, J. C. (2008). Specificity of ARGONAUTE7-miR390 interaction and dual functionality in TAS3 trans-acting siRNA formation. Cell 133(1): 128-141.
- Qi, Y. and Mi, S. (2010). Purification of Arabidopsis argonaute complexes and associated small RNAs. Methods Mol Biol 592: 243-254.
- Varkonyi-Gasic, E., Wu, R., Wood, M., Walton, E. F. and Hellens, R. P. (2007). Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods 3: 12.
- Weiberg, A., Bellinger, M. and Jin, H. (2015). Conversations between kingdoms: small RNAs. Curr Opin Biotechnol 32: 207-215.
- Zeng, J., Gupta, V. K., Jiang, Y., Yang, B., Gong, L. and Zhu, H. (2019). Cross-kingdom small RNAs among animals, plants and microbes. Cells 8(4): 371.
- Zhang, X., Zhao, H., Gao, S., Wang, W. C., Katiyar-Agarwal, S., Huang, H. D., Raikhel, N. and Jin, H. (2011). Arabidopsis Argonaute 2 regulates innate immunity via miRNA393*-mediated silencing of a Golgi-localized SNARE gene, MEMB12. Mol Cell 42(3): 356-366.
- Zhao, H., Lii, Y., Zhu, P. and Jin, H. (2012). Isolation and profiling of protein-associated small RNAs. Methods Mol Biol 883: 165-176.
Article Information
Copyright
![]() Dunker et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Dunker et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Dunker, F., Lederer, B. and Weiberg, A. (2021). Plant ARGONAUTE Protein Immunopurification for Pathogen Cross Kingdom Small RNA Analysis. Bio-protocol 11(3): e3911. DOI: 10.21769/BioProtoc.3911.
- Dunker, F., Trutzenberg, A., Rothenpieler, J. S., Kuhn, S., Pröls, R., Schreiber, T., Tissier, A., Kemen, A., Kemen, E., Huckelhoven, R. and Weiberg, A. (2020). Oomycete small RNAs bind to the plant RNA-induced silencing complex for virulence. Elife 9: e56096.
Category
Plant Science > Plant molecular biology > RNA > RNA-protein interaction
Microbiology > Microbe-host interactions > Protista
Biochemistry > RNA > RNA-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.