- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A mant-GDP Dissociation Assay to Compare the Guanine Nucleotide Binding Preference of Small GTPases

(*contributed equally to this work) Published: Vol 11, Iss 2, Jan 20, 2021 DOI: 10.21769/BioProtoc.3886 Views: 5226
Reviewed by: Ching Yao YangPiero CrespoAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Dec 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Small GTPases are cellular switches that are switched on when bound to GTP and switched off when bound to GDP. Different small GTPase proteins or those with mutations may bind to GTP or GDP with different relative affinities. However, small GTPases generally have very high affinities for guanine nucleotides, rendering it difficult to compare the relative binding affinities for GTP and GDP. Here we developed a method for comparing the relative binding strength of a protein to GTP and GDP using a mant-GDP dissociation assay, whereby the abilities of GTP and GDP to induce the dissociation of bound mant-GDP are compared. This equilibrium type assay is simple, economic, and much faster than obtaining each protein’s affinity for GDP and GTP. The GDP/GTP preference value obtained is useful for comparing the relative GTP/GDP binding preferences of different GTPases or different mutants, even though it is not the real GDP/GTP affinity ratio (but rather an estimation of the ratio).
Keywords: GTPaseBackground
Small GTPases (also known as small G proteins) are a group of functionally important macromolecules that can bind to GTP and hydrolyze it into GDP owing to their intrinsic hydrolase activity. In human, more than a hundred different small GTPases are grouped into five families, namely Ras, Rho, Ran, Rab, and Arf, based on their sequence, structure, and function (Wennerberg et al., 2005).
GTPase proteins are cellular switches that are active when bound to GTP and inactive when bound to GDP (Cherfils and Zeghouf, 2013). Generally, small GTPases have very high affinities towards guanine nucleotides (GDP and GTP) (Bos et al., 2007). To switch on/off precisely, two groups of proteins are often required, namely, the GTPase activating proteins (GAPs), which activate the hydrolase activity (to switch off the GTPase), and guanine nucleotide exchange factors (GEFs), which lower nucleotide affinity to allow exchange of nucleotides (usually switching the GTPase on).
The affinity towards GTP or GDP can vary between wild type and mutant proteins (John et al., 1993; Klebe et al., 1995). Because the affinities for GTP and GDP are high, in the pico- to nano- molar range, they are usually hard to measure directly, and are instead measured separately by determining the association rate constant kon and the dissociation rate constant koff (Goody et al., 1992; Ahmadian et al., 2002). One way to measure koff is to first charge a protein with fluorescently labeled nucleotides, then incubate the protein in an excess of free nucleotide to measure the dissociation rate of fluorescently labeled nucleotides (John et al., 1993; Klebe et al., 1993 and 1995). Usually a stop-flow device is needed to measure kon, whereby different concentrations of apo protein (lacking GDP or GTP) are rapidly mixed with fluorescently labeled nucleotides (John et al., 1990; Ahmadian et al., 2002). For the Ras-related nuclear protein Ran, kon is not measureable because of the difficulty in generating apo Ran (Klebe et al., 1995).
Mant (N-methylanthraniloyl)-GDP and mant-GTP are the most widely used fluorescently labeled nucleotides because they have the following two characteristics (Hiratsuka, 1983; Ahmadian et al., 2002). First, modification of the mant group usually does not alter the interaction of GDP/GTP with the protein (John et al., 1990). Second, mant fluorescence increases by about two-fold after binding to proteins, and the fluorescence of bound mant is often sensitive to environmental changes such as binding of other proteins (Alexandrov et al., 2001; Kanie and Jackson, 2018). In addition to fluorescent labeling methods, filter binding (radioactive labeling) and intrinsic tryptophan fluorescent changes can also be used to measure association/dissociation rates; however, their usage is more limited (Feuerstein et al., 1987; Klebe et al., 1993; Kanie and Jackson, 2018).
Measuring kon and koff separately can be time consuming and laborious. Generating mant-nucleotide-labeled proteins or using an excess of mant-nucleotides to dissociate the bound nucleotide often requires a large amount of mant-nucleotides, which are expensive. Here we present a quick, simple, and economic mant-GDP dissociation assay to estimate the relative GTP/GDP binding affinities or the ‘GTP preference’ of small GTPases. We tested this assay using the small GTPase Ran and one of its mutants RanP180L, which was once reported in the cancer database cBioPortal (https://www.cbioportal.org/). P180 is located at the C-terminal autoinhibitory tail of Ran, and mutation of this residue might affect GTP/GDP preference. The method we developed for estimating ‘GTP preference’ is reproducible and yields results that agree well with those in the literature, as illustrated by the examples provided.
Materials and Reagents
Micro-centrifuge tube (Biosharp, Life sciences, catalog number: BS-15-M )
50 ml centrifuge tube (Biofil®, catalog number: CFT011500 )
15 ml centrifuge tube (Biofil®, catalog number: CFT011150 )
Petri dish (Biofil®, 90 mm Petri dish)
Centrifugal filter (Vivaspin® Turbo 15, 10,000 MWCO PES )
Chromatography column (Sangon Biotech®, catalog number: F506609-0001 )
HiTrap Q HP 5ml(GE Healthcare,catalog number: 17115401 )
Aluminum foil (METRO)
96-well microplate (Sangon Biotech, catalog number: F605033-0001 )
Multichannel pipette tip (Biofil®)
Oak ridge PPCO centrifuge tube (Thermo ScientificTM NalgeneTM, catalog number: 3119-0010PK )
BL21 (Genotype: F-, ompT, hsds (Rbb-Mb-), gal, dcm (DE3))
Plasmids encoding recombinant GTPase with 6x his-tag (Synthesized from TsingKe Biological Technology)
2-mercaptoethanol (BME) (Amresco®, catalog number: 0482-250ML )
Agar (Sangon Biotech, catalog number: A505255-0250 )
Guanosine 5'-triphosphate trisodium salt (GTP·Na3) (Sangon Biotech, catalog number: A620250-0100 )
Guanosine 5'-diphosphate disodium salt (GDP·Na2) (Sangon Biotech, catalog number: A600492-0100 )
Classical laemmli sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel preparation kit (Sangon Biotech, catalog number: C631100-0200 )
Unstained protein marker (Sangon Biotech, catalog number: C600524-0020 )
Coomassie Brilliant Blue R-250 (Sangon Biotech, catalog number: A100472-0025 )
Ni-NTA Sefinose (TM) Resin (Sangon Biotech, catalog number: C600033-0100 )
Milli-Q water
Phenylmethylsulfonyl fluoride (PMSF) (Sangon Biotech, catalog number: A610425-0025 )
Glycerol (Chengdu Kelong Chemical Reagent Factory)
Glycine (Sangon Biotech, catalog number: A100167-0001 )
Mant-GDP triethylammonium salt solution (Jena Bioscience, catalog number: NU-204S ) (chemical structure was shown in Figure S1)
Tris (Sangon Biotech, catalog number: A100193-0005 )
SDS (Beyotime, catalog number: ST626 )
Concentrated HCl (Chengdu Kelong Chemical Reagent Factory)
Glacial acetic acid (Chengdu Kelong Chemical Reagent Factory)
Ethanol (Chengdu Kelong Chemical Reagent Factory)
HEPES (Sigma-Aldrich, catalog number: V900477-500G )
Sodium hydroxide (Chengdu Kelong Chemical Reagent Factory)
Sodium chloride (NaCl) (Sangon Biotech, catalog number: A610476-0001 )
Tryptone (Sangon Biotech, catalog number: A110859-0500 )
Yeast extract (OXOID, catalog number: LP0021 )
Liquid nitrogen
Magnesium chloride hexahydrate (MgCl2·6H2O)
Ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA) (Chengdu Kelong Chemical Reagent Factory)
IPTG (Sangon Biotech, catalog number: A100487-0100 )
Isopropanol (Chengdu Kelong Chemical Reagent Factory)
Ampicillin (Sangon Biotech, catalog number: A610028-0025 )
LB broth (see Recipes)
LB agar plate containing 100 μg/ml ampicillin (see Recipes)
100 mg/ml ampicillin (see Recipes)
1 M IPTG (see Recipes)
1 M Tris-HCl (pH 7.5) (see Recipes)
1 M HEPES-NaOH (pH 7.5) (see Recipes)
5 M NaCl (see Recipes)
100x PMSF (see Recipes)
50% glycerol (see Recipes)
1 M MgCl2 (see Recipes)
2 M imidazole-HCl (pH 7.5) (see Recipes)
0.5 M EDTA (pH 8) (see Recipes)
10 M NaOH (see Recipes)
10x running buffer (see Recipes)
5x loading buffer (see Recipes)
Coomassie Brilliant Blue staining solution (see Recipes)
Coomassie Brilliant Blue destaining solution (see Recipes)
Lysis buffer (see Recipes)
Wash buffer (see Recipes)
Elution buffer (see Recipes)
Stock buffer (see Recipes)
Mant-GDP exchange buffer (see Recipes)
Equipment
Milli-Q water (Elix® Advantage Water Purification System)
250 ml culture flask (Chengdu Shuniu, catalog number: sjsp24624 )
2 L culture flask (Chengdu Shuniu, catalog number: sjsp4637 )
500 ml beaker (Chengdu Shuniu, catalog number: sb56448 )
Incubator shaker (Shanghai Zhichu Instrument Co., Ltd, model: ZQZY-80CH )
Low-speed and high-capacity refrigerated centrifuge (Wuxi Ruijiang Analysis Instrument Co., LTD, model: RJ-LD-5000C )
Refrigerated centrifuge (SCILOGEX, model: SNJK-D3024R )
1 L centrifuge bottle (Thermo ScientificTM NalgeneTM PPCO, catalog number: DS3132-0063 )
Benchtop pH meter (Ohaus®, model: STARTER 2100 )
AKTA Pure Chromatography System (GE Healthcare, model: AKTA Pure )
-80 °C freezer (Haier, model: DW-86L626 )
Refrigerated centrifuge (Beckman Coulter, model: Avanti J-26S XP , catalog number: B14536)
Fluorescent microplate reader (BioTek, model: SynergyTM H1 )
Sonifier (BioSafer, model: 650-92 )
360° vertical rotator (SCILOGEX, model: MX-RL-E )
Lab stand (Chengdu Shuniu, catalog number: a13104 )
Electrophoresis tank (Bio-Rad Mini-PROTEAN® Tetra, model: 1658000 )
4 °C and -20 °C refrigerator (Haier, model: BCD-571WDPF )
Vortex mixer (Crystal Technology & Industries, Inc., ScilentShake®, model: VM-01U, serial number: 3222046)
Multichannel electronic pipette (Dragonmed toppette)
Autoclave (Zealway, model: GR85DA )
Mental bath (SCILOGEX, model: HB120-S )
NanoPhotometer (IMPLEN, model: IMN50/N60 )
SuperdexTM 200 Increase 10/300 GL Prepacked TricornTM Columns (GE Healthcare)
Software
Microsoft Excel (Microsoft)
GraphPad Prism 7 software (GraphPad Software)
Procedure
Expression and purification of recombinant Ran in Escherichia coli
Recombinant Ran and its mutant RanP180L were separately cloned into a pET-15b expression vector, incorporating an N-terminal 6x his-tag fusion and a thrombin digestion site. The vector map including the inserted gene is shown in Figure S2. The following purification protocol for Ran and RanP180L is modified from a previous report (Bibak et al., 2004). For the purification of Ras and RCC1, please refer to these studies (Pu and Dasso, 1997; Burd et al., 2014).
Transform the vector encoding 6x his-Ran (or 6x his-RanP180L or the 6x his-tagged small GTPase of interest) into E. coli BL21 (DE3) cells. Amplify the bacteria in liquid broth medium in 2.5 L culture flask. Induce protein expression by adding 200 μl 1 M IPTG (Isopropyl β-d-1-thiogalactopyranoside), and shake the flask at 37 °C at 220 rpm for 3 h.
Place the flask on ice before harvesting the cells. Transfer the cells into a 1 L centrifuge bottle. Harvest bacteria by centrifuging at 4,700 × g at 4 °C for 15 min.
Discard the supernatant and resuspend the cell pellet with 30 ml of pre-chilled lysis buffer. Transfer the resuspended cells into a 50 ml centrifuge tube.
Sonicate the cells in an ice-water bath for 10 min (4 s on/6 s off at 50% amplitude). Centrifuge at 39,000 × g at 4 °C in a Beckman refrigerated centrifuge for 30 min.
Fill a chromatography column with 3 ml bed volume of Ni-NTA resin. Equilibrate the column with 10 ml of wash buffer. Repeat the equilibration three times. Incubate the clarified lysate with the resin in the chromatography column at 4 °C with rotation for 15 min.
Wash the resin with 10 ml of wash buffer 7-10 times.
Elute the protein with 7 ml elution buffer. Repeat three times.
Concentrate the eluate at 3,200 × g at 4 °C in a refrigerated centrifuge using a centrifugal filter. Transfer the concentrated eluate into a 1.5 ml micro-centrifuge tube and centrifuge at 21,000 × g for 5 min. Discard the pellet.
Load the supernatant (1 ml) onto a size exclusion chromatography column (Superdex 200 Increase) to exchange the buffer to stock buffer. Collect the peak fractions (usually from 15.5 ml to 17.5 ml) at a volume of 0.5 ml/tube. Run an SDS-PAGE gel with 5 μl of the peak fraction sample mixed with 2 μl of loading buffer to assess purity of the protein. An SDS-PAGE gel that shows the final pure proteins is shown in Figure S3.
Concentrate the protein by centrifugation in a centrifugal filter at 3,200 × g at 4 °C until the final protein concentration is 5 to 10 mg/ml when measured with a NanoDrop spectrophotometer. Aliquot 1 mg of protein per tube. Freeze the tubes in liquid nitrogen and store the protein in a -80 °C freezer.
Pilot experiment to test mant-GDP loading efficiency (optional)
Mant-GDP loading efficiency in this assay can vary depending on the type of GTPase or the mutation. This step may be skipped if the GTPase is known to have high GDP binding preference.
Thaw 1 mg of purified Ran. It does not matter if the protein is charged with GDP (or GTP) or not.
Incubate 3 nmol of Ran, 5 μg of RCC1, and 3 nmol of mant-GDP in a total volume of 800 μl in a 1.5 ml centrifuge tube for 20 min at RT (room temperature). The RanGEF RCC1 helps to accelerate nucleotide exchange and shorten the time requried to reach equilibrium.
Add 20 μl of exchange buffer, exchange buffer plus 25 nmol GTP, and exchange buffer plus 25 nmol GDP, into three separate wells in a 96-well plate (i.e., three wells per buffer for a total of nine wells).
Aliquot the incubated Ran mixture from step 2 into the nine wells prepared in step 3 (80 μl/well). Incubate the plate at RT for 30 min to reach equilibrium.
Read the fluorescent signal using a fluorescence microplate reader at Ex/Em = 360 nm/440 nm.
Analyze the data. To proceed, there must be a difference between the buffer and GTP sample, or between the buffer and GDP sample, preferably more than 20 times higher than the standard deviation.
Note: A low difference relative to standard deviation indicates that the protein has relatively low affinity for GDP. In this case, mant-GTP instead of mant-GDP should be used to test the loading efficiency before proceeding to the dissociation assay. One may also pre-charge the protein with mant-GDP according to a pervious protocol (Kanie et al., 2018), preferably with more than 50% of the protein being charged with mant-GDP, before proceeding to the following steps.
Mant-GDP dissociation assay
To estimate the relative GTP/GDP binding affinities or the ‘GTP preference’, we devised the following mant-GDP dissociation assay, whereby wild-type Ran (RanWT) or RanP180L were first charged with mant-GDP, then the bound-mant-GDP was dissociated by adding increasing concentrations of free GTP or GDP. Two competitor-dissociation response curves can be plotted, from which the IC50 values, termed IC50GDP and IC50GTP can be determined. BiasGTP, which is defined as IC50GDP divided by IC50GTP, is then calculated as a unified measure of the GTP preference for a small GTPase that describes how biased the GTPase is toward GTP.
Thaw 1.0 mg of Ran, 0.1 mg of RCC1, 100 mM GTP, and 100 mM GDP on ice.
Note: GTP should be checked for possible GDP contamination, especially after long-term storage. GTP-containing buffer should be kept on ice or stored at -80 °C. Purify using an anion exchange Q column if needed. GTP and GDP stocks may be prepared in exchange buffer at 100 mM concentrations.
Dilute GDP or GTP to 1.25 mM using the exchange buffer. Make eight 2.5-fold serial dilutions of 1.25 mM GTP or GDP with the exchange buffer. The lowest concentration should be ~ 0.8 μM.
Note: When generating BiasGTP for only one protein, the experiment should be repeated several times with different serial dilutions (not prepared at the same time), so that the reported BiasGTP value does not reflect dilution errors. At least 60 μl of solution (for three repeats) for each GDP or GTP concentration is required. When comparing a few proteins, a serial dilution stock should be prepared and used for all proteins (even if there are dilution errors, the errors will affect all BiasGTP values to the same extent). We recommend preparing 1 ml for each concentration for comparing five proteins.
Mix 264 μg of Ran, 10 μg of RCC1, and 10 nmol of mant-GDP with mant-GDP exchange buffer to a total volume of 4 ml. Incubate for 20 min at RT.
Notes:
Mant-GTP should be used for GTP-preferring small GTPases. If mant-GDP or mant-GTP is chosen correctly, incubating an equal molar ratio of protein and mant would in theory result in at least 50% mant-labeled proteins. Our unpublished data showed that the ratio of GDP-, GTP- or mant-GDP/GTP-loaded proteins is not important, as long as the labeling efficiency is well above the level of noise (see Procedure B). A higher concentration of mant-GDP or mant-GTP does increase the labeling level and the observed signal; however, the background noise would be even greater (mostly due to pipetting errors), and thus may decrease the signal-to-noise ratio. Due to an unknown reason, desalting the sample to reduce background noise sometimes fails to improve the signal-to-noise ratio. Hence we do not recommend an extra desalting step in this assay.
The minimum volume of the protein mix for one protein is 3.84 ml (14 concentrations × 3 repeats, and 6 buffer samples, 80 µl each).
Add 100 μl exchange buffer to all the peripheral unused wells of a 96-well plate (as indicated in Figure 1A) to avoid edge effects.
Note: Edge wells tend to have slightly smaller readings. Using only the center wells can prevent these edge effects, especially when one wishes to increase the assay time.
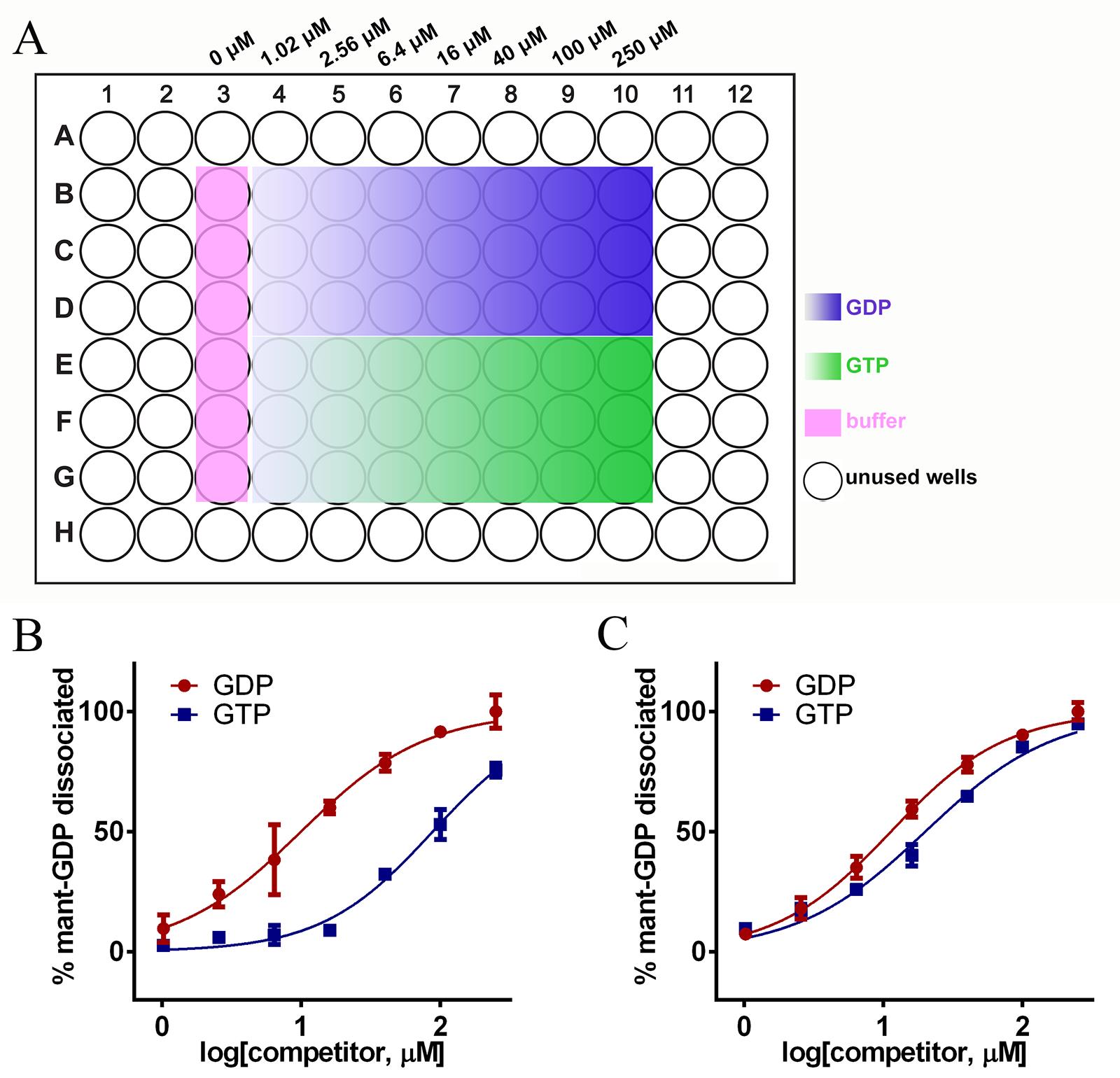
Figure 1. Mant-GDP dissociation profiles of Ran and a Ran mutant RanP180L. A. Typical layout of a dissociation plate. Unused wells are each filled with 100 µl of exchange buffer. The buffer samples (pink) contain proteins but not GDP or GTP. The final concentrations of GDP or GTP are indicated at the top. B. RanWT dissociation from GDP (red) and GTP (blue). Error bars represent standard deviations of triplicates. IC50 values are shown in Table 1. C. RanP180L dissociation profile. P180 is located at the C-terminal autoinhibitory tail of Ran.Add 20 μl of different concentrations of GTP or GDP or buffer to 48 wells in the middle (as indicated in Figure 1A). Each concentration is tested in triplicate. There are six buffer samples (Figure 1A, colored in pink).
Note: The assay may be conducted with quadruplicate readings for better precision.
Aliquot 80 μl/well of the protein mixture into the 48 wells to make a total volume of 100 μl (final concentrations: 2 μM Ran-nucleotide, 0.04 μM RCC1, and seven concentrations of GTP or GDP ranging from 250 μM to 1.0 μM).
Shake the plate gently three times for 30 s in a fluorescence microplate reader. Incubate the plate at RT for 30 min.
Read the fluorescent signal using the fluorescence microplate reader at Ex/Em = 360 nm/440 nm. Shake the plate and repeat the reading. Do this three to five times. An example of raw data is shown in the Supplemental Excel file.
Data analysis
Export the raw data to a Microsoft Excel spreadsheet.
Generate the standard deviations and averages for the replicate readings. Check for data inconsistency. Use the average for the following calculations.
Calculate standard deviations and averages for each concentration of GDP or GTP, which is performed in triplicate. Check for any obvious outliers. Please refer to the Supplemental Excel file for calculations.
The background (smallest readings) should be the highest GDP concentration readings if the protein shows a higher preference for GDP, and vice versa. Subtract the average of the background, which is calculated in step 3, from each of the average values from step 2.
Normalize the data from step 4 by dividing each of the subtracted values by the average of the subtracted exchange buffer sample readings, in which no mant-GDP is dissociated.
Convert the data to the percentage of protein dissociated from mant-GDP by subtracting each normalized value from 100%.
Note: The y-axis of unconverted data may be labeled as ‘mant-GDP bound %’. However, this is potentially ambiguous/misleading as the initial mant-GDP bound % would always be 100% in the figure, but the initial sample always contains some portion of bound GDP or GTP. After data conversion, labeling the y-axis with ‘mant-GDP dissociated %’ no longer causes problems for interpretation. The conversion does not change the BiasGTP values calculated.
Fit the converted data using four-parameter logistic regression in Graphpad software. Remember to use the log scale of concentration values for data plotting. Display standard deviations for each data point. An example of plotted data is shown in Figures 1B and 1C.
Record the IC50 values and confidence intervals for GTP and GDP. Calculate the BiasGTP values (Table 1) by dividing IC50GDP by IC50GTP.
Note: It should be noted that the IC50GTP or IC50GDP values are not directly comparable between proteins since the initial mant-GDP charging levels are variable. For one protein, GDP and GTP at their IC50 concentrations are equally effective at dissociating 50% of the bound mant-GDP under the same conditions. Thus, each IC50GDP/IC50GTP ratio (referred to as BiasGTP) represents a protein’s preference for GTP compared with GDP. A BiasGTP ratio of one means that the protein is not biased toward GDP or GTP. The larger the BiasGTP number, the more biased the protein is toward GTP. It should also be noted that the BiasGTP value may not be the real ratio of GTP/GDP affinity, but rather an approximation of the real ratio. BiasGTP values may not be used to calculate the real affinity ratio because the starting ratio of GDP/GTP/mant-GDP charged proteins is unknown. As mentioned earlier, it might be hard to obtain the real affinity ratio. BiasGTP values are much easier and faster to acquire, yet are reproducible and appropriate when used to compare GTPases or their mutants. The obtained Ran BiasGTP values are generally consistent with previously reported GDP/GTP affinity ratios (Table 1) (Klebe et al., 1995).
Table 1. GDP/GTP binding preference (BiasGTP) by different Ran mutants. A larger value of BiasGTP indicates that the protein is more preferred to bind GTP. Ran mutant P180L is more biased to bind GTP compared to Ran WT.
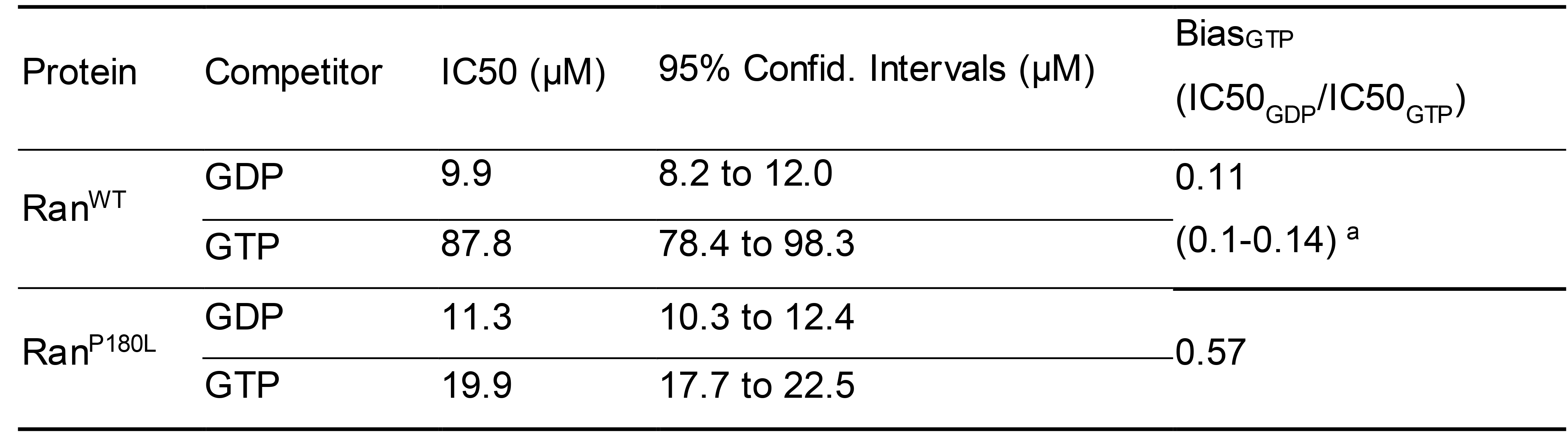
a This is the estimated value by Klebe et al. (1995).
Recipes
LB broth (1 L)
Dissolve 10 g NaCl, 10 g tryptone and 5 g yeast extract in 900 ml of Milli-Q water and bring volume with Milli-Q water to 1 L
Sterilize by autoclave and store at RT
LB agar plate containing 100 μg/ml ampicillin (1 L)
Dissolve 10 g NaCl, 10 g tryptone, 10 g Agar and 5 g yeast extract in 900 ml of Milli-Q water and bring volume with Milli-Q water to 1 L
Sterilize by autoclave and cool down to 50 °C
Add 1 ml of 100 mg/ml ampicillin and mix well
Pour into plates and store at 4 °C
100 mg/ml ampicillin (10 ml)
Dissolve 1 g of ampicillin in 8 ml of Milli-Q water and bring volume with Milli-Q water to 10 ml
Sterilize with a 0.22 μm syringe filter
Dispense 1 ml liquid into 1.5 ml centrifuge tube and store at -20 °C
1 M IPTG (10 ml)
Dissolve 2.383 g of IPTG in 8 ml of Milli-Q water and bring volume with Milli-Q water to 10 ml
Sterilize with a 0.22 μm syringe filter
Dispense 1 ml liquid into 1.5 ml centrifuge tube and store at -20 °C
1 M Tris-HCl (pH 7.5) (1 L)
Dissolve 121.14 g of Tris in 800 ml Milli-Q water
Adjust pH with concentrated HCl to 7.5 and bring volume with Milli-Q water to 1 L
Filter to sterilize
1 M HEPES-NaOH (pH 7.5) (1 L)
Dissolve 238.30 g of HEPES base in 800 ml of Milli-Q water
Adjust pH to 7.5 with 10 M NaOH and bring volume with Milli-Q water to 1 L
Filter sterilize through a 0.2 μm disposable bottle top filter
Store at 4 °C
5 M NaCl (1 L)
Dissolve 292.2 g of NaCl in 700 ml of Milli-Q water and bring volume with Milli-Q water to 1 L
Sterilize by filter and store at room temperature
50% glycerol (1 L)
Mix 500 ml glycerol and 500 ml Milli-Q water
2 M imidazole (pH 7.5) (50 ml)
Dissolve 6.808 g imidazole in 40 ml of Milli-Q water
Adjust pH to 7.5 and bring volume with Milli-Q water to 50 ml
Sterilize by filter and store at room temperature
1 M MgCl2 (100 ml)
Dissolve 20.33 g MgCl2·6H2O in 80 ml of Milli-Q water and bring volume with Milli-Q water to 100 ml
Sterilize by filter and store at room temperature
0.5 M EDTA (pH 8) (50 ml)
Dissolve 9.306 g of EDTA·2H2O in 40 ml of Milli-Q water
Adjust pH to 8.0 with NaOH, and bring volume with Milli-Q water to 50 ml
Sterilize by filter and store at room temperature
10 M NaOH (50ml)
Slowly add 20 g of sodium hydroxide into 40 ml of Milli-Q water while stirring continuously
Bring volume with Milli-Q water to 50 ml
100x PMSF (50 ml)
Dissolve 0.87 g into 40 ml isopropanol, and bring volume with isopropanol to 50 ml
Dispense 1 ml liquid into 1.5 ml centrifuge tube and store at -20 °C
10x running buffer (1 L)
Dissolve 30.2 g of Tris base, 144.0 g of glycine and 10.0 g of SDS in 1 L of Milli-Q water
The pH of the buffer should be 8.3 and no pH adjustment is required
Store at RT and dilute to 1x before use
5x loading buffer (10 ml)
Dissolve 25 mg Bromothymol Blue, 1 g SDS, 2 ml 2 M Tris-HCl pH 6.8, 5 ml glycerol and 0.5 ml BME and bring volume with Milli-Q water to 10 ml
Dispense 1 ml liquid into 1.5 ml centrifuge tube and store at -20 °C
Coomassie Brilliant Blue staining solution (1 L)
In a 1 L glass bottle, add 1 g of Coomassie Brilliant Blue R250, 400 ml ethanol and mix to dissolve
Add 100 ml of glacial acetic acid, 500 ml of Milli-Q water, and mix well
Store at RT
Coomassie Brilliant Blue destaining solution (1 L)
Mix 300 ml ethanol, 100 ml acetic acid and 600 ml Milli-Q water
Store at RT
Lysis buffer (50 ml)
Volume Final concentration
1 M Tris-HCl (pH 7.5) 2.5 ml 50 mM
5 M NaCl 4 ml 400 mM
1 M MgCl2 100 μl 2 mM
50% glycerol 10 ml 10% (v/v)
2M imidazole (pH 7.5) 250 μl 10 mM
100x PMSF 500 μl 1x
Milli-Q water 32.65 ml
Wash buffer (500 ml)
Volume Final concentration
5 M NaCl 40 ml 400 mM
1 M MgCl2 1 ml 2 mM
50% glycerol 100 ml 10% (v/v)
2 M imidazole (pH 7.5) 2.5 ml 10 mM
Milli-Q water 356.5 ml
Elution buffer (50 ml)
Volume Final concentration
5 M NaCl 3 ml 300 mM
1 M MgCl2 100 μl 2 mM
50% glycerol 10 ml 10%
2M imidazole (pH 7.5) 7.5 ml 300 mM
Milli-Q water 29.4 ml
Stock buffer (100 ml)
Volume Final concentration
1 M Tris-HCl (pH 7.5) 2 ml 20 mM
5 M NaCl 2 ml 100 mM
1 M MgCl2 200 μl 2 mM
50% glycerol 20 ml 10%
Milli-Q water 75.8 ml
Mant-GDP exchange buffer (50 ml)
Volume Final concentration
1 M HEPES-NaOH (pH 7.5) 2 ml 40 mM
5 M NaCl 2 ml 200 mM
1 M MgCl2 250 μl 5 mM
Milli-Q water 45.75 ml
References
- Ahmadian, M. R., Wittinghofer, A. and Herrmann, C. (2002). Fluorescence methods in the study of small GTP-binding proteins. Methods Mol Biol 189: 45-63.
- Alexandrov, K., Scheidig, A. J. and Goody, R. S. (2001). Fluorescence methods for monitoring interactions of Rab proteins with nucleotides, Rab escort protein, and geranylgeranyltransferase. Methods Enzymol 329: 14-31.
- Bibak, N., Paul, R. M., Freymann, D. M. and Yaseen, N. R. (2004). Purification of RanGDP, RanGTP, and RanGMPPNP by ion exchange chromatography. Anal Biochem 333(1): 57-64.
- Bos, J. L., Rehmann, H. and Wittinghofer, A. (2007). GEFs and GAPs: critical elements in the control of small G proteins. Cell 129(5): 865-877.
- Burd, C. E., Liu, W., Huynh, M. V., Waqas, M. A., Gillahan, J. E., Clark, K. S., Fu, K., Martin, B. L., Jeck, W. R., Souroullas, G. P., Darr, D. B., Zedek, D. C., Miley, M. J., Baguley, B. C., Campbell, S. L. and Sharpless, N. E. (2014). Mutation-specific RAS oncogenicity explains NRAS codon 61 selection in melanoma. Cancer Discov 4(12): 1418-1429.
- Cherfils, J. and Zeghouf, M. (2013). Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev 93(1): 269-309.
- Feuerstein, J., Goody, R. S. and Wittinghofer, A. (1987). Preparation and characterization of nucleotide-free and metal ion-free p21"apoprotein. J Biol Chem 262(18): 8455-8458.
- Goody, R. S., Pai, E. F., Schlichting, I., Rensland, H., Scheidig, A., Franken, S. and Wittinghofer, A. (1992). Studies on the structure and mechanism of H-ras p 21. Philos Trans R Soc Lond B Biol Sci 336(1276): 3-10; discussion 10-11.
- Hiratsuka, T. (1983). New ribose-modified fluorescent analogs of adenine and guanine nucleotides available as substrates for various enzymes. Biochim Biophys Acta 742(3): 496-508.
- John, J., Rensland, H., Schlichting, I., Vetter, I., Borasio, G. D., Goody, R. S. and Wittinghofer, A. (1993). Kinetic and structural analysis of the Mg2+-binding site of the guanine nucleotide-binding protein p21H-ras. J Biol Chem 268(2): 923-929.
- John, J., Sohmen, R., Feuerstein, J., Linke, R., Wittinghofer, A. and Goody, R. S. (1990). Kinetics of interaction of nucleotides with nucleotide-free H-ras p 21. Biochemistry 29(25): 6058-6065.
- Kanie, T. and Jackson, P. K. (2018). Guanine nucleotide exchange assay using fluorescent MANT-GDP. Bio Protoc 8(7): e2795.
- Klebe, C., Bischoff, F. R., Ponstingl, H. and Wittinghofer, A. (1995). Interaction of the nuclear GTP-binding protein Ran with its regulatory proteins RCC1 and RanGAP. Biochemistry 34(2): 639-647.
- Klebe, C., Nishimoto, T. and Wittinghofer, F. (1993). Functional expression in Escherichia coli of the mitotic regulator proteins p24ran and p45rcc1 and fluorescence measurements of their interaction. Biochemistry 32(44): 11923-11928.
- Pu, R. T. and Dasso, M. (1997). The balance of RanBP1 and RCC1 is critical for nuclear assembly and nuclear transport. Mol Biol Cell 8(10): 1955-1970.
- Wennerberg, K., Rossman, K. L. and Der, C. J. (2005). The Ras superfamily at a glance. J Cell Sci 118(Pt 5): 843-846.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Tan, Y. and Sun, Q. (2021). A mant-GDP Dissociation Assay to Compare the Guanine Nucleotide Binding Preference of Small GTPases. Bio-protocol 11(2): e3886. DOI: 10.21769/BioProtoc.3886.
Category
Biochemistry > Protein > Quantification
Cancer Biology > Cancer biochemistry > Protein
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.