- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Multiplex T-cell Stimulation Assay Utilizing a T-cell Activation Reporter-based Detection System
(*contributed equally to this work) Published: Vol 11, Iss 2, Jan 20, 2021 DOI: 10.21769/BioProtoc.3883 Views: 7725
Reviewed by: Alessandro DidonnaShuiqing HuShalini Low-Nam
Original research article
The authors used this protocol in:

Apr 2020
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Immune tolerance and response are both largely driven by the interactions between the major histocompatibility complex (MHC) expressed by antigen presenting cells (APCs), T-cell receptors (TCRs) on T-cells, and their cognate antigens. Disordered interactions cause the pathogenesis of autoimmune diseases such as type 1 diabetes. Therefore, the identification of antigenic epitopes of autoreactive T-cells leads to important advances in therapeutics and biomarkers. Next-generation sequencing methods allow for the rapid identification of thousands of TCR clonotypes from single T-cells, and thus there is a need to determine cognate antigens for identified TCRs. This protocol describes a reporter system of T-cell activation where the fluorescent reporter protein ZsGreen-1 is driven by nuclear factor of activated T-cells (NFAT) signaling and read by flow cytometry. Reporter T-cells also constitutively express additional pairs of fluorescent proteins as identifiers, allowing for multiplexing of up to eight different reporter T-cell lines simultaneously, each expressing a different TCR of interest and distinguishable by flow cytometry. Once TCR expression cell lines are made they can be used indefinitely for making new T-cell lines with just one transduction step. This multiplexing system permits screening numbers of TCR-antigen interactions that would otherwise be impractical, can be used in a variety of contexts (i.e., screening individual antigens or antigen pools), and can be applied to study any T-cell-MHC-antigen trimolecular interaction.
Keywords: AntigensBackground
The interactions between T-cells, antigen-presenting cells (APCs), and their cognate antigens are central events in autoimmune diseases such as type 1 diabetes (Michels et al., 2017; Mann et al., 2020), as well as other immune-based responses including tumor suppression. High-throughput sequencing of the α and β chain genes for T-cell receptors (TCRs) from single T-cells via next-generation sequencing (NGS) allows for the rapid identification of thousands of TCRs that are potentially involved in disease onset and progression. Determining the biologically relevant antigens recognized by a large number of TCRs is an important need towards understanding disease pathogenesis and ultimately developing antigen-specific immunotherapies and diagnostics for personalized medicine. Techniques that enable screening of immense numbers of potential antigens, such as combinatorial peptide libraries, require stable T-cell lines that can be expanded reliably without losing sensitivity or reproducibility. The system described here achieves this purpose by generating T-cell ‘avatars’ expressing human TCRs in a murine T-cell hybridoma line that can be easily grown while retaining response to cognate antigens.
Flow cytometric methods, in which a fluorescent reporter gene is driven by response-specific transcription factors such as nuclear factor of activated T-cells (NFAT), sensitively detect T-cell activation (Figure 1) and can distinguish each cell population by marking with fluorescent proteins (FPs), allowing for the simultaneous evaluation of multiple cell populations. This multiplexing approach reduces the labor and reagents needed for detection of response to antigen when large numbers of TCRs are to be analyzed (Mann et al., 2020). Here we describe an NFAT-driven fluorescent reporter system and T-cell multiplexing strategy for large-scale screening of TCR and peptide-MHC complex interactions, to surmount this limiting step in determination of TCR antigen specificity.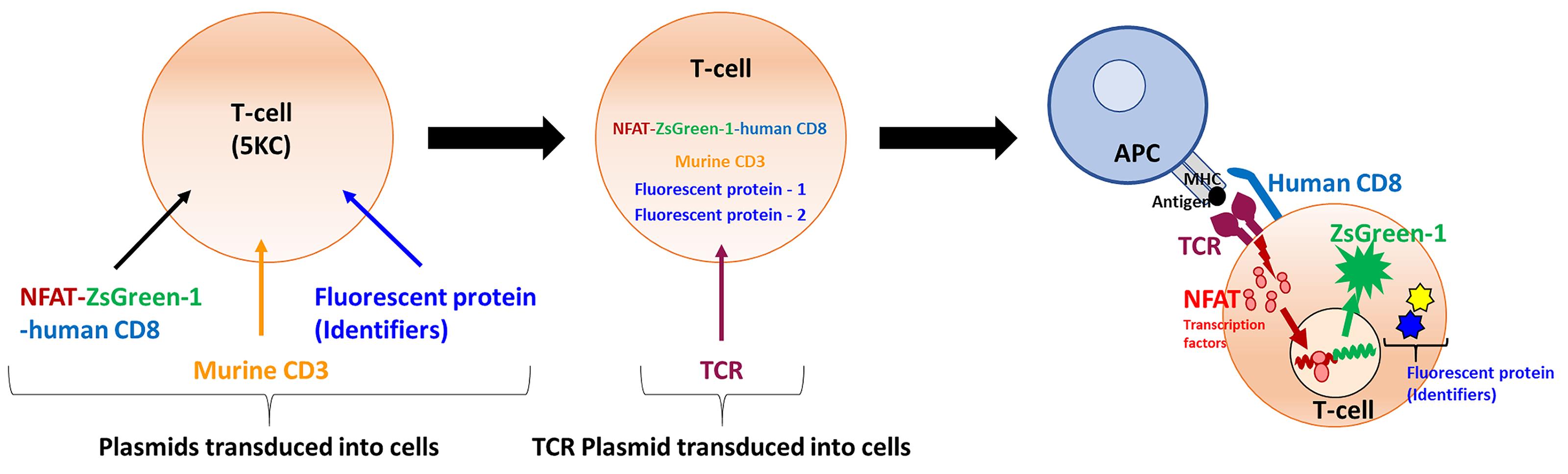
Figure 1. Principle of the assay – strategy of reporter T-cell generation. Four vectors for making TCR expression cell lines are transduced into 5KC α-β- cells (T-hybridoma cells lacking endogenous TCR expression). The first vector contains the NFAT-binding sequence followed by the fluorescent ZsGreen-1 gene, and the human CD8 co-receptor gene driven by the PGK promoter. The second vector encodes the Murine CD3. The third and fourth vectors contain fluorescent protein (FP) genes as identifiers. The TCR expression cell line is then transduced with a vector encoding TCR alpha and beta chain genes to generate functional TCR reporter cells. When the TCR reporter cell is activated upon the recognition of a peptide presented by an antigen presenting cells (APC), NFAT transcription factor proteins bind to the NFAT-binding site and promote the ZsGreen-1 gene expression, which is detected by flow cytometry. By using 8 combinations of FP identifiers, mixtures of 8 different TCRs can by assayed in the same reaction well.
In this procedure, murine T-hybridoma cells without endogenous TCR expression (5KC α-β-, White et al., 1993) are used to express the TCR α and β chains corresponding to a specific clonotype. 5KC α-β- cells are derived from murine CD4 T-cells and express the murine CD4 antigen. Therefore, human CD8 (hCD8) or human CD4 (hCD4) genes need to be introduced into 5KC α-β- for the screening of human TCRs. In this protocol, in which we describe experiments to screen hCD8 TCR antigen specificity, T-cell lines are created by introduction of the following components: (1) NFAT-driven ZsGreen-1 reporter vector that also contains hCD8 genes, (2) additional murine CD3 genes to enhance TCR expression, (3) fluorescent protein identifiers, allowing up to eight cell lines to be multiplexed in one reaction well and distinguished via flow cytometry, and (4) chimeric TCR α and β chain genes composed of the human TCR variable region of interest and the mouse constant region (Figure 1). Of note, once cell lines that are introduced with the first three components without TCR genes are generated, such parental cell lines can be used indefinitely to express different TCRs. By assaying eight T-cell lines together, requirements of time, labor and reagents for antigen screening (media, peptides) are substantially reduced, permitting the analysis of large numbers of TCRs for the response to thousands of antigens. This approach can be applied to human or murine T-cells, as well as those with different major histocompatibility complex (MHC) restrictions, by replacing hCD8 with other molecules, such as hCD4.
Materials and Reagents
5-ml serological pipettes (Fisherbrand, catalog number: 13-678-11D )
10-ml serological pipettes (Fisherbrand, catalog number: 13-678-11E )
25-ml serological pipettes (Fisherbrand, catalog number: 13-678-11 )
Filtered 10 μl micropipette tips (MultiMax, catalog number: JM2015-108 )
Filtered 200 μl micropipette tips (MultiMax, catalog number: B-2104-SH )
Filtered 1,250 μl micropipette tips (MultiMax, catalog number: B-2105-L )
15-ml conical centrifuge tubes (TrueLine, catalog number: TR2000 )
50-ml conical centrifuge tubes (TrueLine, catalog number: TR2003 )
1.5-ml Optimum Microcentrifuge tubes (Life Science Products, catalog number: 8215-0500N )
96-well PCR plates (Fisherbrand, catalog number: 14230232 )
Sterile 2 mil Seal Film Tape for PCR (Life Science Products, catalog number: ST-3099 )
Foil PCR plate seals (Fisher Scientific, Axygen, catalog number: 14-222-343 )
96-well cell culture plates (Falcon, catalog number: 353077 )
12-well cell culture plates (Falcon, catalog number: 353043 )
6-well cell culture plates (Falcon, catalog number: 353046 )
10-cm tissue culture dishes (Falcon, catalog number: 353003 )
T25 tissue culture flasks (Thermo Scientific Nunc, catalog number: 156367 )
T75 tissue culture flasks (Thermo Scientific Nunc, catalog number: 156499 )
5-ml disposable syringes (BD, catalog number: 1482945 )
Millex HV syringe filter, 0.45 μm (EMD Millipore, catalog number: SLHV033RS )
Syringe filters, 0.2 μm (Corning, catalog number: 431219 )
Wide-bore filter tips (ART, catalog number: 212362C )
1.8-ml cryotubes (Thermo Fisher Nunc, catalog number: 377267 )
Sterile 14-ml round-bottom tubes (Falcon, catalog number: 352059 )
Caplugs sterile polypropylene culture tubes, 4 ml (Evergreen Scientific, catalog number: 05-551-2 )
Rapid-Flow Sterile Disposable Filter Units, 500 ml, 0.2 μm (Nalgene, catalog number: 569-0020 )
Blunt cannulas, 15-gauge (Covidien Monoject, catalog number: 22-652-085 )
100 μm cell strainer (Falcon, catalog number: 352360 )
10 cm Petri dishes (Fisherbrand, catalog number: 08-757-12 )
Synthetic double-stranded DNA fragments (TWIST Bioscience or Integrated DNA Technologies, custom)
Cell lines
Vectors
8xNFAT-ZsG-hCD8 (Addgene, catalog number: 153417 )
Murine CD3 WTdelta-F2A-gamma-T2A-epsilon-P2A-zeta pMIA II [CD3-AM] (Addgene catalog number: 52093 )
CD3-2A_pMI-LO [CD3-LO] (Addgene, catalog number: 153418 )
pMSCVII (Addgene, catalog number: 162750 )
pMSCVII-AM [pMSCV-AM] (Addgene, catalog number: 162751 )
pMSCVII-LO [pMSCV-LO] (Addgene, catalog number: 162754 )
pMSCVII-BFP [pMSCV-BFP] (Addgene, catalog number: 162755 )
pMSCVII-tdTomato [pMSCV-TM] (Addgene, catalog number: 162752 )
pMSCVII-E2Crimson [pMSCV-CR] (Addgene, catalog number: 162756 )
pMSCVII-mChe [pMSCV-mChe] (Addgene, catalog number: 162753 )
MBC1-null (Addgene, catalog number: 162748 )
MBC2-null (Addgene, catalog number: 162749 )
CaP2AIII_pK18 [CaP2AIII] (Addgene, catalog number: 153421 )
pUS (Addgene, catalog number: 153416 )
A2_uSFFV (Addgene, catalog number: 153415 )
pCL-ECO (Addgene, catalog number: 12371 )
pMD2.G (Addgene, catalog number: 12259 )
psPAX2 (Addgene, catalog number: 12260 )
Restriction enzymes (New England Biolabs)
BspEI (NEB, catalog number: R0540S )
HindIII (NEB, catalog number: R0104S )
MfeI (NEB, catalog number: R0589S )
HpaI (NEB, catalog number: R0105S )
EcoRI (NEB, catalog number: R0101S )
XhoI (NEB, catalog number: R0146S )
MscI (NEB, catalog number: R0534S )
AleI-v2 (NEB, catalog number: R0685S )
SalI (NEB, catalog number: R0138S )
BglII (NEB, catalog number: R0144S )
MluI-HF (NEB, catalog number: R3198S )
SbfI (NEB, catalog number: R0642S )
Primers (Integrated DNA Technologies)
BspEI-F (GCGGGCGCCCGAAGGTCCT)
Hind Seq-R (GGCTTCAGCTGGTGAAGCTT)
CD3-Seq (ATGGCCTTTACCAGGGTCTC)
SalI-Seq (GCATGCTAGCTATAGTTCTAGAG)
5pMIG (GCCTCCTCTTCCTCCATCCG)
3LTR-2 (AATTTGCGCATGCTAGCTATAG)
TRAC1-R (CAGGCAGAGGGTGCTGTCCTG)
5MAC3 (AGAGACCAACGCCACCTACC)
3TRBC (CAAGGAGACCTTGGGTGGAG)
Cas9-S14 (CACATAGCGTAAAAGGAGC)
Cas9-S16 (AAGAGCTCACAACCCCTCAC)
P2A-R (GAAGTTCGTGGCTCCGGAG)
PGK-F2 (CATTCCACATCCACCGGTAG)
P2A-F (CTCCGGAGCCACGAACTTC)
P2A-R2 (GGGACCGGGGTTTTCTTCCAC)
3LTR (GCAAAATGGCGTTACTTAAGC)
IRES-Seq (CTGATCTGGGGCCTCGGTGC)
LB broth (Gibco, catalog number: 10855-021 )
LB agar (BD Difco, catalog number: 244520 )
Ampicillin Ready Made Solution (Sigma-Aldrich, catalog number: A5354-10ML )
QIAQuick Gel Extraction Kit (Qiagen, catalog number: 28704 )
QIAQuick PCR Clean-up Kit (Qiagen, catalog number: 28104 )
Qiagen Minelute Gel Extraction Kit (Qiagen, catalog number: 28604 )
NEBuilder HiFi DNA Assembly Master Mix (NEB, catalog number: E2621L )
NEB 5-alpha Competent E. coli (HE) (NEB, catalog number: C2987H )
Agarose (Fisherbrand, catalog number: BP160-500 )
Ethidium bromide, 10 mg/ml (Fisher Scientific, Invitrogen, catalog number: 15-585-011 )
PreMix D (Lucigen, catalog number: MO7205D )
Taq DNA Polymerase (Sigma, catalog number: D1806-20X250UN )
100 bp DNA ladder (Fisher Scientific, Invitrogen, catalog number: 15628-019 )
1 Kb Plus DNA ladder (Fisher Scientific, Invitrogen, catalog number: 10-787-018 )
Qiagen Plasmid Plus Midi Kit (Qiagen, catalog number: 12943 )
Fetal bovine serum (FBS) (Atlanta Biologicals, catalog number: S11150H )
Lipofectamine 2000 (Invitrogen, catalog number: 11668-500 )
Molecular biology grade water (HyClone, catalog number: 3053801 )
Poly-D-lysine (PDL) hydrobromide lyophilized powder (Sigma-Aldrich, catalog number: P6407-10x5MG )
Sterile cell culture grade water (HyClone Water, Cell Culture Grade (Endotoxin-Free), catalog number: SH3052901 )
Phosphate-buffered saline (PBS), pH 7.4 (Gibco, catalog number: 10010023 )
Penicillin/streptomycin (pen/strep), sterile filtered (Sigma-Aldrich, catalog number: P0781 )
Trypsin-EDTA, 0.25% (Gibco, catalog number: 25200-056 )
Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, catalog number: 11965-092 )
Iscove’s Modified Dulbecco’s Medium (IMDM) (Gibco, catalog number: 12440079 )
RPMI 1640 medium, no phenol red (Gibco, catalog number: 11835030 )
MEM Non-essential Amino Acids Solution (MEM-NEAA) (100x) (Gibco, catalog number: 11140-050 )
HEPES Buffer, 1 M (Gibco, catalog number: 15630-080 )
Sodium Pyruvate, 100 mM (Gibco, catalog number: 11360-070 )
2-mercaptoethanol [14.3 M] (Sigma-Aldrich, catalog number: M3148 )
Dimethyl sulfoxide (DMSO) (Fisher Bioreagents, catalog number: BP231-100 )
Protamine sulfate powder (MP Biomedicals, catalog number: ICN19472901 )
PE-labeled anti-human CD8 antibody (Biolegend, clone SK1, catalog number: 344706 )
Anti-mouse CD3e antibody, functional grade (BD, clone 145-2C11, catalog number: 553057 )
PE-labeled anti-mouse CD3e antibody (BD, clone 145-2C11, catalog number: 553064 )
PE-labeled anti-HLA-ABC antibody (BD, clone G46-2.6, catalog number: 560964 )
Mouse CD3e microbead kit (Miltenyi Biotec, catalog number: 130-094-973 )
Zombie NIR Fixable Viability Kit (BioLegend, catalog number 423105 )
Transfection medium (see Recipes)
5KC medium (see Recipes)
Phoenix medium (see Recipes)
5KC Freezing Medium (see Recipes)
Phenol red-free RPMI (see Recipes)
Protamine sulfate, 5 mg/ml solution (see Recipes)
PDL, 0.1 mg/ml (see Recipes)
LB-ampicillin Broth (see Recipes)
LB-ampicillin plates (see Recipes)
Equipment
Pipet controller (Fisherbrand, catalog number: FB14955202 )
Micropipettes (Fisherbrand Elite Kit, catalog number: 14-388-100 )
Multichannel pipettors (Fisherbrand Elite series)
Water bath (Fisher Scientific, Corning, 6-L digital waterbath, catalog number: 07-202-156 )
Ultra-low freezer (Thermo Scientific, TSX series, catalog number: TSX50086A )
Dry bath (Fisherbrand, Isotemp digital heat double-stranded DNA fragment, catalog number: 88-860-021 )
Electrophoresis gel boxes (Fisher Scientific, Owl D4 Horizontal Electrophoresis System, catalog number: 09-528-205 )
Electrophoresis power supply (Fisherbrand, catalog number: FB200Q )
PCR Thermal Cycler (Thermo Fisher Scientific, Applied Biosystems ProFlex 96-well PCR System, catalog number: 4484075 )
NanoDrop 2000 spectrophotometer (Thermo Scientific, model: NanoDropTM 2000 )
Shaking incubator (New Brunswick Scientific, model: Innova 43 )
GelDoc Go Gel Imaging System (Bio-Rad, catalog number: 12009077 )
Microcentrifuge (Fisherbrand accuSpinTM Micro 17, catalog number: 13-100-675 )
Countess II automated cell counter (Invitrogen, catalog number: AMQAX1000 )
Countess II slides (Invitrogen, catalog number: C10228 )
Inverted light microscope (Olympus, model: IX53 )
Bench centrifuge (Thermo Scientific, SorvallTM LegendTM XT/XF Centrifuge and Rotor Packages, catalog number: 75-333-839 )
Biosafety BSL2 cabinet (NuAire, model: NU-425-400 )
CO2 Incubator (Fisherbrand Isotemp CO2 Incubators, catalog number: 11-676-600 )
Cytoflex (Beckman Coulter, https://www.beckman.com/flow-cytometry/instruments/cytoflex)
Mr. Frosty freezing container (Thermo Scientific, catalog number: 15-350-50 )
MS Columns (Miltenyi Biotec, catalog number: 130-042-201 )
OctoMACS Separator (Miltenyi Biotec, catalog number: 130-042-109 )
MilliQ water machine (Millipore-Sigma, lot number: F8BA18025 )
Software
Sequencher, Gene Codes Corp (http://www.genecodes.com/)
SnapGene version 5.0.4 (https://www.snapgene.com/)
FlowJo v10 (https://www.flowjo.com/)
Prism GraphPad (https://www.graphpad.com/)
NCBI nucleotide website (https://www.ncbi.nlm.nih.gov/nucleotide/)
FP database (https://www.fpbase.org/)
IMGT website (http://www.imgt.org/IMGTrepertoire/Proteins/index.php#B)
HLA database (https://www.ebi.ac.uk/ipd/imgt/MHC/allele.html)
Procedure
Overview of the Procedure
The workflow for this protocol is illustrated in Figure 2. In the first step, TCR expression cell lines are generated using vectors made as described in Steps A1 to A4. Transfection and transduction are used to introduce the vectors (Figure 1) into 5KC α-β- hybridoma T-cells to generate TCR expression cell lines having all components of the reporter system (along with eight distinct fluorescent identifiers) except TCR α and β chains (Steps B1 to B2c). These TCR expression cell lines can be expanded and frozen, and then used repeatedly for multiple rounds of TCR reporter cell line generation. In the second step, vectors with TCR α and β chains are generated (Step A5) and transduced into the TCR expression cell lines, resulting in functional reporter T-cells (Step B2d). In step 3, APC cell lines are generated expressing MHC genes of biological relevance, depending on the origin of TCRs being analyzed. Step A6 describes the generation of MHC expression vectors, and vectors are then transduced into APC cell lines (Step B3). In step 4, the multiplex stimulation assay is conducted by combining 8 different FP-identified T-cell reporter lines and co-culturing with APCs and antigens (Steps C1-C4). Finally, flow cytometry is used to measure positively stimulated cells by induction of the reporter color ZsGreen-1 (Step C5).
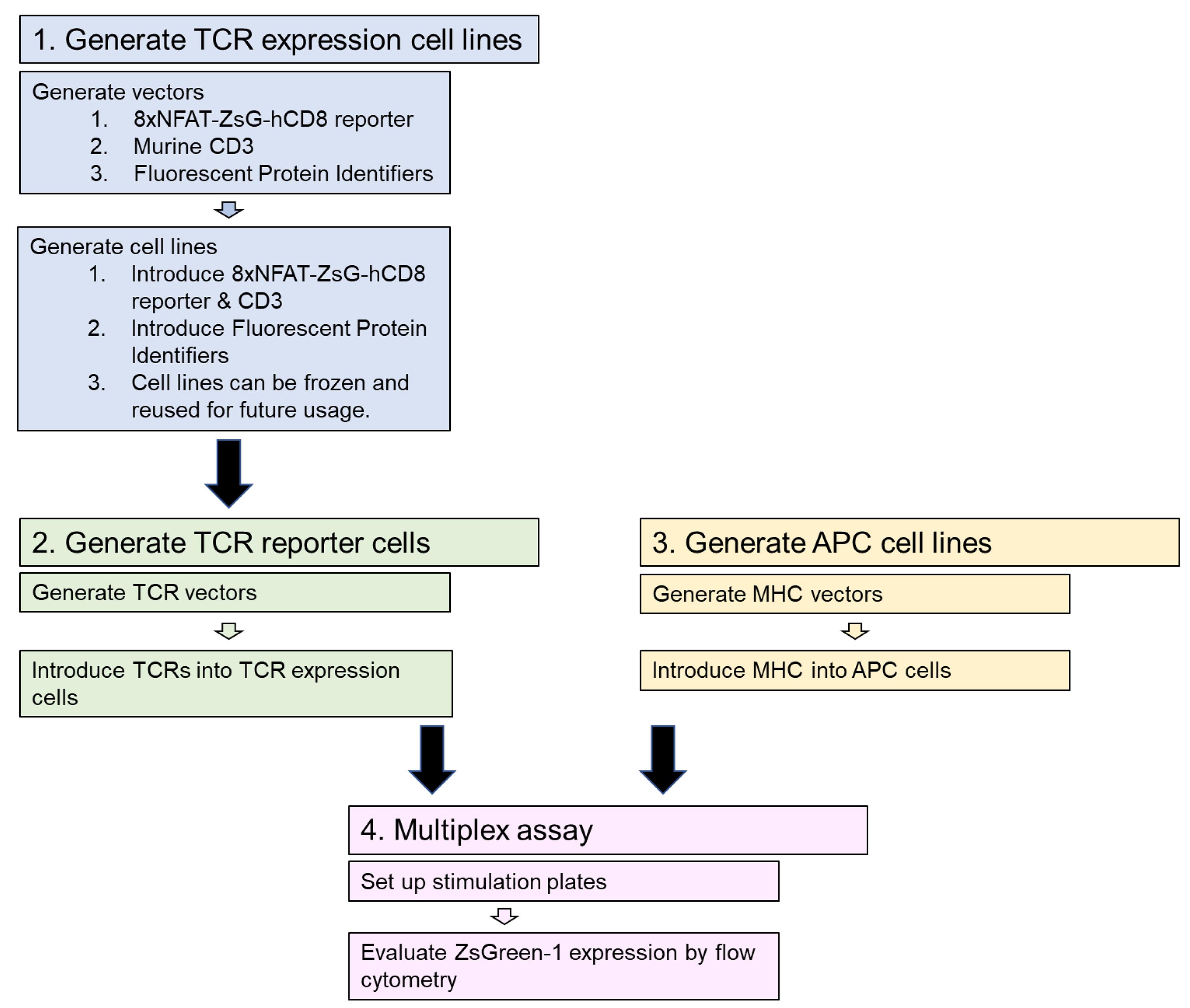
Figure 2. Procedure workflow. The protocol proceeds in four major steps. The first step generates TCR expression cells lines that are then used in step 2 to make TCR reporter cell lines. Step 3, generating APC cell lines, can be done concurrently with steps 1 and 2. In step 4, the multiplex stimulation assay is performed, using the TCR reporter lines co-cultured with APC cells and antigens. Flow cytometry is used to measure ZsGreen-1 expression in positively stimulated cells.
Vector Description and Construction
Refer to Table 1 for a list of all vectors mentioned in Section A, which are available from Addgene. Table 2 lists all primers used in Section A for vector construction and verification by PCR and sequencing.
Table 1. Vectors used in the following protocol, their source, and catalog numbers
Table 2. Primers used for PCR and sequencing
*Locations of primers in each figure are shown.
hCD8-8xNFAT reporter vector. This vector is to introduce the T-cell activation reporter gene as well as TCR co-receptor genes such as human CD8. As shown in Figure 3, the 8xNFAT-ZsG-hCD8 reporter vector is a replication-incompetent and self-inactivating retroviral vector with the 8xNFAT-ZsG reporter construct (8 repeats of NFAT binding sites in a row, followed by a TATA box and the ZsGreen-1 gene) inserted into the site behind truncated gag gene. When transduced into 5KC α-β- cells with reconstituted TCR genes, the 8xNFAT-ZsG reporter gene will induce the expression of fluorescent protein ZsGreen-1 when cells are stimulated by the appropriate antigen. The vector contains the human CD8 (hCD8) α and β genes downstream of the PGK promoter for constitutive hCD8 expression. The hCD8 genes can be replaced with other TCR co-receptor genes such as human CD4 (hCD4). Multi-subunit gene insertions, such as for hCD8α and hCD8β in this example, employ a porcine teschovirus-1 2A cleaving peptide sequence (P2A), which joins subunit gene sequences.
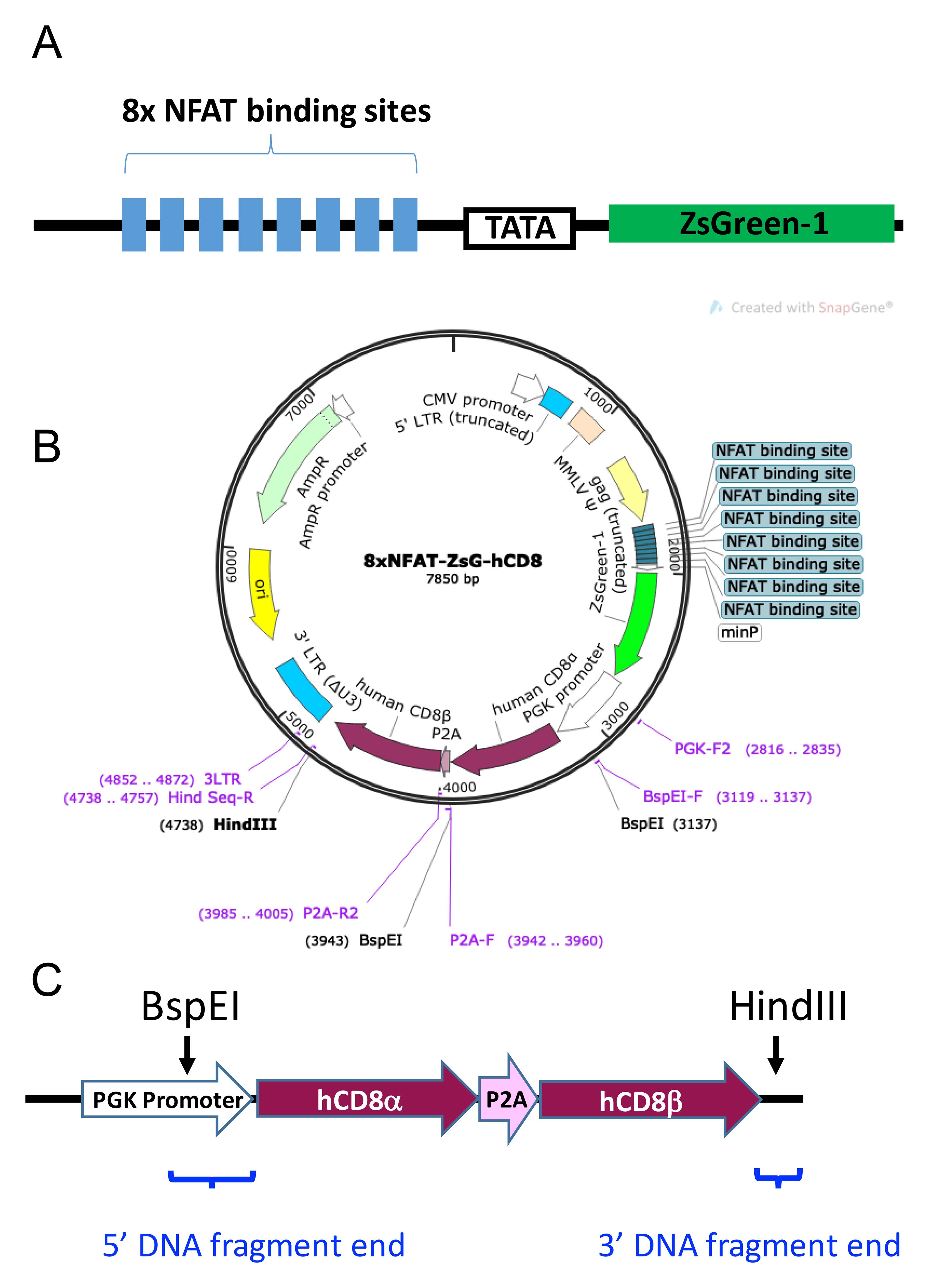
Figure 3. 8xNFAT-ZsG-hCD8 reporter vector. A. Schematic diagram of the 8xNFAT-ZsG construct. The 8xNFAT-ZsG reporter construct consists of 8 repeats of NFAT binding sites (GGAGGAAAAACTGTTTCATACAGAAGGCGT), a TATA box, and the ZsGreen-1 gene. B. Plasmid map of 8xNFAT-ZsG-hCD8. The 8xNFAT-ZsG-hCD8 reporter vector backbone has BspEI and HindIII restriction enzyme sites that can be used to remove the hCD8α-P2A-hCD8β gene construct. A TCR co-receptor gene fragment containing sequences overlapping the linearized vector ends can then be inserted by Gibson assembly. C. Schematic diagram of the hCD8α-P2A-hCD8β gene construct. Sequences for hCD8α and hCD8β genes (maroon) are joined by P2A (pink). The portion of PGK promoter (white) excised when the vector is digested with BspEI is restored by addition to the 5’ end of hCD8α as part of the 5’ DNA fragment end. Blue brackets indicate the positions of the 5’ and 3’ DNA fragment ends. Panel A is adapted from the article published by Mann et al. (2020) in Frontiers in Immunology (doi 10.3389/fimmu.2020.00633), which is licensed under by CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/). The sequence of the vector is also available in Mann et al. (2020).Optional step: Replace the hCD8 genes with hCD4 genes or another TCR co-receptor. Refer to the NCBI nucleotide website (https://www.ncbi.nlm.nih.gov/nucleotide/) to obtain the coding sequence for hCD4 or the desired genes.
Design a nucleotide sequence for the DNA fragment to be inserted into the vector. For multi-subunit gene insertions, such as for hCD4 α and β in this example, use a P2A cleaving peptide to join subunit gene sequences.
P2A (added between the 3′ end of the first gene sequence, omitting the stop codon, and the starting ATG of the second gene sequence):
5′-GGCTCCGGAGCCACGAACTTCTCTCTGTTAAAGCAAGCAGGAGACGTGGAAGAAAACCCCGGTCCC-3′
Amino Acid sequence of P2A: GSGATNFSLLKQAGDVEENPGP
Add the 5’ and 3’ DNA fragment ends, each of which contains an overlapping sequence with the linearized vector, to the gene sequences:
5’ DNA fragment end (includes partial sequence of PGK promoter that is removed when the vector is linearized by restriction digest with BspEI; placed in front of gene start codon):
5′-GGGCGGGCGCCCGAAGGTCCTCCGGAGGCCCGGCATTCTGCACGCTTCAAAAGCGCACGTCTGCCGCGCTGTTCTCCTCTTCCTCATCTCCGGGCCTTTCGACCTGCATCCCGCCACC-3′
3′ DNA fragment end (placed after gene stop codon):
5′-AAGCTTCACCAGCTGAAGCCTA-3′
Each portion of sequence should be in the following order to consist of the entire DNA fragment: The 5’ DNA fragment end, the first gene (e.g., hCD4α) excluding the stop codon, P2A, the second gene (e.g., hCD4β) including the stop codon, and the 3’ DNA fragment end. Ensure that the entire coding sequence from the start codon of the first gene through to the stop codon of the second gene is in-frame and gives the correct amino acid sequence.
Order synthetic double-stranded DNA fragments from TWIST Bioscience or Integrated DNA Technologies. Reconstitute DNA fragments in molecular biology grade water.
Digest 5 μg 8xNFAT-ZsG-hCD8 with BspEI and HindIII according to manufacturer’s instructions.
Isolate the 6,249 bp vector backbone fragment by separation on a 1% agarose gel followed by gel purification using a Qiagen QiaQuick Gel Extraction Kit according to manufacturer’s instructions.
Re-purify the 6,249 bp vector backbone fragment using QIAQuick PCR Clean-up Kit according to the manufacturer’s instructions, and elute in 20 μl molecular biology grade water.
Perform a Gibson assembly with NEBuilder HiFi DNA Assembly Master Mix according to manufacturer’s instructions. Use a 1:2 molar ratio of vector backbone to gene fragment insert.
Transform NEB 5-alpha E. coli.
Add 2 μl of Gibson assembly reaction to 25 μl chemically competent NEB 5-alpha cells and transform cells by heat shock method according to manufacturer’s instructions.
Note: When NEBuilder HiFi DNA Assembly Master Mix is used for Gibson assembly, NEB 5-alpha Competent E. coli are recommended for transformation for best transformation efficiency.
Warm LB-ampicillin plates (see Recipes) in 37 °C incubator.
Plate the entire transformation mixture onto pre-warmed LB-ampicillin plates and incubate overnight at 37 °C.
Grow mini-cultures of individual colonies and perform PCR to verify presence of the DNA fragment insert. Sequences for all primers used for PCR are given in Table 2.
Choose 4-8 well-isolated colonies per vector and inoculate into 50 μl LB-ampicillin broth (see Recipes) in individual wells of a 96-well PCR plate.
Cover the mini-cultures with a foil plate seal and incubate for 4 h at 37 °C, shaking at 250 rpm.
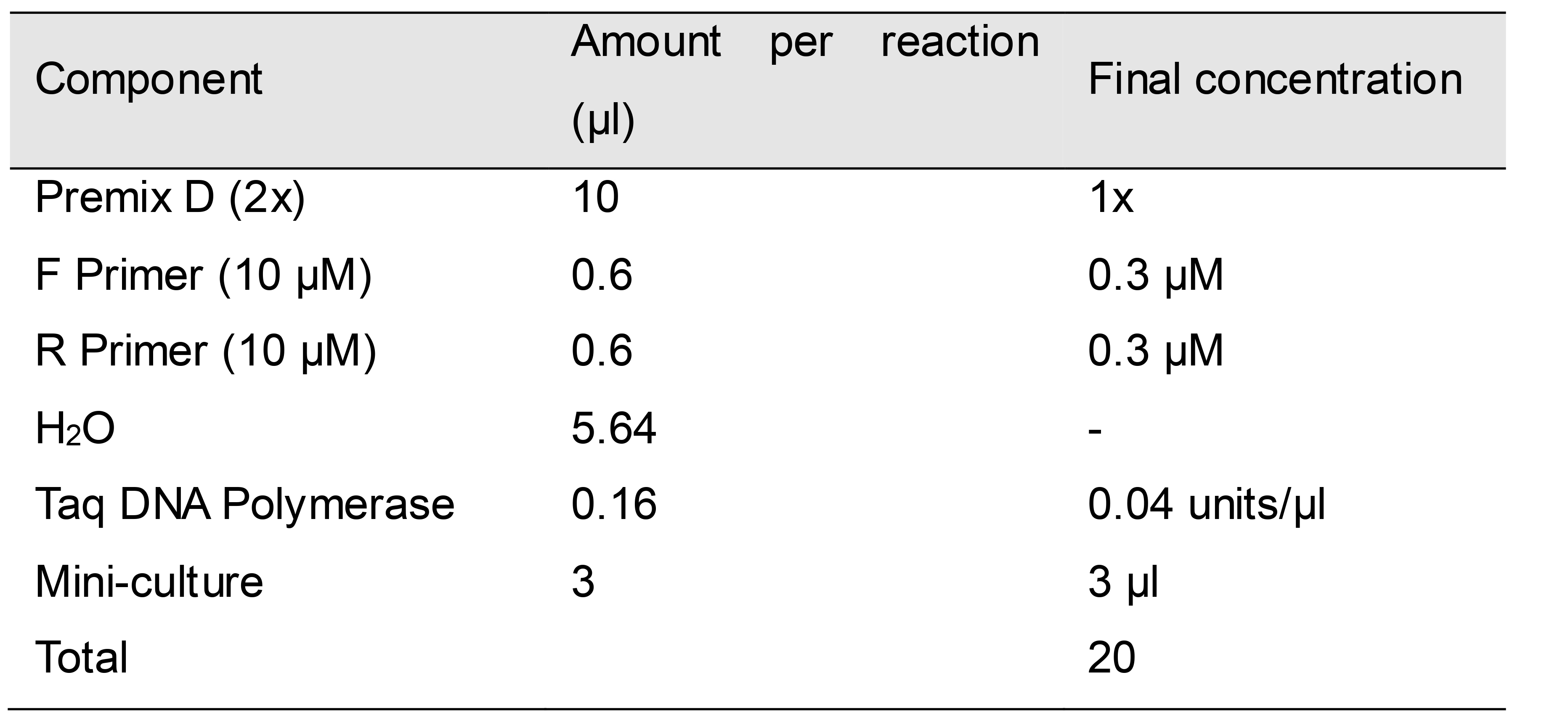
Prepare a PCR master mix with the component ratios given in Table 3. Mini-culture (3 μl) is added directly to 17 μl of reaction master mix containing the primers listed below. The F Primer (BspEI-F) and the R Primer (Hing Seq-R) locate before the BspEI recognition site and behind the HindIII recognition site in the 6,249 bp vector backbone fragment, respectively (Figure 3).
BspEI-F (F Primer) 5’-GCGGGCGCCCGAAGGTCCT-3’
Hind Seq-R (R Primer): 5’-GGCTTCAGCTGGTGAAGCTT-3’
Table 3. PCR master mix for mini-culture PCR
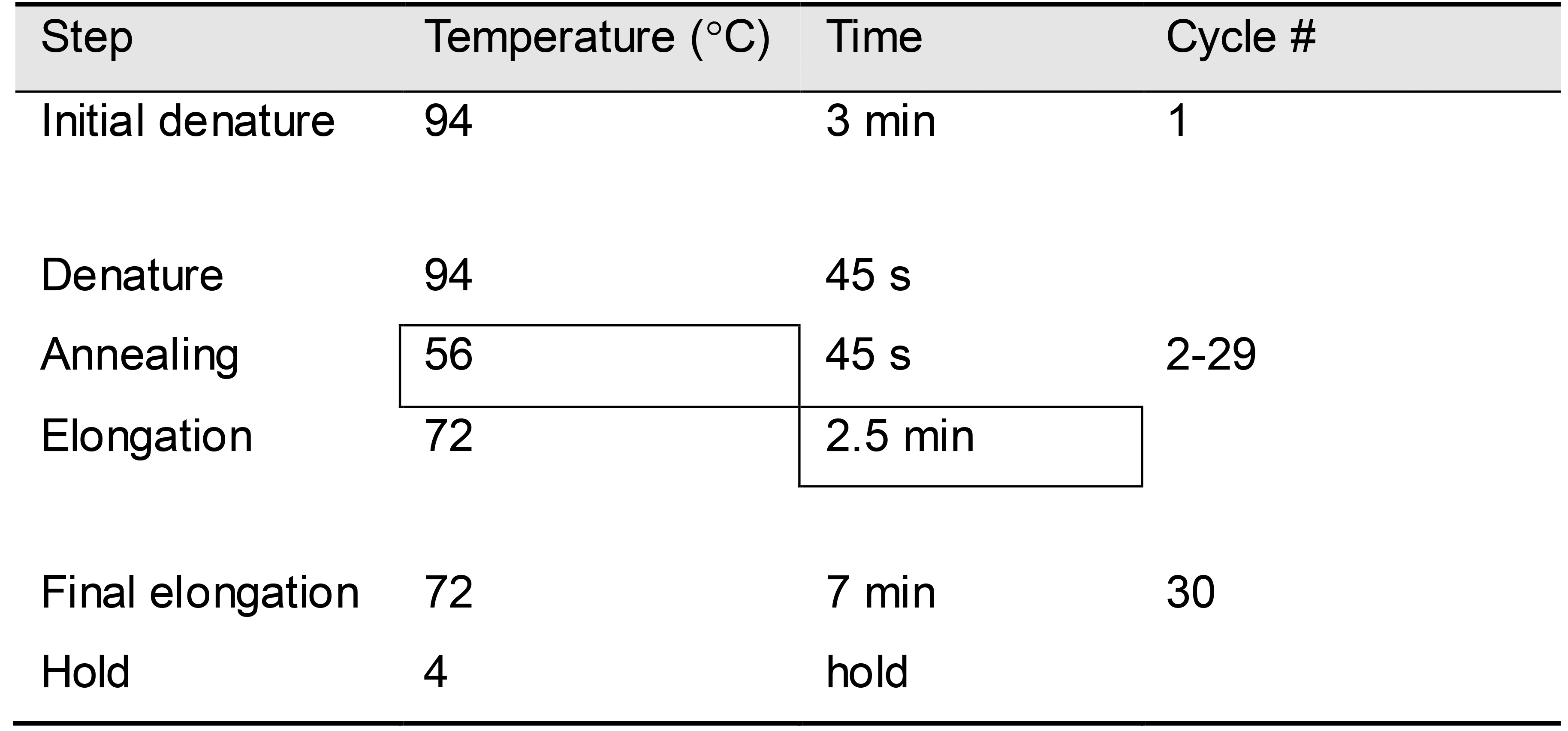
Carry out PCR according to the conditions given in Table 4, using an annealing temperature of 56 °C and an elongation time of 2.5 min.
Table 4. PCR protocol for mini-culture PCR
Visualize the PCR products by separation on a 1% agarose gel with 0.5 μg/ml ethidium bromide. Correct DNA band size will be dependent upon the length of the gene fragment inserted. For hCD8α-P2A-hCD8β the correct size is 1,639 bp (i.e., 1,503 bp of the synthetic double-stranded DNA fragment inserted plus 136 bp contained in the vector backbone).
Prepare vector for transfection. Proceed to large-scale culture plasmid preparation from the mini-culture of one colony having the correct-sized PCR product as confirmed by mini-culture PCR.
Inoculate 40 ml LB-ampicillin broth with 20 μl mini-culture. Grow overnight at 37 °C, shaking at 225 rpm.
Extract plasmid DNA using Qiagen Plasmid Plus Midi Kit according to manufacturer’s instructions. Elute plasmid DNA in 150 μl molecular biology grade water.
Quantitate plasmid DNA by NanoDrop spectrophotometry and adjust DNA concentration to 1,000 ng/μl with molecular biology grade water. Plasmid preps can be stored at -20 °C.
Check the vector for correct sequence and alignment of inserted DNA fragment by submitting samples for Sanger sequencing with the primers shown in Table 2 for 8xNFAT-ZsG-hCD8.
Note: Vector sequences must align perfectly with the designed, in-frame nucleotide sequence, without mutations, which can occur due to ligation error, PCR-introduced mutation, or DNA fragment synthesis errors. If a mutation is found, select another colony for Midi-prep and sequencing. Vectors that are confirmed to have the correct sequence can be used directly for cell transfection.
CD3 Vector. Shown in Figure 4, the CD3-2A_pMI-LO (CD3-LO) vector is a murine stem cell virus (MSCV) retroviral vector for expressing all four murine CD3 δγϵζ subunits, as well as an LSSmOrange (LO) fluorescent protein (FP) gene construct downstream of the internal ribosomal entry site (IRES) for plasmid tagging. Transducing 5KC α-β- cells with additional murine CD3 genes ensures that TCR expression is not limited by insufficient expression of endogenous CD3 (Williams et al., 2018). Because CD3 will not be expressed by T-cells in the absence of TCR genes, a fluorescent tag is required as a marker to identify successfully transduced cells. To increase flexibility of the multiplexing scheme, the LSSmOrange FP gene can be replaced with other FP genes. In the protocol described here, Murine CD3 WTdelta-F2A-gamma-T2A-epsilon-P2A-zeta pMIA II (CD3-AM, Holst et al., 2008), which contains Ametrine (AM) instead of LO and from which CD3-LO was generated, is also used.
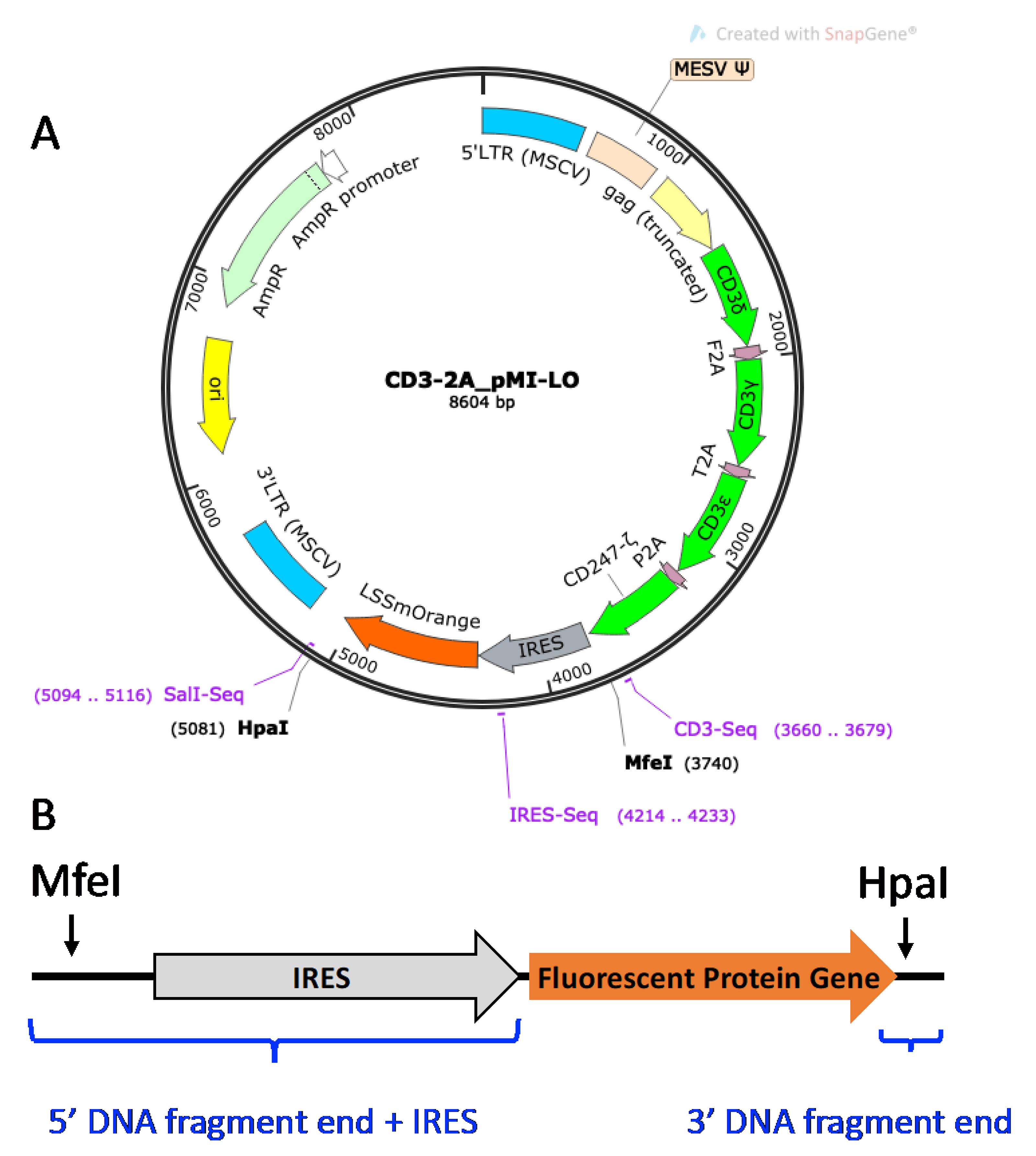
Figure 4. CD3-2A_pMI-LO vector. A. Plasmid map of CD3-2A_pMI-LO. Locations of the murine CD3 subunits (green), IRES (grey), and LO FP gene (orange) are shown. Restriction enzyme sites MfeI and HpaI can be used to excise the IRES-FP gene cassette. B. Schematic diagram of the DNA fragment with IRES-FP gene fragment insert and flanking vector sequence. Blue brackets indicate the vector-overlapping DNA fragment ends added to the FP gene sequence. IRES excised with the original FP gene is replaced as a DNA segment added to the 5’ end of the new FP gene fragment.Optional step: Replace LO FP gene with another FP gene. Refer to the FPbase website (https://www.fpbase.org/) to obtain the coding sequence for the desired FP gene.
Design a sequence for the double-stranded DNA fragment to be inserted into the vector. Add the following 5’ and 3’ end sequences, each of which contains an overlapping sequence with the linearized vector, to the FP coding gene sequence:
5’ DNA fragment end (vector overlap plus IRES; placed in front of the FP gene start codon):
AGACCCTGGCCCCTCGCTAACAATTGTCGAGCGGGATCAATTCCGCCCCCCCCCTAACGTTACTGGCCGAAGCCGCTTGGAATAAGGCCGGTGTGCGTTTGTCTATATGTTATTTTCCACCATATTGCCGTCTTTTGGCAATGTGAGGGCCCGGAAACCTGGCCCTGTCTTCTTGACGAGCATTCCTAGGGGTCTTTCCCCTCTCGCCAAAGGAATGCAAGGTCTGTTGAATGTCGTGAAGGAAGCAGTTCCTCTGGAAGCTTCTTGAAGACAAACAACGTCTGTAGCGACCCTTTGCAGGCAGCGGAACCCCCCACCTGGCGACAGGTGCCTCTGCGGCCAAAAGCCACGTGTATAAGATACACCTGCAAAGGCGGCACAACCCCAGTGCCACGTTGTGAGTTGGATAGTTGTGGAAAGAGTCAAATGGCTCTCCTCAAGCGTATTCAACAAGGGGCTGAAGGATGCCCAGAAGGTACCCCATTGTATGGGATCTGATCTGGGGCCTCGGTGCACATGCTTTACATGTGTTTAGTCGAGGTTAAAAAAACGTCTAGGCCCCCCGAACCACGGGGACGTGGTTTTCCTTTGAAAAACACGATAATACC3’ DNA fragment end (placed after the FP gene stop codon):
AACAACCGGTACCTCTAGAACOrder synthetic double-stranded DNA fragments from TWIST Bioscience or Integrated DNA Technologies. Reconstitute DNA fragments in molecular biology grade water.
Digest CD3-2A_pMI-LO with MfeI and HpaI according to manufacturer’s instructions.
Isolate the 7,263 bp vector backbone fragment by separation on a 1% agarose gel followed by gel purification using a Qiagen Qiaquick Gel Extraction Kit according to manufacturer’s instructions.
Re-purify the 7,263 bp vector backbone fragment using QIAQuick PCR Clean-up Kit according to the manufacturer’s instructions, and elute in 20 μl molecular biology grade water.
Perform a Gibson assembly with NEB HiFi Gibson Assembly Master Mix according to manufacturer’s instructions. Use a 1:2 molar ratio of vector backbone to gene insert.
Transform NEB 5-alpha E. coli as in Step A2a.ix above.
Grow mini-cultures of individual colonies and perform PCR to verify presence of the DNA fragment insert as in Step A2a.x above. Prepare mini-culture PCR reactions as in Table 3, with the following primers:
CD3-Seq (F Primer): 5’-ATGGCCTTTACCAGGGTCTC-3’
SalI-Seq (R Primer): 5’-GCATGCTAGCTATAGTTCTAGAG-3’
The F Primer (CD3-Seq) and the R Primer (SalI-Seq) locate before the MfeI recognition site and behind the HpaI recognition site in the 7,263 bp vector backbone fragment, respectively (Figure 4). Carry out PCR using the conditions given in Table 4, except using an annealing temperature of 50 °C with an elongation time of 2.5 min.
Visualize PCR products by separation on a 1% agarose gel with 0.5 μg/μl ethidium bromide. Correct DNA fragment insertion will result in a PCR product equal to the length of the FP gene plus 746 bp.
Prepare vector for transfection. Proceed to large-scale culture plasmid preparation from the mini-culture of one colony having the correct-sized PCR product as confirmed by mini-culture PCR as in Step A2a.xi.
Check the vector for correct sequence and alignment of inserted DNA fragment by submitting samples for Sanger sequencing with the primers shown in Table 2 for CD3-2A_pMI-LO.
Fluorescent Protein (FP) Vector. As seen in Figure 5, which shows FP mCherry [pMSCVII-mChe (pMSCV-mChe), Figure 5A] as an example, the FP genes are sub-cloned into a non-replicating MSCV-based retroviral vector (pMSCVII, Figure 5B) for constitutive expression of the FP gene. Six different FP vectors are required for the multiplexing strategy described in this protocol: ametrine (AM), LSSmOrange (LO), TagBFP2 (BFP), tdTomato (TM), E2Crimson (CR), and mCherry (mChe). Other FP genes may be used to increase the flexibility of the multiplexing scheme.
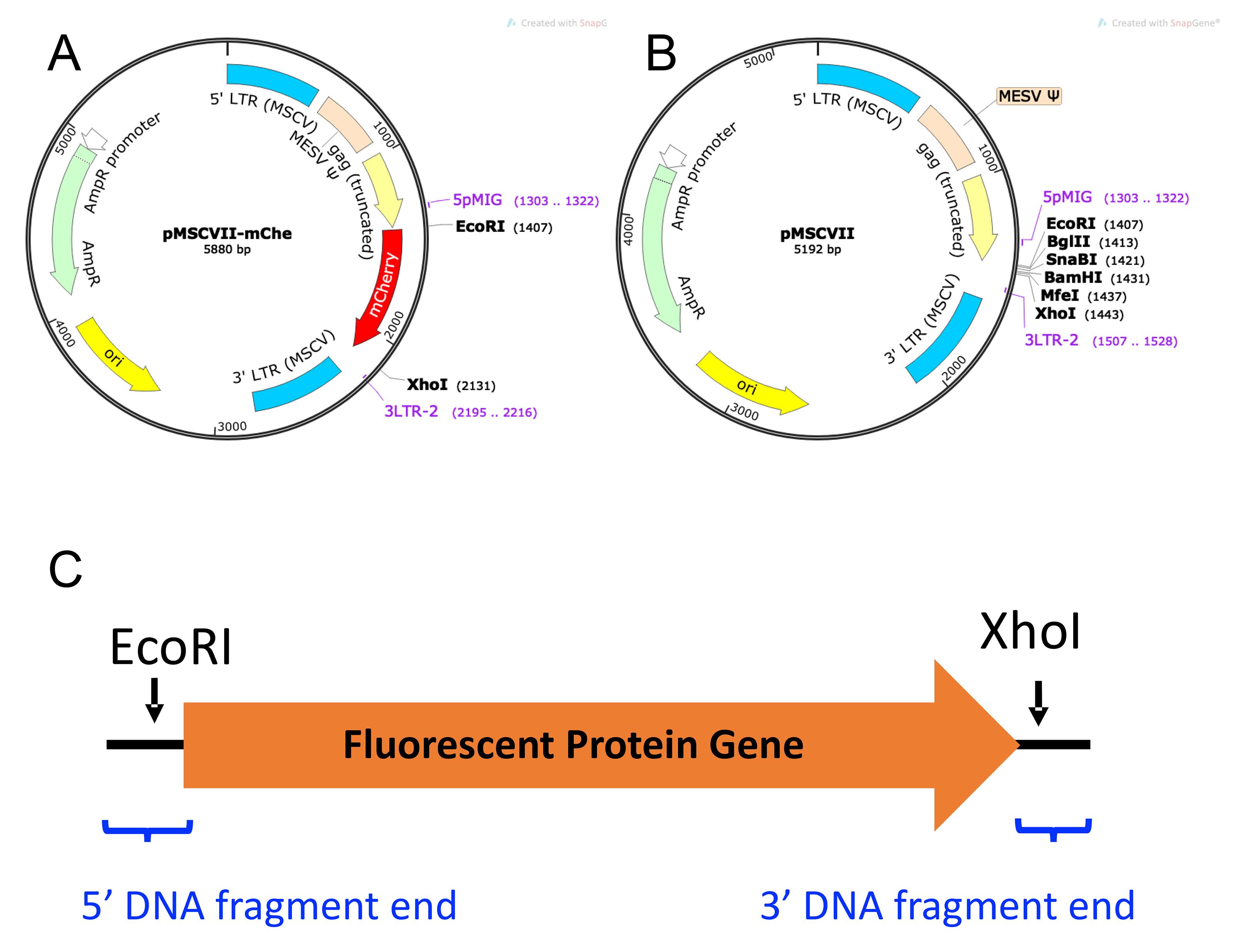
Figure 5. Fluorescent Protein Vectors. A. Plasmid map of pMSCVII-mChe (pMSCV-mChe). B. Plasmid map of pMSCVII. C. Schematic diagram of DNA fragment with FP gene sequence and flanking vector sequence (black line). EcoRI and XhoI restriction enzyme sites are indicated by arrows, and positions of the DNA fragment end vector-overlapping sequences by blue brackets.Design a sequence for the desired FP gene fragment to be inserted in the vector. Refer to the FPbase website (https://www.fpbase.org/) to obtain the FP coding gene sequence for the desired FP.
Add the following 5’ and 3’ end vector-overlapping sequences to the coding gene sequence.
5’ DNA fragment end (placed in front of the gene start codon):
CACTCCTTCTCTAGGCGCCGGAATTCAGCCACC3’ DNA fragment end (placed after the gene stop codon):
CTCGAGCCTGCAGGCATGCAAGCTTC
Order synthetic double-stranded DNA fragments from TWIST Bioscience or Integrated DNA Technologies. Reconstitute the DNA fragments in molecular biology grade water.
Linearize 5 μg pMSCVII by digestion with EcoRI and XhoI according to manufacturer’s instructions.
Purify the linearized pMSCVII using a Qiagen PCR Purification kit according to the manufacturer’s instructions.
Perform a Gibson assembly with NEB HiFi Gibson Assembly Master Mix according to manufacturer’s instructions. Use a 1:2 molar ratio of vector backbone to double-stranded DNA fragment insert.
Transform NEB 5-alpha E. coli as in Step A2a.ix above.
Grow mini-cultures of individual colonies and perform PCR to verify presence of the DNA fragment insert as in Step A2a.x above. Prepare mini-culture PCR reactions as in Table 3, with the following primers:
5pMIG (F Primer): 5’-GCCTCCTCTTCCTCCATCCG-3’
3LTR-2 (R Primer): 5’-AATTTGCGCATGCTAGCTATAG-3’
The F Primer (5pMIG) and the R Primer (3LTR-2) located before the EcoRI recognition site and behind the XhoI recognition site in pMSCVII, respectively (Figure 5).
Carry out PCR using the conditions given in Table 4, except using an annealing temperature of 54 °C and an elongation time of 1.5 min.
Visualize PCR products by separation on a 2% agarose gel with 0.5 μg/μl ethidium bromide. Correct DNA fragment insertion will result in a PCR product equal to the length of the FP gene plus 226 bp.
Prepare vector for transfection. Proceed to large-scale culture plasmid preparation from the mini-culture of one colony having the correct-sized PCR product from mini-culture PCR as in Step A2a.xi.
Check the vector for correct sequence and alignment of inserted DNA fragment by submitting samples for Sanger sequencing with the primers shown in Table 2 for pMSCVII.
TCR Vectors. To express human TCRs in murine 5KC α-β- cells, murine TCR α and β variable region signal peptide sequences (TRAV signal peptide and TRBV signal peptide, respectively) and murine TCR α and β constant regions (TRAC and TRBC, respectively) are used to flank the human TCR α and β variable gene segments, (V-α and V-β, respectively), resulting in murine-human chimeric TCR α and β chains. The chimeric TCR α and β chains are joined by a P2A cleaving peptide sequence (P2A; GGCTCCGGAGCCACGAACTTCTCTCTGTTAAAGCAAGCAGGAGACGTGGAAGAAAACCCCGGTCCC).
TCR-expressing vectors are constructed by the insertion of three DNA fragments into a non-replicating MSCV-based retroviral backbone vector. The vector backbone contains a murine TRAV5D-4 signal peptide sequence (ATGAAAACATATGCTCCTACATTATTCATGTTTCTATGGCTGCAGCTGGATGGGATGAGCCAA) and a murine TRBC region (Figure 6A). TCR vector backbone MBC1-null contains murine TRBC1*01, and MBC2-null contains murine TRBC2*03. MBC1-null or MBC2-null vector backbone is linearized by digestion with SalI and BglII, and Gibson assembly is used to ligate the three fragments and vector backbone.
The three inserted DNA fragments are: 1) the human TCR α variable gene segment (V-α), 2) the murine TRAC*01 gene segment plus P2A sequence (Cα-P2A), and 3) the human TCR β variable gene segment (V-β), as illustrated in Figures 6B and 6C. The Cα-P2A fragment also contains the murine TRBV5 signal peptide sequence (ATGAGCTGCAGGCTTCTCCTCTATGTTTCCCTATGTCTTGTGGAAACAGCACTCATG)after P2A (Figure 6C).
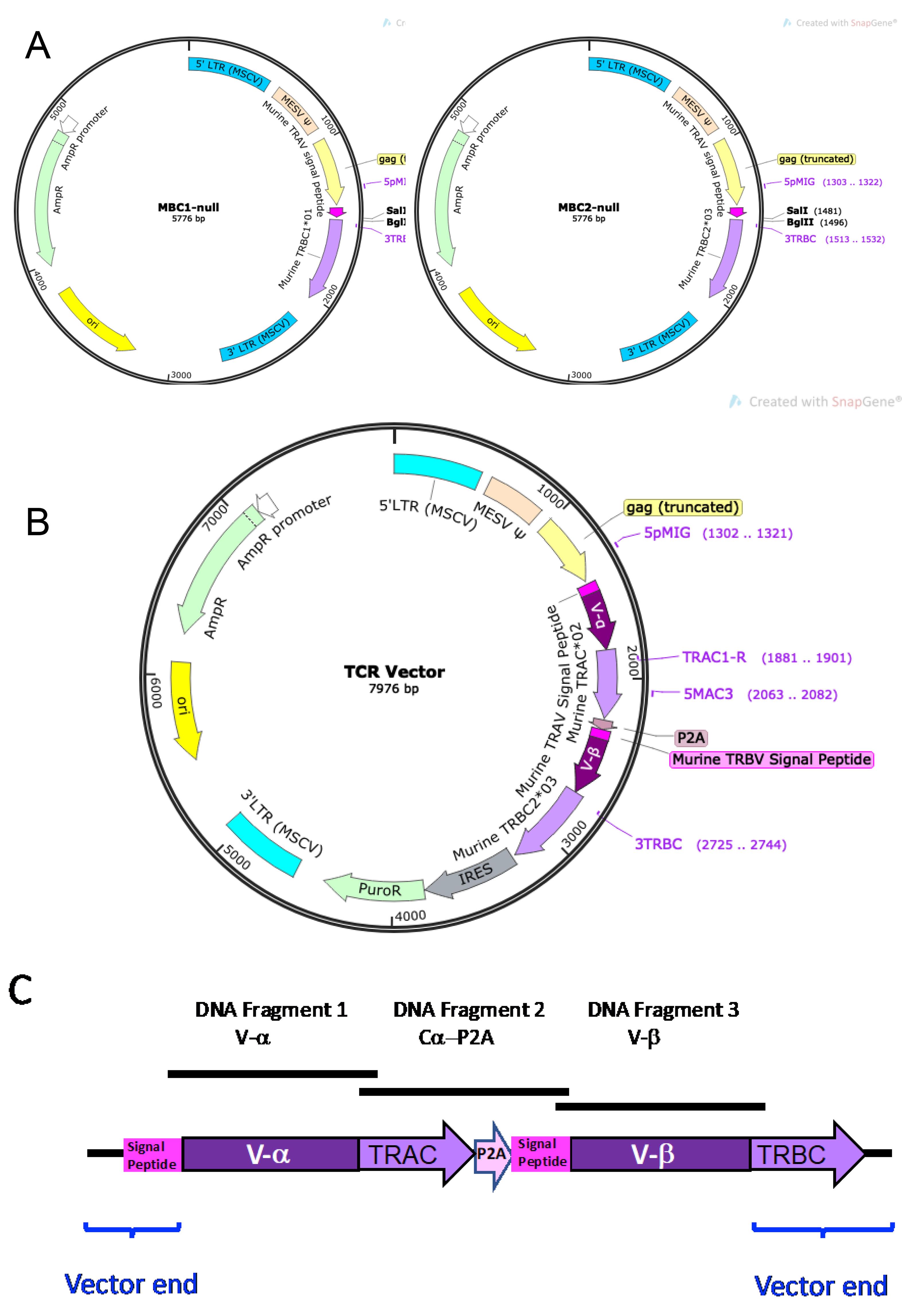
Figure 6. TCR vectors. A. Plasmid maps for MBC1-null and MBC2-null. Each vector contains murine TRAV signal peptide sequence and either murine β constant region TRBC1*01 (MBC1-null) or TRBC2*03 (MBC2-null) in an MSCV-based retroviral vector. The multi-cloning site (SalI-BglII) is located between murine TRAV signal peptide sequence (dark pink) and the TRBC gene (light purple). B. Example plasmid map of a completed TCR vector after insertion of the 1) human V-α DNA fragment (dark purple), 2) Cα-P2A DNA fragment (light purple-light pink-dark pink), and 3) human V-α DNA fragment (dark purple). C. Schematic diagram of the TCR region inserted into the MBC2-null vector. Three DNA fragments, 1) human V-α gene segment (DNA Fragment 1; V-α), 2) the murine TRAC*01-P2A-murine TRBV signal peptide sequence (DNA Fragment 2; Cα-P2A), and 3) human V-β gene segment (DNA Fragment 3; V-β), are inserted between the murine TRAV signal peptide and murine TRBC sequences of the backbone vector.Synthesize V-α and V-β DNA inserts.
Design a nucleotide sequence for DNA Fragment 1 V-α. This fragment consists of the 5’ overlapping sequence with the linearized vector, the human TCR α variable gene segment (V-α), and the 3’ overlapping sequence with DNA Fragment 2 Cα-P2A. The signal peptide sequence is supplied from the vector, and thus the V-α segment starts with the first amino acid after the signal peptide sequence of TRAV. Also, the mouse TRAC segment is supplied from DNA Fragment 2 Cα-P2A, and thus the V-α segment ends with the last amino acid of the joining region (TRAJ). An example DNA Fragment 1 V-α is shown in Figure 7.
Prepare a human TCR α chain sequence of interest from a single cell sequencing or equivalent method. The V-α segment sequence is from the first codon after the signal peptide sequence of TRAV through the last codon of TRAJ. Refer to the IMGT website (http://www.imgt.org/IMGTrepertoire/Proteins/index.php#B) to identify 1) the first codon after the signal peptide sequence of the TRAV segment and 2) the last codon of the TRAJ segment. Note that the TRAJ gene segment contains only the first nucleotide of the last amino acid codon. The last two nucleotides are provided from the TRAC segment, and thus confirmed to include the last nucleotide of TRAJ in the V-α segment.
Add 5’ and 3’ DNA fragment end sequences, corresponding to sequences that overlap the 3’ end of the linearized vector and 5’ end of the Cα-P2A fragment, to the V-α segment sequence.
V-α 5’ DNA fragment end sequence (overlaps with linearized vector end that contains the mouse TRAV signal peptide sequence; placed before the V-α segment sequence):
5’-CTGCAGCTGGATGGGATGAGCCAA-3’
V-α 3’ DNA fragment end sequence (overlaps with Cα-P2A fragment; placed after the V-α segment sequence):
5’-ACATCCAGAACCCAGAACCTGCTGTGTAC-3’
Ensure that the entire V-α fragment is in-frame with respect to the vector backbone TRAV signal peptide sequence.

Figure 7. Example of a DNA fragment 1 V-α. DNA fragment 1 starts with the last 8 codons of the murine TRAV signal peptide sequence (green, 5’ DNA fragment end sequence), immediately followed by human TRAV12-3*01 sequence (red), starting with the first codon after the signal peptide sequence, a recombination junction sequence (TC, purple), and TRAJ4*01 sequence (blue). The first nucleotide of the final TRAJ codon (T) will join the first two nucleotides of the constant region (AC, black) to make the last codon of TRAJ. These two AC nucleotides plus subsequent 9 codons of the TRAC segment (black, 3’ DNA fragment end sequence) is added to overlap with DNA fragment 2 Cα-P2A.
Design a nucleotide sequence for DNA Fragment 3 V-β. This fragment consists of the 5’ overlapping sequence with DNA Fragment 2 Cα-P2A, the human TCR β variable gene segment (V-β), and the 3’ overlapping sequence with the linearized vector. The signal peptide sequence is supplied from DNA Fragment 2 Cα-P2A, and thus the V-β segment starts with the first amino acid after the signal peptide sequence of TRBV. Also, the mouse TRBC segment is supplied from the vector, and thus the V-β segment ends with the last amino acid of the joining region (TRBJ). An example DNA Fragment 3 V-β is shown in Figure 8.
Prepare a human TCR β chain sequence of interest from a single cell sequencing or equivalent method. The V-β segment sequence is from the first codon after the signal peptide sequence of TRBV through the last codon of TRBJ. Refer to the IMGT website (http://www.imgt.org/IMGTrepertoire/Proteins/index.php#B) to identify 1) the first codon after the signal peptide sequence of the TRBV segment and 2) the last codon of the TRBJ segment. Note that the TRBJ gene segment contains only the first nucleotide of the last amino acid codon. The last two nucleotides are provided from the TRBC segment, and thus confirmed to include the last nucleotide of TRBJ in the V-β segment.
Add 5’ and 3’ DNA fragment end sequences, corresponding to sequences that overlap the 3’ end of Cα-P2A and the 5’ end of the linearized vector.
V-β 5’ DNA fragment end sequence (overlaps with Cα-P2A that contains the mouse TRBV signal peptide sequence; placed before the V-β segment sequence): GTCTTGTGGAAACAGCACTCATG
V-β 3’ DNA fragment end sequence (overlaps with linearized vector end that contains the mouse TRBC segment; placed after the V-β segment sequence): AGGATCTGAGAAATGTGACTCCACCCAAG
Ensure that the entire V-β fragment is in-frame with respect to the vector backbone TRBV signal peptide sequence.

Figure 8. Example of a DNA fragment 3 V-β. DNA fragment 3 starts with the last 9 codons of the murine TRBV signal peptide sequence (green, 5’ DNA fragment end sequence), immediately followed by human TRBV2*01 sequence (red), starting with the first codon after the signal peptide sequence, a recombination junction sequence (CG, purple), and TRBJ2-5*01 sequence (blue). The first nucleotide of the final TRBJ codon (G) will join the first two nucleotides of the constant region (AG, black) to make the last codon of TRBJ. These two AG nucleotides plus the subsequent 9 codons of the TRBC segment (black, 3’ DNA fragment end sequence) is added to overlap with the linearized vector.
Order V-α and V-β synthetic double-stranded DNA fragments from TWIST Bioscience or Integrated DNA Technologies.
Prepare DNA fragment 2 Cα-P2A.
To make DNA fragment 2 Cα-P2A, digest 5 μg of CaP2AIII_pK18 (Figure 9) with NEB restriction enzymes MscI and AleI-v2 at 37 °C for 4 h to overnight, according to manufacturer’s instructions.
Purify the 523 bp fragment insert (DNA fragment 2 Cα-P2A) by separation on a 2% agarose gel using the QiaQuick Gel Extraction kit according to the manufacturer’s instructions. Elute the Cα-P2A fragment in 25 μl molecular biology grade water.
Column purify the eluted Cα-P2A fragment (second purification) using a QIAGEN MiniElute kit according to manufacturer’s directions, eluting the final products in 10 μl molecular biology grade water.
Check DNA purity and concentration by NanoDrop spectrophotometer and store the purified DNA fragment at -20 °C until needed.
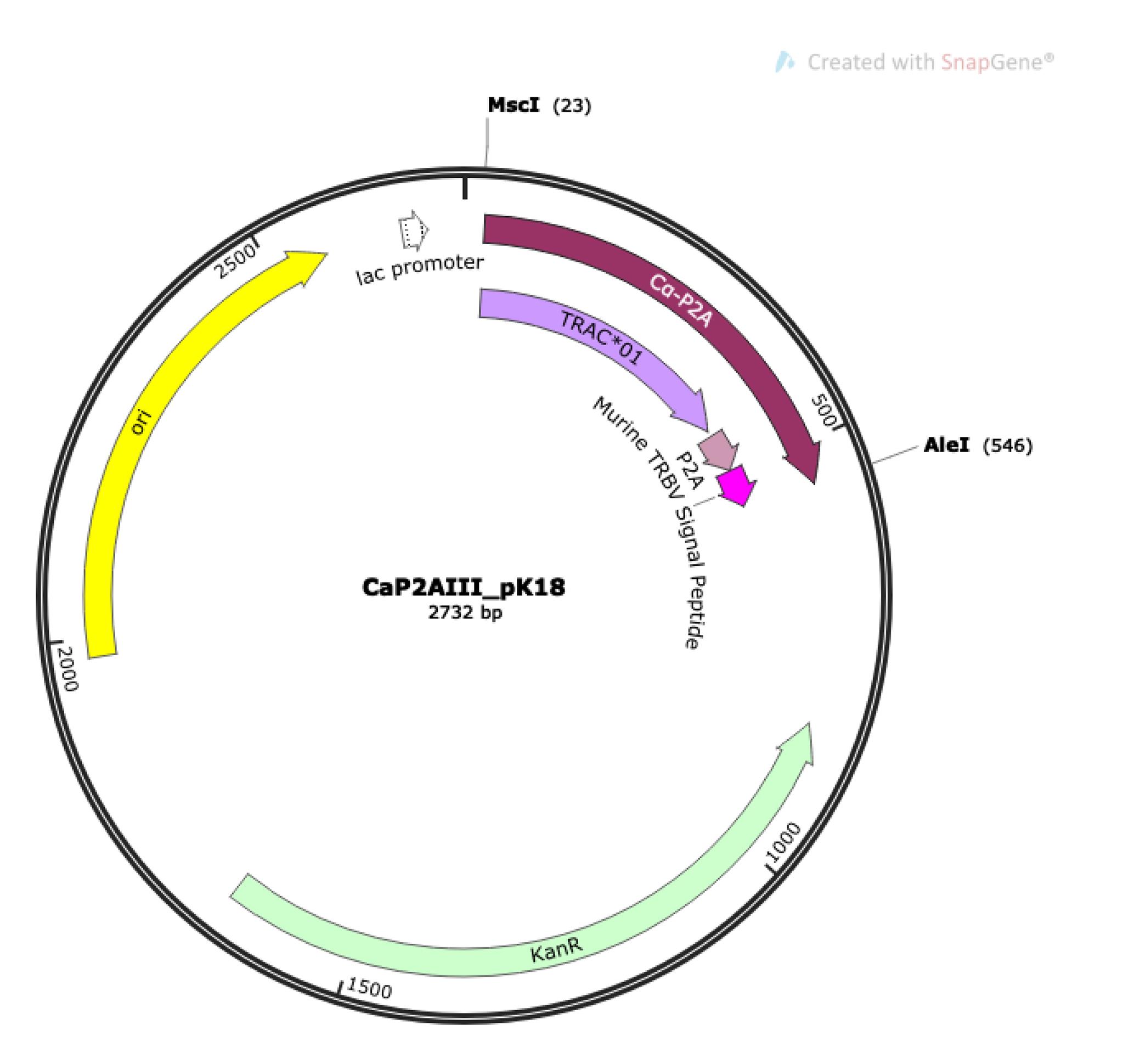
Figure 9. Plasmid map for CaP2AIII_pK18. DNA fragment 2 Cα-P2A (maroon) contains murine TRAC*01 (light purple)-P2A (light pink)-murine TRBV signal peptide sequence (dark pink), inserted between the MscI and AleI restriction digest sites of CaP2AIII_pK18.Prepare MBC1-null or MBC2-null vector backbone. Use MBC1-null for V-β that has TRBJ1-# and MBC2-null for V-β that has TRBJ2-#.
Digest 10 μg of appropriate backbone vector, MBC1-null or MBC2-null (Figure 6A), with NEB SalI and BglII 37 °C for 4 h to overnight, according to manufacturer’s instructions.
Column purify the digested MBC1-null or MBC2-null backbone vector using a QIAGEN MiniElute kit according to manufacturer’s directions, eluting the final products in 10 μl molecular biology grade water.
Check DNA purity and concentration by NanoDrop spectrophotometer and store the purified linearized vector at -20 °C until needed.
Gibson Assembly
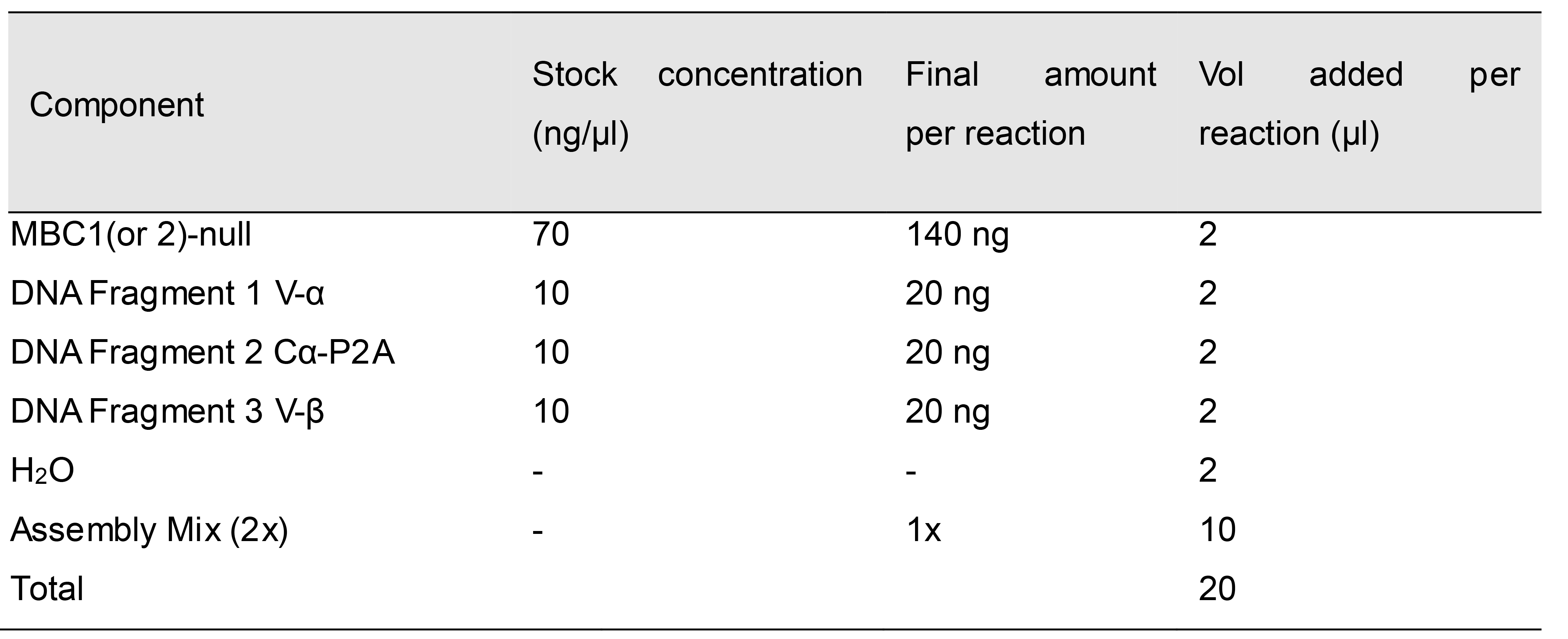
Set up a Gibson assembly reaction according to the reaction ratios in Table 5.
Incubate reactions at 50 °C for 1.5 h, and then hold at 4 °C.
After incubation, reactions can be stored at 4 °C overnight and used to transform NEB 5-alpha Competent E. coli the next day.
Table 5. Gibson assembly reaction master mix
- Transform NEB 5-alpha E. coli as in Step A2a.ix above.
- Grow mini-cultures of individual colonies and perform PCR to verify presence of the DNA fragment insert as in Step A2a.x above. Prepare mini-culture PCR reactions for both α and β chains, as shown in Table 3, with the following primers:
α chain primers:
5pMIG (F Primer): 5’-GCCTCCTCTTCCTCCATCCG-3’
TRAC1-R (R Primer): 5’-CAGGCAGAGGGTGCTGTCCTG-3’
The F Primer (5pMIG) and the R Primer (TRAC1-R) locate before the SalI recognition site in MBC1(or 2)-null and within DNA Fragment 2 Cα-P2A, respectively.
β chain primers:
5MAC3 (F Primer): 5’-AGAGACCAACGCCACCTACC-3’3
TRBC (R Primer): 5’-CAAGGAGACCTTGGGTGGAG-3’
The F Primer (5MAC3) and the R Primer (3TRBC) locate within DNA Fragment 2 Cα-P2A and behind the BglII recognition site in MBC1(or 2)-null, respectively. Carry out PCR according to the conditions given in Table 4, except using an annealing temperature of 60 °C and an elongation time of 1.0 min.
- Visualize PCR products by separation on a 2% agarose gel with 0.5 μg/μl ethidium bromide. Correct DNA fragment insertion will result in a PCR product 600 ± 20 bp for α and 680 ± 20 bp for β.
- Prepare vector for transfection. Proceed to large-scale culture plasmid preparation from the mini-culture of one colony having the correct-sized PCR products from mini-culture PCR for both α and β chains, as in Step A2a.xi.
- Check the vector for correct sequence and alignment of inserted DNA fragment by submitting samples for Sanger sequencing with the primers shown in Table 2 for TCR vector.
- MHC Vectors. Vectors expressing the Major Histocompatibility Class I (MHC I) or Class II (MHC II) genes are transduced into immortalized human myelogenous leukemic cells (K562 cells, ATCC catalog number CCL-243 ) to generate antigen presenting cells (APCs) for use in TCR stimulation assays. As shown in Figure 10, MHC gene cassettes are inserted into the multi-cloning site (MCS) of a non-replicating lentiviral vector containing a ubiquitous chromatin opening element (UCOE)-preceded spleen focus forming virus (SFFV) promoter (pUS, Figure 10A). Figure 10B illustrates MHC I gene vector A2_uSFFV, which is used in the example stimulation assay in part B.
MHC Class I genes require human β2 microglobulin for expression on the cell surface. Porcine teschovirus-1 2A cleaving peptide sequence (P2A; GGCTCCGGAGCCACGAACTTCTCTCTGTTAAAGCAAGCAGGAGACGTGGAAGAAAACCCCGGTCCC) is used to join the MHC I gene to human β2 microglobulin gene sequence. Thus, the MHC Class I gene cassettes consist of the MHC I gene-P2A-β2 microglobulin gene (Figure 10C). For MHC Class II genes, the nucleotide sequence for MHC II α and MHC II β chain genes are joined by P2A. MHC Class II cassettes consist of the MHC II α gene-P2A-MHC II β gene (Figure 10D). MHC Class I and Class II gene cassettes are flanked by DNA fragment ends that overlap with the linearized vector for ligation into pUS by Gibson assembly.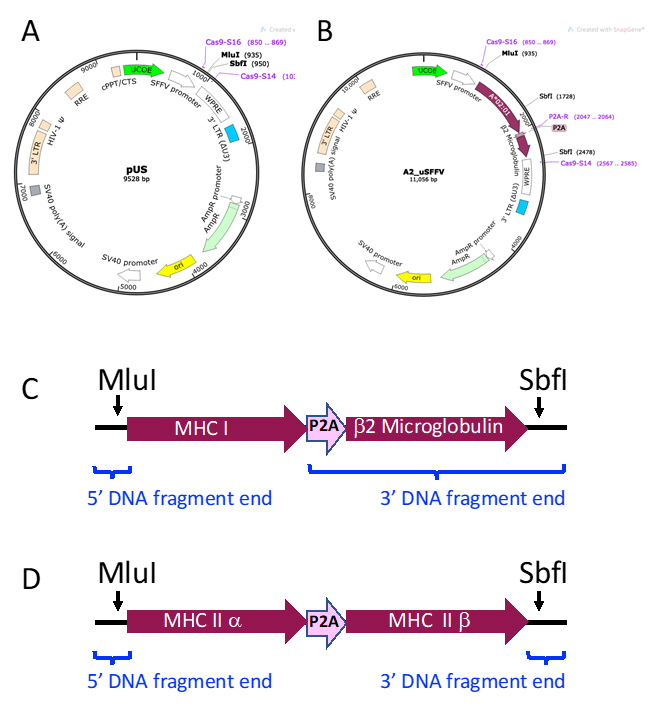
Figure 10. MHC Vector Construction. A. Plasmid map of non-replicating lentiviral backbone vector pUS. A ubiquitous chromatin opening element (UCOE) precedes the spleen focus forming virus (SFFV) promoter (Adamson et al., 2016). An MluI-SbfI cloning site is located after the SFFV promoter. B. Plasmid map of an example MHC I vector, A2_uSFFV. A2_uSFFV vector was constructed by digesting the pUS backbone vector with MluI and SbfI and inserting the HLA-A*02:01 gene-P2A-human β2 microglobulin gene cassette by Gibson assembly. C. Schematic diagram of an MHC I gene-P2A-human β2 microglobulin gene cassette. D. Schematic diagram of an MHC II α gene-P2A-MHC II β gene cassette. The blue brackets indicate the positions of the 5’ and 3’ DNA fragment ends added to MHC gene sequences.Synthesize the MHC gene cassette DNA fragment.
Design a nucleotide sequence for the desired MHC I gene cassette, consisting of the MHC I gene-P2A-human β2 microglobulin gene. Refer to the Immuno Polymorphism Database (https://www.ebi.ac.uk/ipd/) to obtain sequence information for the desired MHC I gene.
- Add the following 5’ and 3’ DNA fragment end sequences before the start codon and after the last amino acid-encoding codon of the MHC I gene sequence (omitting the stop codon), respectively:
5’ MHC Class I DNA fragment end (overlaps with the linearized vector; place before the start codon of the MHC I gene): CTGGAGCTCTCGAGAATTCTCACGCGTCTGCCACC3’ MHC Class I DNA fragment 3’ end (contains P2A followed by the human β2 microglobulin nucleotide sequence; place after the last amino acid codon of the MHC I gene, omitting the MHC I stop codon):
GGCTCCGGAGCCACGAACTTCTCTCTGTTAAAGCAAGCAGGAGACGTGGAAGAAAACCCCGGTCCCATGTCTCGCTCCGTGGCCTTAGCTGTGCTCGCGCTACTCTCTCTTTCTGGCCTGGAGGCTATCCAGCGTACTCCAAAGATTCAGGTTTACTCACGTCATCCAGCAGAGAATGGAAAGTCAAATTTCCTGAATTGCTATGTGTCTGGGTTTCATCCATCCGACATTGAAGTTGACTTACTGAAGAATGGAGAGAGAATTGAAAAAGTGGAGCATTCAGACTTGTCTTTCAGCAAGGACTGGTCTTTCTATCTCTTGTACTACACTGAATTCACCCCCACTGAAAAAGATGAGTATGCCTGCCGTGTGAACCATGTGACTTTGTCACAGCCCAAGATAGTTAAGTGGGATCGAGACATGTAACGACCTGCAGGCATGCAAGCTTGATATCAAG
- For MHC Class II gene expression, nucleotide sequence is needed both for α and β genes, connected by P2A. Design a nucleotide sequence for the desired MHC II gene cassette, consisting of the MHC II α gene-P2A-MHC II β gene (shown schematically in Figure 10D) by adding the following DNA sequences to the MHC II α and β genes:
5’ MHC Class II α DNA fragment end (overlaps with the linearized vector; placed before the MHC II α gene start codon): CTGGAGCTCTCGAGAATTCTCACGCGTCTGCCACC
P2A (placed between the 3’ end of the MHC II α gene, omitting the stop codon, and the start codon of the MHC II β gene):
5’-GGCTCCGGAGCCACGAACTTCTCTCTGTTAAAGCAAGCAGGAGACGTGGAAGAAAACCCCGGTCCC-3’
Amino Acid sequence of P2A: GSGATNFSLLKQAGDVEENPGP
3’ MHC Class II DNA fragment end (overlaps with the linearized vector; placed after the MHC II β gene stop codon): CGACCTGCAGGCATGCAAGCTTGATATCAAG Order the MHC gene cassette DNA fragment from TWIST Biosciences or Integrated DNA Technologies. Reconstitute the DNA fragment in molecular biology grade water at 10 ng/μl.
- Digest 5 μg pUS with NEB MluI-HF and SbfI according to the manufacturer’s instructions.
- Purify the digested pUS backbone vector using a QIAGEN MiniElute kit according to manufacturer’s directions, eluting the final product in 10 μl molecular biology grade water.
- Quantitate the backbone vector by NanoDrop spectrophotometry and adjust concentration to 75 ng/μl with molecular biology grade water.
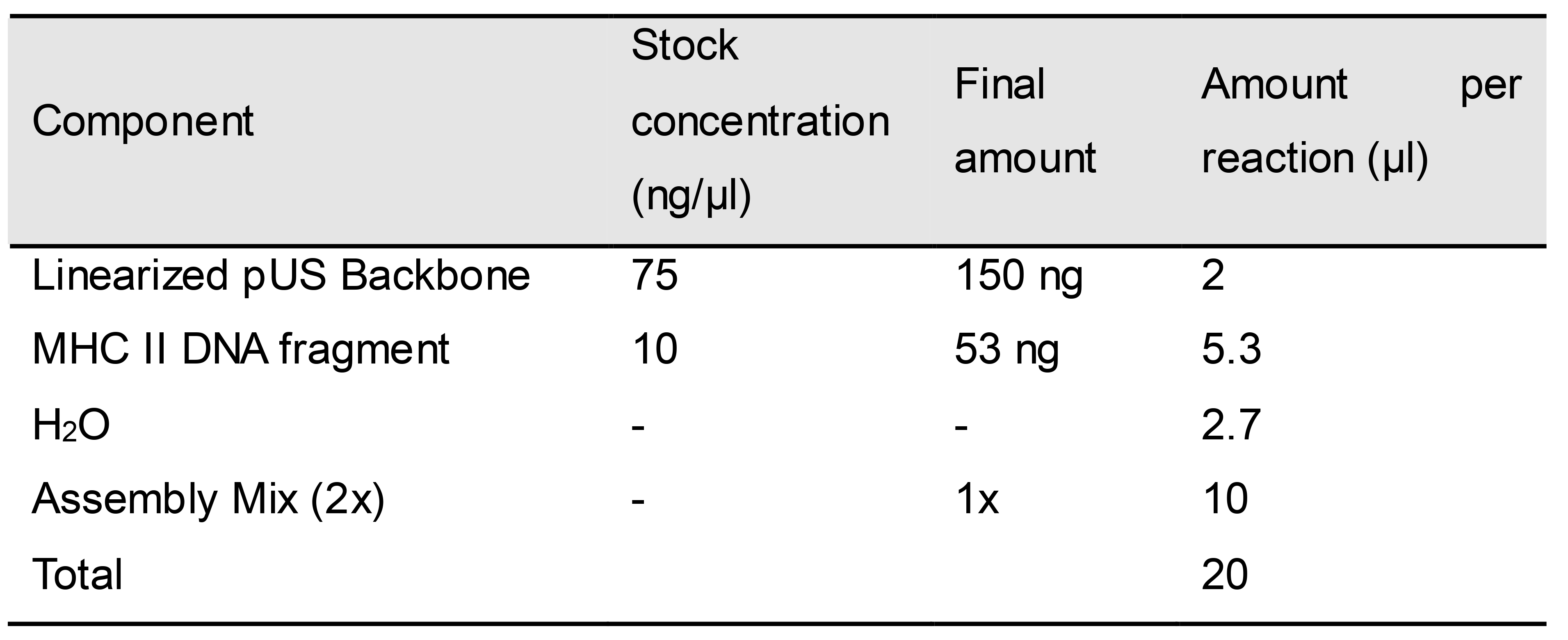
- Set up a Gibson assembly reaction according to the reaction ratios shown in Tables 6 and 7.
Table 6. Gibson assembly master mix for MHC Class I DNA fragment
Table 7. Gibson assembly master mix for MHC Class II DNA fragment
Incubate the Gibson assembly reactions at 50 °C for 1.5 h, and then hold at 4 °C.
After incubation, this reaction can be stored at 4 °C overnight and used to transform NEB 5-alpha Competent E. coli the next day.
Transform NEB 5-alpha E. coli as in Step A2a.ix above.
Grow mini-cultures of individual colonies and perform PCR to verify presence of the DNA fragment insert as in Step A2a.x above. Prepare mini-culture PCR reactions as in Table 3, with the following primers:
Cas9-S16 (F Primer): 5’-AAGAGCTCACAACCCCTCAC-3’
Cas9-S14 (R Primer): 5’-CACATAGCGTAAAAGGAGC-3’
The F Primer (Cas9-S16) and the R Primer (Cas9-S14) locate within before and behind the MCS of the pUS vector, respectively.
Carry out PCR using the conditions given in Table 4, except using an annealing temperature of 50 °C and an elongation time of 2.5 min.
Visualize PCR products by separation on a 1% agarose gel with 0.5 μg/μl ethidium bromide. Correct DNA fragment insertion will result in a PCR product 1,740 ± 20 bp for MCH I vectors and 1,840 ± 20 bp for MCH II vectors.
Prepare vector for transfection. Proceed to large-scale culture plasmid preparation from the mini-culture of one colony containing the correct-sized PCR products from mini-culture PCR, as in Step A2a.xi.
Check the vector for correct sequence and alignment of inserted DNA fragment by submitting samples for Sanger sequencing with the primers shown in Table 2 for MHC vectors.
Cell Lines
Overview
Cell lines are generated using transfection and transduction with both retroviral and lentiviral systems. All vectors for the generation of TCR cell lines are retroviral, while the vectors used for MHC cell lines are lentiviral. The MSCV promoter in the retrovial vectors used here provides robust expression in 5KC cells, with reduced silencing of transgene expression found with other retroviral systems in hematopoietic cells. The lentiviral vector uses a spleen focus forming virus (SFFV) promoter preceded by a ubiquitous chromatin opening element (UCOE) to reduce gene silencing. We have empirically found that UCOE-SFFV lentiviral system gives higher transgene expression than the MSCV system in K562 cells. Tables 8 and 9 give an overview of the cells and reagents used in both retroviral and lentiviral transfections and transductions. Figure 11 provides a graphical representation of the steps and timeline for transfections and transductions.
Note that all transfections and transductions should be handled at biosafety level 2 in the United States. Investigators in other countries should confirm biosafety restrictions for their institutions.
We strongly recommend optimizing cell line growth conditions before starting transfection and transduction experiments, including selecting well-qualified FBS lots and culturing cells at the proper concentration. See Notes 7 and 8 in the Notes section for detailed recommendations.
Table 8. Reagents and cell types needed for retroviral and lentiviral transfections
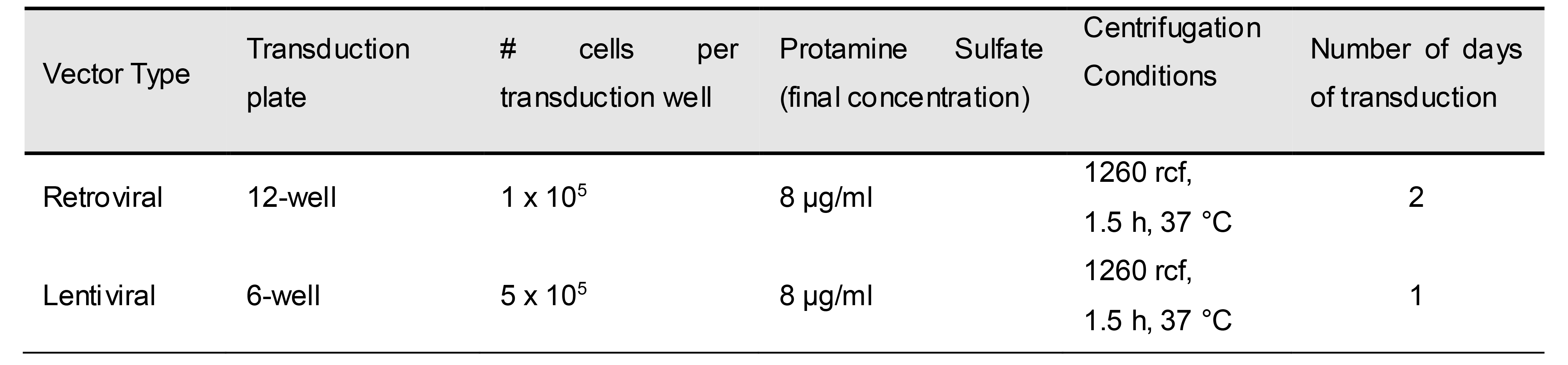
Table 9. Transduction conditions for retroviral and lentiviral vectors
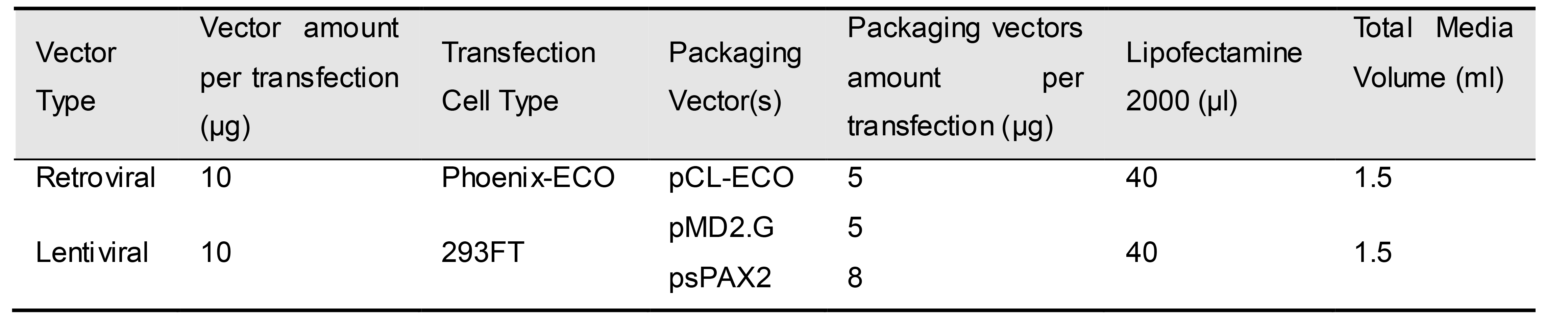
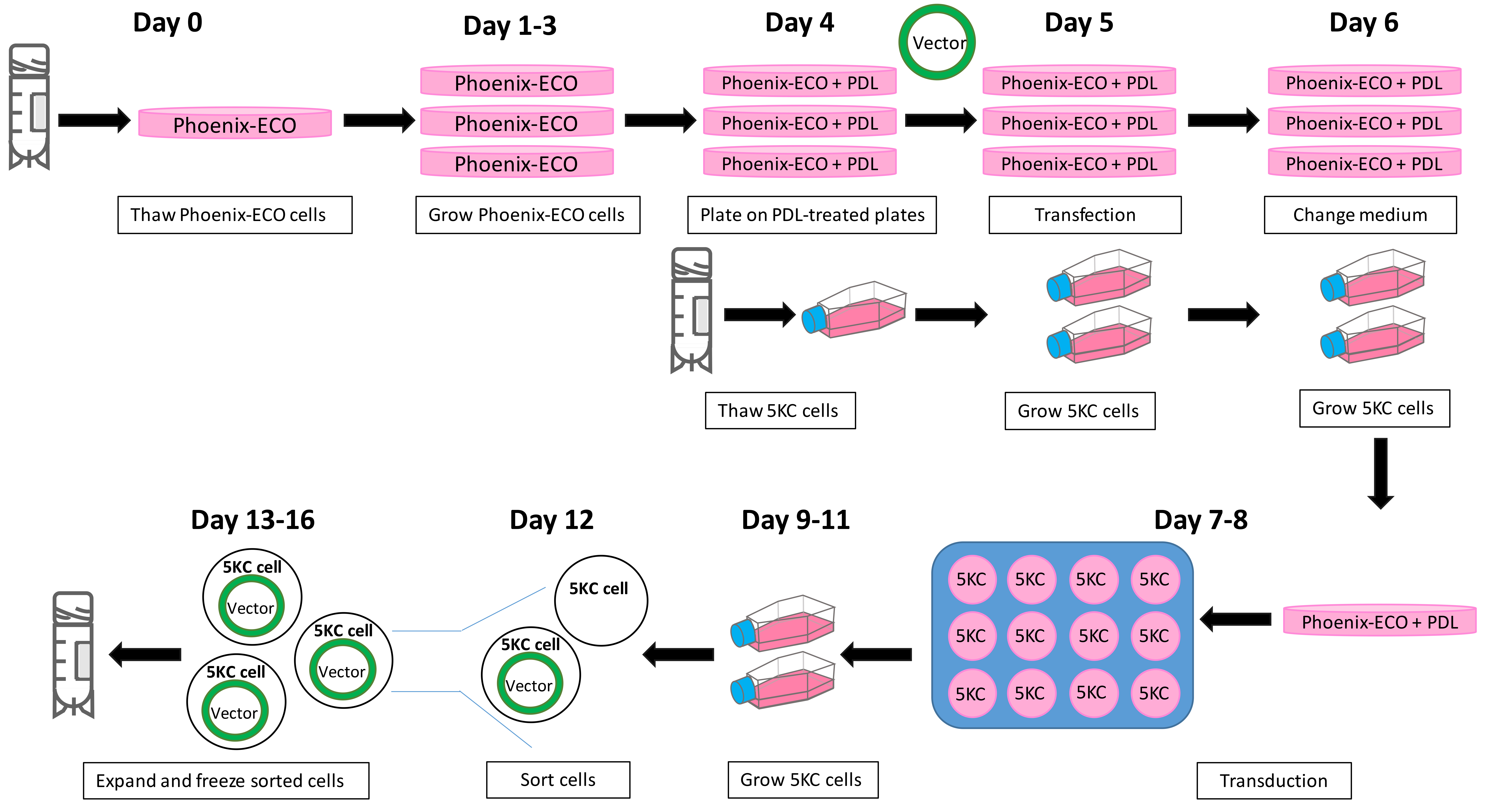
Figure 11. Overview of the timeline and workflow to generate cell lines via transfection and transduction. The retroviral system is shown as an example.
T-cell lines
Figure 12 illustrates the vectors added during each round of transduction. The final products of these transductions are color-coded reporter T-cell lines.
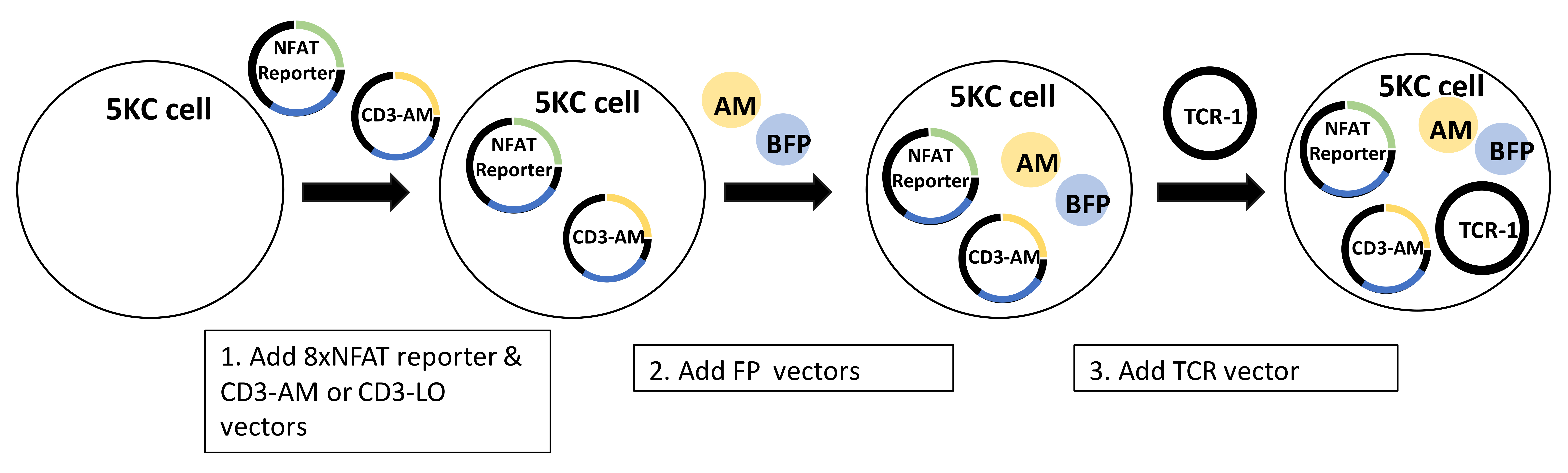
Figure 12. Overview of the procedure for creating TCR-expressing NFAT-reporter 5KC α-β- cells for multiplexing. In step 1, the hCD8-8xNFAT reporter vector (8xNFAT-ZsG-hCD8, generated in Step A2) and CD3 vectors containing either the ametrine (AM) or LSSmOrange (LO) fluorescent protein gene (CD3-AM or CD3-LO, generated in Step A3) are co-transduced into 5KC α-β- cells. In the second step, either AM or LO fluorescent protein vectors (pMSCV-AM or pMSCV-LO) are transduced with one of four other fluorescent protein vectors (generated in Step A4). Finally, a TCR vector (generated in Step A5) is added to the cells containing the 8xNFAT reporter, hCD8, CD3, and fluorescent identifiers.Make cell lines expressing the 8xNFAT reporter, human CD8, and CD3 genes along with either LSSmOrange (LO) or Ametrine (AM). This is achieved by introducing two vectors, 8xNFAT-ZsG-hCD8 and CD3-AM or CD3-LO, into 5KC α-β- cells. (Figure 12, step 1).
Transfection. Note that Table 8 lists the cells and reagents used in retroviral transfections.
Grow Phoenix-ECO cells (ATCC CRL-3214 ) in 10-cm tissue culture dishes with Phoenix medium (Recipe 3) at 37 °C, 5% CO2. Cells should be seeded at a density that will not exceed 80% confluency in 24 h. Subculture cells daily according to ATCC directions.
Treat 10-cm tissue culture dishes with poly-D-lysine (PDL) as follows on the day before transfection: Add 3 ml of 0.1 mg/ml PDL solution (Recipe 7) to dish, covering the bottom of the dish completely, and incubate at room temperature for 5 min.
Remove PDL solution and wash plate two times with 10 ml PBS. Leave second PBS wash on plate until cells are ready to be plated. Alternatively, follow instructions for PDL plate preparation recommended by Sigma (https://www.sigmaaldrich.com/catalog/product/sigma/p6407?lang=en®ion=US).
Collect Phoenix cells by removing with Trypsin-EDTA and centrifuging at 168 rcf at room temperature for 2 min. Remove medium above the cell pellet and resuspend cells in fresh Phoenix media at 1.5 x 106 cells per ml.
Aspirate PBS from PDL-treated plates and replace with 8 ml fresh Phoenix medium.
Add 2 ml Phoenix cells per plate, for a final concentration of 3 x 106 cells in 10 ml, and distribute by gently rocking plate.
Return cells to CO2 incubator and culture overnight. Perform transfection the following day.
Note: For each reporter cell line to be made, prepare two PDL-treated plates of Phoenix: one for transfection with 8xNFAT-ZsG-hCD8 and the other with CD3-AM or CD3-LO vector.Add 1.5 ml of transfection medium (Recipe 1) to two 14-ml round-bottom tubes for each transfection.
Add 5 μg of the packaging vector pCL-ECO and 10 μg of the retroviral vector to one tube. Mix 3x by pipetting.
Add 40 μl Lipofectamine 2000 to the other tube and mix 3 x by slow, gentle pipetting with a P1000 pipettor. Incubate at room temperature for 5 min.
Add the lipofectamine solution into the DNA solution dropwise using a P1000 pipettor, not allowing drops to touch the side of the tube. Mix 3 x by slow, gentle pipetting.
Incubate for 30 min at room temperature.
Aspirate the culture medium from each PDL-treated Phoenix cell plate and wash with 4 ml of prewarmed transfection medium. Gently rock the plates once and aspirate transfection medium. Add 7 ml transfection medium and return plates to CO2 incubator.
Note: This step should be performed during the 30-minute incubation.Add the DNA-lipofectamine solution directly onto the prepared Phoenix plate by slow, drop-wise pipetting.
Return plate to CO2 incubator for 6 h.
Add 1 ml FBS to each plate dropwise. Rock plate gently 1-2 times to distribute FBS and return to incubator overnight.
Remove medium from each plate and replace with 5 ml prewarmed Phoenix medium the following morning. Return to CO2 incubator and culture overnight.
- Transduction. Note that Table 9 lists the conditions used in retroviral transductions.
Thaw 5KC α-β- cells on the same day that Phoenix cells are plated on PDL plates. Place a cryovial containing 1 x 106 cells in 37 °C water bath until the last bit of ice is gone. Carefully transfer cells into a 15-ml centrifuge tube containing 10 to 12 ml 5KC medium (Recipe 2) using wide bore pipette tip.
Centrifuge cells at 315 rcf at room temperature for 3 min.
Remove medium above the cell pellet, gently resuspend cells, and transfer to a T25 flask containing a total of 10 ml of medium. Grow in CO2 incubator overnight.
Subculture 5KC α-β- cells every day prior to transduction. Seed cells at 5 x 104 cells per ml in an appropriately sized (T25 or T75) culture flask.
Pre-warm table-top centrifuge to 37 °C while preparing transduction plate with 5KC α-β- cells and retroviral supernatants:
Collect 5KC α-β- cells by centrifugation and resuspend in fresh 5KC medium at 1x105 cells per ml.
Add 1 ml of cell suspension to one well of a 12-well plate. Prepare one well for each cell line to be made.
Add 4.8 μl of 5 mg/ml protamine sulfate (Recipe 6) to each well for a final concentration of 8 μg/ml in 3 ml.
Remove 5 ml of medium from each transfected Phoenix cell plate using a disposable syringe and filter through a 0.45 μm syringe filter into a 14-ml round-bottom culture tube. This is now referred to as the retroviral supernatant. To continue producing retroviruses for the second day of transduction, add 5 ml of prewarmed Phoenix medium back to each Phoenix cell plate and return to incubator.
Add 1 ml of 8xNFAT-ZsG-hCD8 and 1 ml of CD3-AM or CD3-LO retroviral supernatants into 5KC α-β- cells in each transduction well on the 12-well plate made in step (ii). An example plate map is shown in Figure 13.
Centrifuge the plate with cells and retroviral supernatants at 1,048 rcf, 37 °C for 1.5 h in pre-warmed table-top centrifuge.
Remove 1.5 ml of medium from each transduction well, avoiding the removal of cells as much as possible. Replace with 1.5 ml of fresh 5KC medium and return to incubator overnight.
Repeat transduction the next day by removing 1.5 ml of medium from each well of the transduction plate and adding 5.6 μl of protamine sulfate (for final concentration of 8 μg/ml in 3.5 ml) directly to each well. Collect and filter retroviral supernatants as before and add 1 ml each of 8xNFAT-ZsG-hCD8 and CD3-AM or CD3-LO retroviral supernatants to each well as before. Discard Phoenix transfection plates in biosafety level 2 waste.
Centrifuge plate and replace media as described in Steps B2b.ii.6 and B2b.ii.7 above.
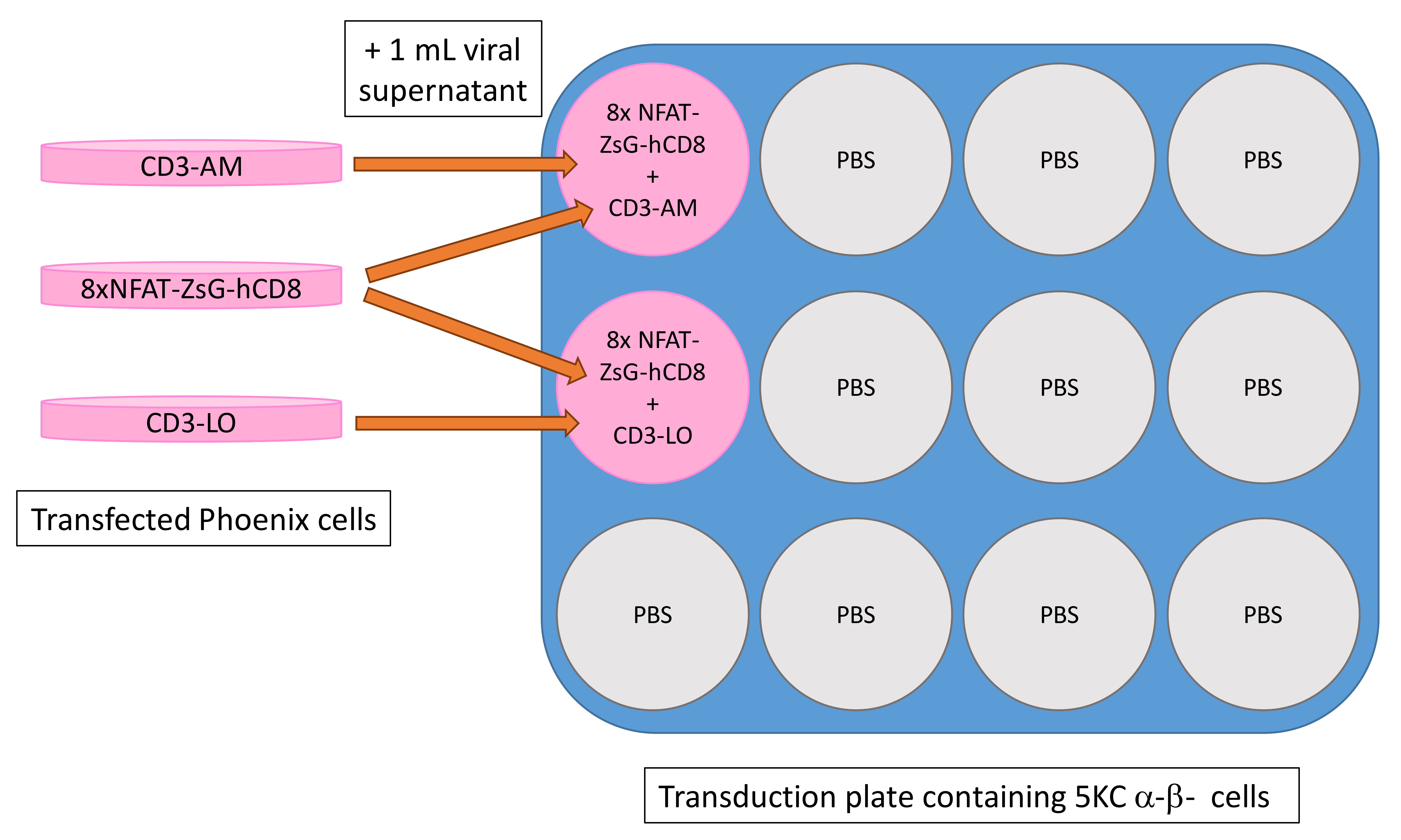
Figure 13. Schematic of plate layout for transduction with 8xNFAT-ZsG-hCD8 and CD3-LO or CD3-AM. Note that each transduction well receives retroviral supernatants from two transduction plates. Orange arrows represent the addition of retroviral supernatant from transfected Phoenix plates to 5KC α-β- cells on transduction plate. Unused wells are filled with 3 ml phosphate-buffered saline (PBS).
- Expand and sort transduced cells
Subculture transduced 5KC α-β- cells every day for 3 days, keeping all cells and seeding at 5 x 104 cells per ml in an appropriately sized (T25 or T75) culture flask. Ensure three complete media exchanges so that retroviral particles are sufficiently removed from the medium.
Collect all cells and stain with PE-labeled anti-human CD8 antibody according to manufacturer’s instructions.
Resuspend cells in 500 to 1,000 μl of Phenol red-free RPMI (Recipe 5) and flow sort to collect PE+AM+ or PE+LO+ double positive cells, as shown in Figure 14.
Note: Use stringent gating to ensure purity at this step.Centrifuge the sorted cells at 315 rcf at room temperature for 3 min and remove sorting buffer. Resuspend cells at 1 × 105 cells per ml in 5KC medium and place in appropriately sized culture vessel.
Subculture cells each day for 3 days, expanding to larger culture vessels as necessary.
Evaluate expression of AM or LO and hCD8 by staining with PE-labeled anti-human CD8 antibody followed by flow cytometry analysis. Greater than 97% of cells should be expressing both the CD8 protein and the CD3 fluorescent marker protein (AM or LO). 5KC α-β- cells transduced with 8xNFAT-ZsG-hCD8 and CD3-AM or CD3-LO are henceforth referred as 8xNFAT-AM and 8xNFAT-LO cells, respectively.
[Break point and optional freezing] Freeze cells once they have grown sufficiently to make several vials at 1 x 106 cells per vial.
Collect cells and spin at 315 rcf at room temperature for 3 min. Remove medium and gently tap tube to dislodge cell pellet.
Resuspend at 1 x 106 cells/ml in freezing medium (Recipe 4).
Aliquot 1 ml of cell suspension into each labeled cryovial and immediately place in Mr. Frosty cell freezing device. Allow cells to slow-freeze in an ultra-low (-80 °C) freezer, according to manufacturer’s instructions before moving to permanent storage location.
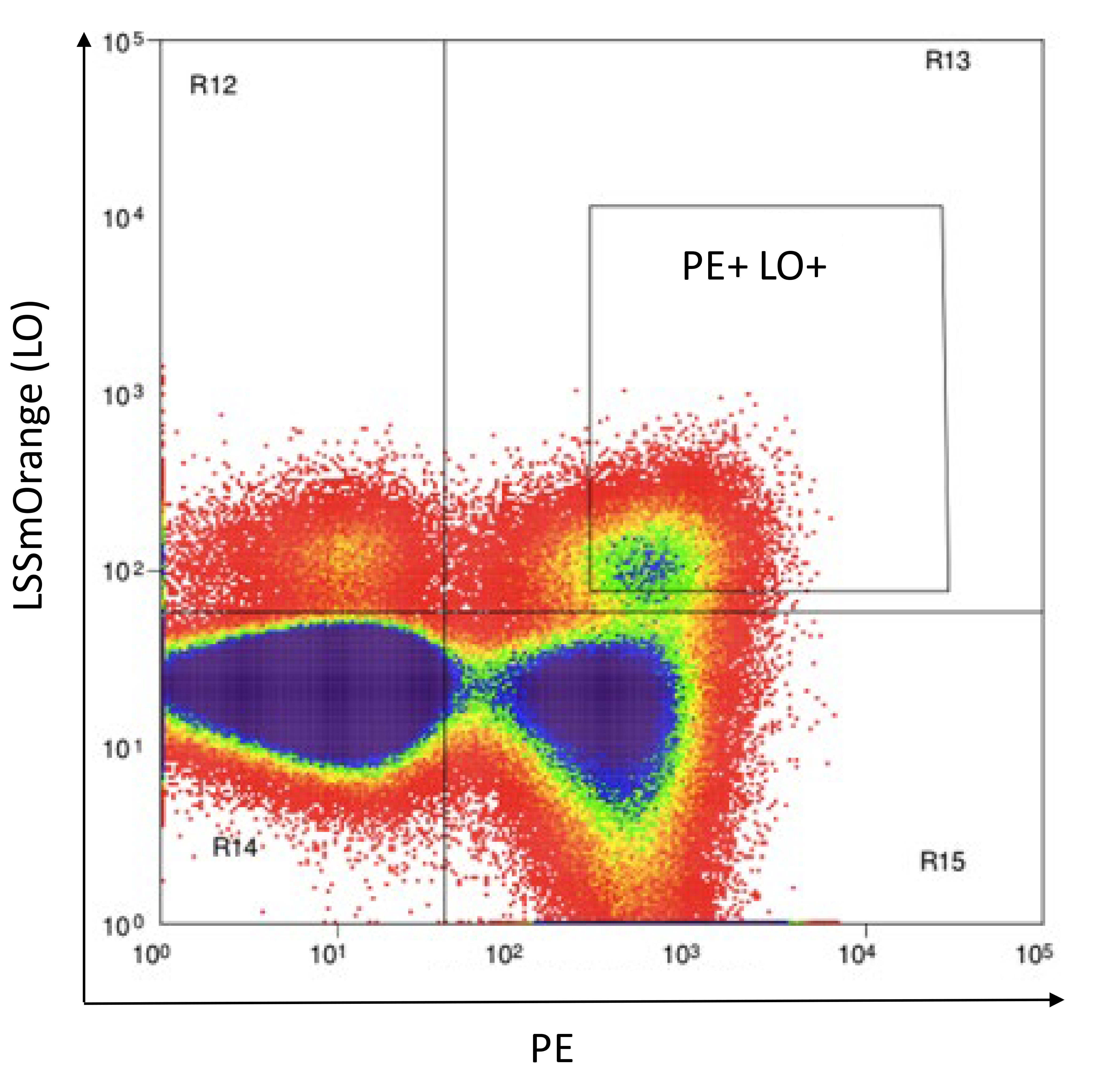
Figure 14. Example flow sorting plot for the transduction represented in Figure 13. 5KC α-β- cells transduced with 8xNFAT-ZsG-hCD8 and either CD3-LO or CD3-AM are stained with PE-labeled anti-hCD8 antibody. After gating on live cells using the FSC vs. SSC scatter plot, double positive cells (PE and AM or PE and LO) are sorted. LSSmOrange is shown in the figure as an example.Adding fluorescent protein identifiers to 8xNFAT-AM and 8xNFAT-LO 5KC α-β- cell lines to generate color-coded TCR expression cell lines (Figure 12, step 2).
Note that AM and LO expression by CD3-AM and CD3-LO is relatively weak. Therefore, transducing cells with additional vectors encoding the AM or LO gene, pMSCVII-AM (pMSCV-AM) or pMSCVII-LO (pMSCV-LO), drives intense fluorescent protein expression. Another fluorescent protein is co-transduced, resulting in eight color combinations (Figure 15).
Transfection
Prepare one PDL-treated plate of Phoenix cells for each of the following fluorescent protein vectors and transfect according to the steps described in Step B2b.i.Fluorescent protein vectors: pMSCV-AM, pMSCV-LO, pMSCVII-BFP (pMSCV-BFP), pMSCVII-tdTomato (pMSCV-TM), pMSCVII-E2Crimson (pMSCV-CR), pMSCVII-mChe (pMSCV-mChe).
Note: Reagents for retroviral transfections can be found in Table 8.Transduction
Transduce the 8xNFAT-AM and 8xNFAT-LO cell lines with fluorescent protein vectors according to the plate map shown in Figure 15. The 8xNFAT-AM cell line is transduced with pMSCV-AM along with pMSCV-BFP, pMSCV-TM, pMSCV-CR, or pMSCV-mChe. Likewise, the 8xNFAT-LO cell line is transduced with pMSCV-LO along with each of the remaining four fluorescent protein vectors. Follow steps in Step B2b.ii to perform transduction.
Note: Reagents for retroviral transductions can be found in Table 9.Repeat transduction the following day.
Expand transduced 5KC α-β- cells for 3 days, sub-culturing each day as described in Step B2b.iii.
After 3 days, collect all cells by centrifugation and remove medium above pellet. Resuspend cells in 500 to 1,000 μl Phenol red-free RPMI and flow sort to purify cells that are double positive for both fluorescent proteins (e.g., AM+BFP+).
Note: There will likely be two AM-positive or LO-positive populations due to the preexisting AM or LO expression from the CD3 vectors. Collect only the AM-High or LO-High population. An example of LO-High cells is shown in Figure 16.Subculture cells for 3 days, expanding to larger culture vessels as necessary.
Evaluate fluorescent protein expression by flow cytometry analysis. Greater than 97% of cells should express AM or LO and the secondary color (BFP, TM, CR, or mChe).
Freeze 12-16 vials of each color-coded reporter cell line at 1 x 106 cells per vial, following procedure described in Step B2b.iii.7, above.
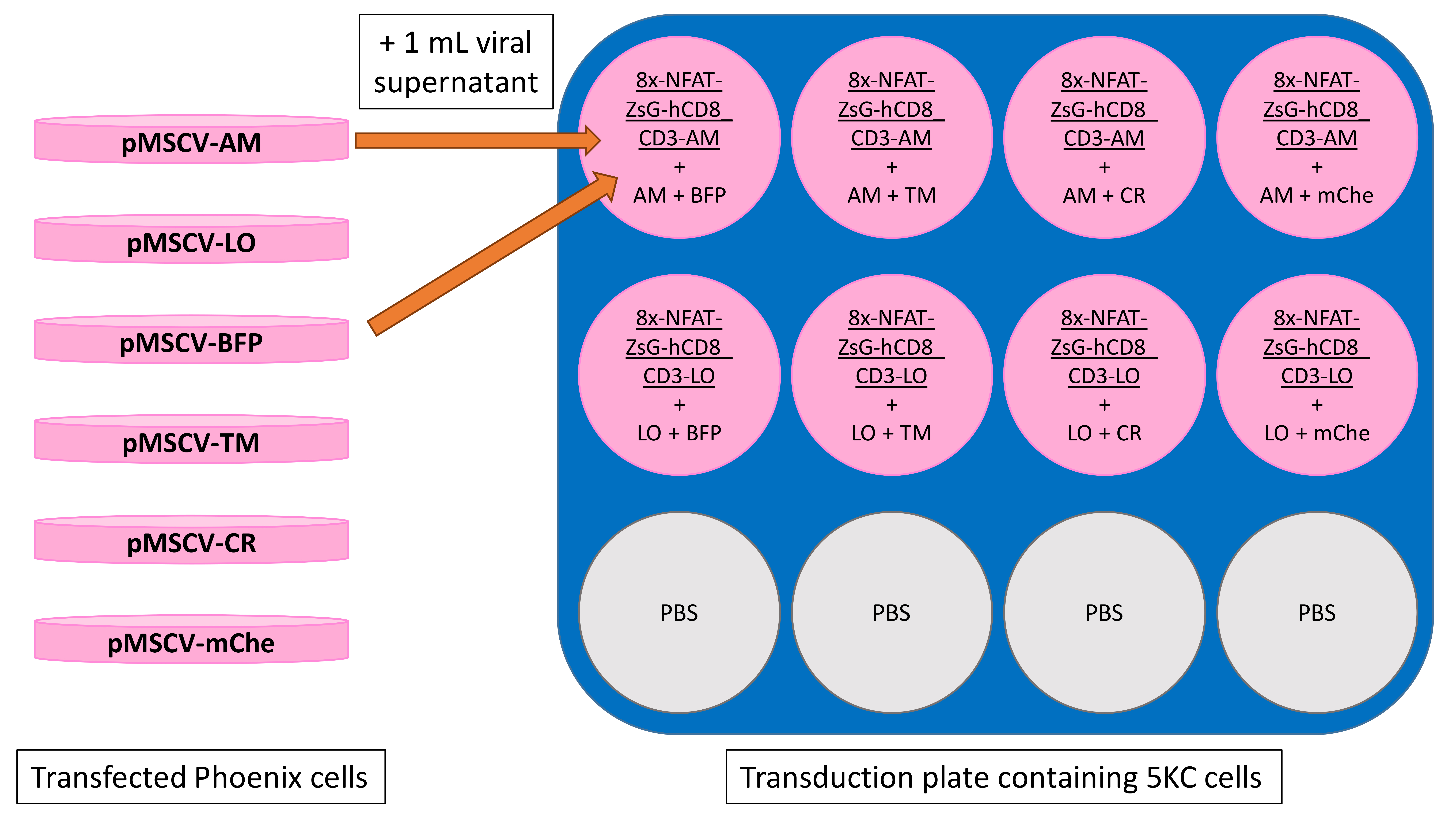
Figure 15. Plate layout for transduction of identifying fluorescent proteins. Note that each transduction well receives 1 ml of each retroviral supernatant from two transfection plates. Representative arrows demonstrate the addition of both pMSCV-AM and pMSCV-BFP retroviral supernatants to well A1. Underlined text at the tops of wells represents the 8xNFAT-CD3-AM and 8xNFAT-CD3-LO cell lines being transduced.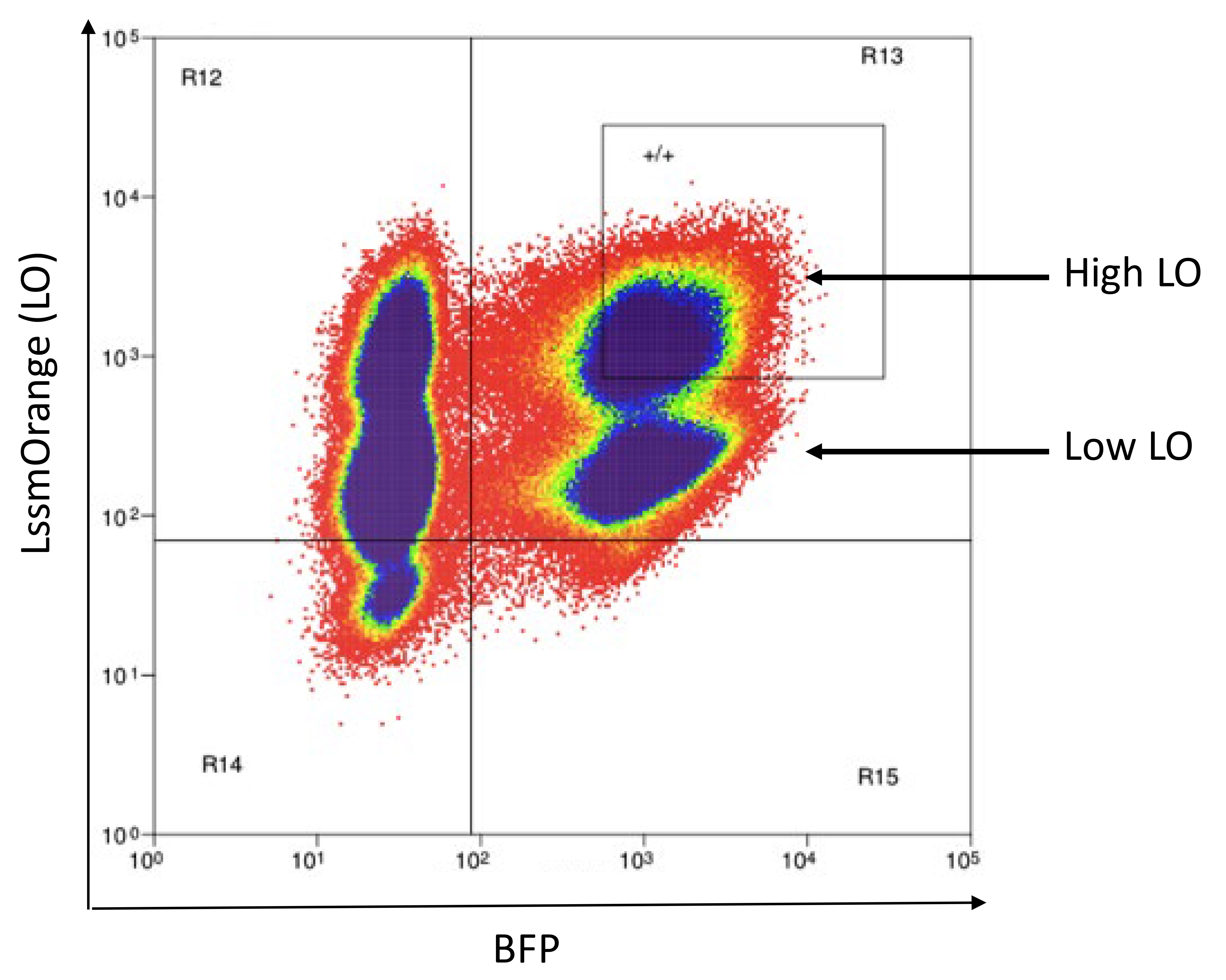
Figure 16. Example flow sorting plot for the LO-High/BFP+ transduction represented in Figure 15. LO is already present in cells as part of the CD3-LO vectors added in Step 1. pMSCV-LO is added in this step to ensure sufficient color expression. LO-High/BFP+ cells are collected, while LO-Low/BFP+ cells are discarded because they were not transduced with the pMSCV-LO vector. See notes 9 and 10 for details.
- Adding TCR vectors to generate 5KC-TCR reporter cell lines (Figure 12, step 3)
Transfection
Prepare one PDL-treated plate of Phoenix cells for each TCR vector to be added and transfect according to the steps described in Step B2b.i.
Note: Reagents for retroviral transfections can be found in Table 8.Transduce the 8 color-coded reporter cell lines with TCR vectors according to the plate map shown in Figure 17. Follow steps in Step B2b.ii, adding 2 ml of retroviral supernatant per well.
Note: Reagents for retroviral transductions can be found in Table 9.Repeat transduction the following day.
Expand transduced 5KC cells for 3 days, sub-culturing each day.
Collect cells and enrich CD3+ cells using a Miltenyi MACS mouse CD3e microbead kit with MS columns, according to the manufacturer’s instructions. Before separating, reserve small portions of pre-enriched cells in a 6-well plate for flow cytometry analysis.
Note: Phenol red-free RPMI can be used in place of MACS buffer to improve cell condition and viability.Stain enriched and pre-enriched 5KC-TCR cells with PE-labeled anti-mouse CD3e antibody according to the manufacturer’s instructions and evaluate CD3 expression via flow cytometry.
Note: Cells successfully transduced with a functional TCR will express CD3 on their surface. Typically, 20%-40% of pre-enriched 5KC cells express CD3, but as low as 10% is still useable. For post-enriched cells, >97% should express CD3.Seed cells at a density of 1x105 cells per ml in an appropriately sized culture vessel for the first culturing period after MACS separation.
Expand and freeze sorted cells by following steps described in B2b.iii.7, above.
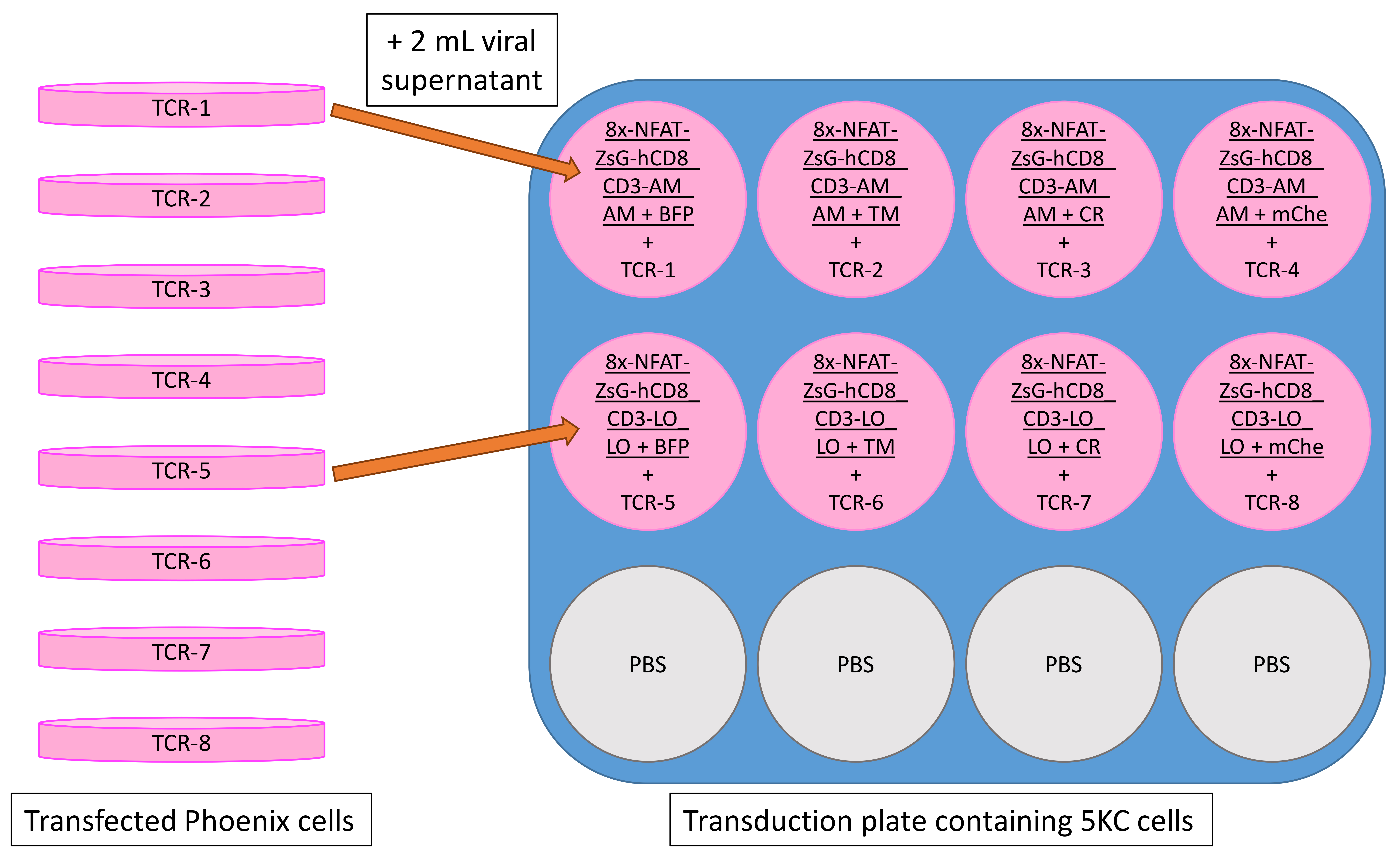
Figure 17. Schematic of transduction plate for introducing TCRs to generate color-coded 5KC TCR reporter cells. Underlined text represents vectors that have already been introduced to parental cells. Note that each TCR is transduced into cells with different color combinations, so that individual TCRs can be distinguished by flow cytometry.
- K562-APC lines
MHC vectors for expression of MHCI or MHCII genes are transduced into human myelogenous leukemic K562 cells (ATCC CCL-243 ) via spin-infection as described above, except using a lentiviral packaging system instead of a retroviral system (Tables 8 and 9). The MHC vector A2_uSFFV is used as an example in this protocol for the generation of K562 cells expressing HLA-A*02:01.
Transfection
Grow 293FT cells (Fisher R70007 ) in 10-cm culture dishes in Phoenix medium. Subculture daily according to manufacturer’s instructions.
Prepare one PDL-treated plate of 293FT cells for each MHC expression vector and transfect using the lentiviral system, reagents for which can be found in Table 8.
Note: The transfection procedure for the lentiviral system follows the steps described in Step B2b.i, except using cell lines and packaging vectors as outlined in Table 8, lentiviral conditions.Grow transfected cells overnight and exchange medium the following morning.
- Transduction
Thaw K562 cells (ATCC CCL-243 ) on the same day as (or a day before) PDL plating as described in Step B2b.ii and subculture each day according to the manufacturer’s instructions.
Prepare a 6-well transduction plate seeded with 5 x 105 K562 cells in each well according to the parameters for lentiviral transduction given in Table 9. Prepare one well for each transduction.
Prepare lentiviral supernatant following the steps described for retroviral transduction, add 2 ml of lentiviral supernatant to each well of transduction plate, and perform one day of transduction according to parameters given in Table 9.
Expand transduced K562 cells for 3 days, seeding cells at 1 x 105 cells per ml each day and transferring to appropriately sized culture vessel.
Stain cells with PE-labeled HLA-ABC antibody according to the manufacturer’s instructions and assess transduction efficiency (usually >95%) by flow.
Note: Sorting is not usually required for K562 cells after transduction because the lentiviral system often results in a transduction efficiency of >95%.Freeze cells as soon as there are enough to make 8 to 12 vials at 2 x 106 cells/vial, following steps described in B2b.iii.7, above.
Stimulation assay
Overview
Up to 8 different 5KC-TCR reporter lines, identified by their expression of different combinations of fluorescent proteins, are mixed and co-cultured with APCs in the presence or absence of antigens or anti-CD3 antibody. Flow cytometry is used to detect cell response to stimulation (represented by ZsGreen-1 expression) 18 to 32 h after addition of 5KC-TCR cells. Figure 18 shows the schedule for a stimulation assay experiment and a schematic for plate set up.
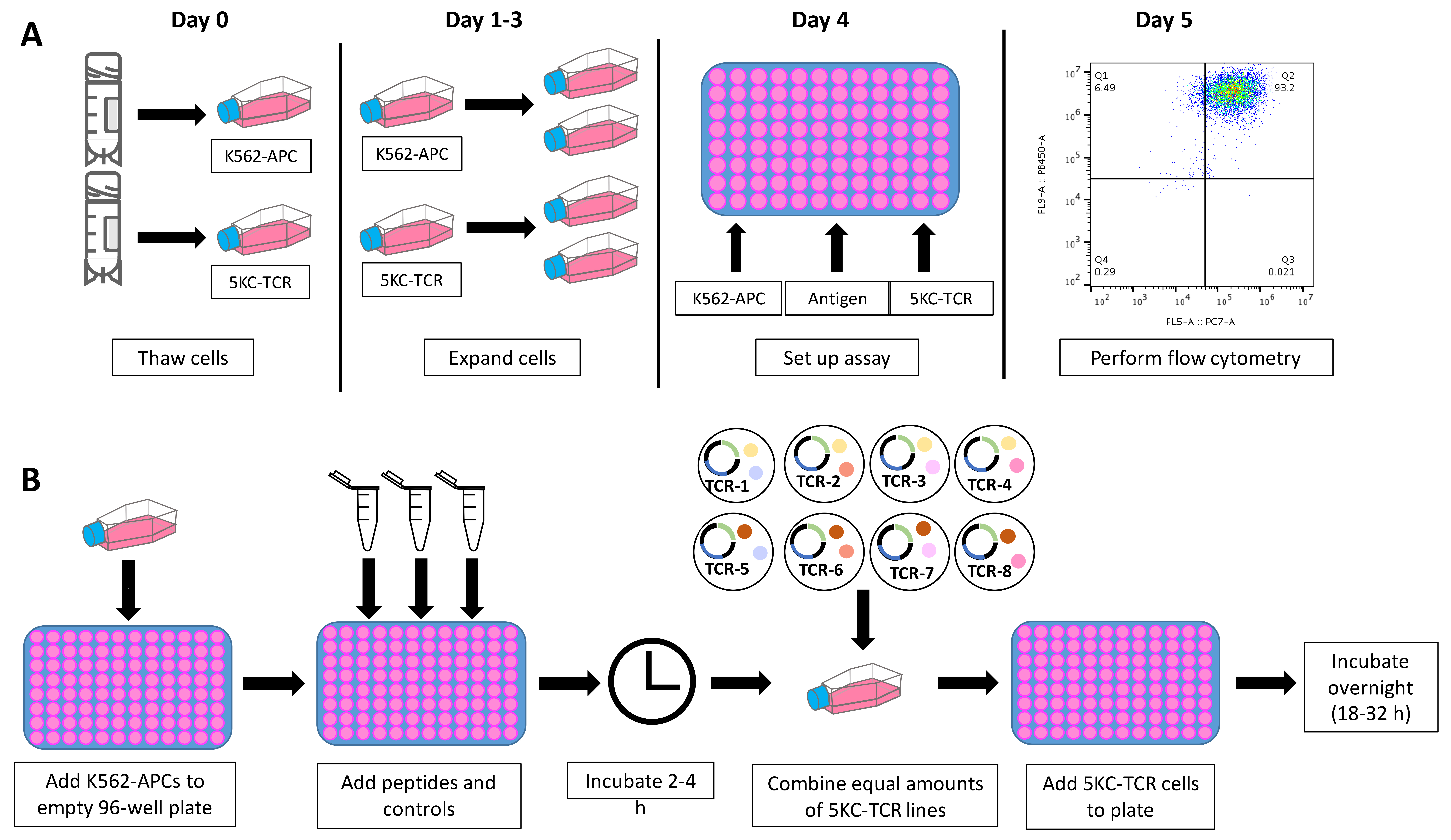
Figure 18. Stimulation assay workflow and plate setup. A. Schedule of stimulation assay experiments, beginning with thawing all 5KC-TCR reporter cell lines and their corresponding K562-APC lines at day 0. Stimulation assays must be set up no earlier than 4 days and no later than 7 days after 5KC-TCR reporter cells are thawed. Stimulation assay plate should be read between 18 and 32 h after the addition of 5KC-TCR cells to ensure sufficient time for response to be mounted. B. Workflow of stimulation plate setup on Day 4. Note that if antigens are proteins, rather than short peptides, K562-APCs and antigens should be cultured overnight before adding 5KC-TCR reporter cells.
Controls: When stimulating TCRs, three controls must be assessed.
Non-activated cell control
5KC-TCR cells are cultured in 5KC medium without antigens or APCs to determine the intensity of signals by non-activated cells in the channel for ZsGreen-1 detection. This control is required because some 5KC-TCR cells occasionally respond to APCs expressing certain MHC molecules without antigen added.No antigen control
5KC-TCR cells are cultured in the presence of APCs without antigen to be compared with cells cultured with antigen and APCs.Positive control
5KC-TCR cells are stimulated with anti-CD3 antibody. Greater than 80% of cells typically express ZsGreen-1 upon anti-CD3 antibody stimulation.
- Thaw 5KC-TCR cell lines and corresponding K562-APC cell lines four days prior to setting up a stimulation assay. Other types of cells such as autologous B cells can be used as APCs if wished.
Note: 5KC-TCR cells require three to four days to regain function after thawing, but begin to lose T-cell function after 7 days in culture.On day 0, thaw frozen cells (approximately 1 x 106 cells per cryovial for 5KC-TCR cells, approximately 2 x 106 cells for K562-APCs).
- Subculture each day, seeding 5KC-TCR lines at 5 x 104 cells per ml and K562-APC lines at 1 x 105 cells per ml.
- Stimulate 5KC-TCR cells.
Collect K562-APC cells by centrifugation and resuspend at 1 x 106 cells per ml in 5KC medium. Add 50 μl of cell suspension (5 x 104 cells) per well to a 96-well round-bottom plate.
- Prepare antigen solutions in 5KC medium at 4x desired final concentration (e.g., to achieve 100 nM final antigen concentration, make a 400 nM solution). Add 50 μl of antigen solution to designated wells containing APCs.
- Prepare control solutions in 5KC medium.
Prepare no antigen control solution by including PBS in place of antigen in 5KC medium. If stock antigen contains organic solvent such as DMSO, include the same concentration of the solvent in the no antigen control solution.
- For the non-activated cell control, add 50 μl of the no antigen control solution and 50 μl of 5KC medium to a well.
- For the no antigen control, add 50 μl of the no antigen control solution and 50 μl of the K562-APC cell suspension (5 x 104 cells) to a well.
- Prepare positive control solution at 10 μg/ml of functional grade anti-CD3 antibody (for 5 μg/ml final concentration) in 5KC medium. Add 100 μl of the anti-CD3 positive control solution to a well. If stock antigen contains organic solvent, include the same concentration of solvent in positive control solution.
- Incubate stimulation plates at 37 °C, 5% CO2 for 2 to 4 h.
Note: If antigens are proteins, rather than peptides, overnight culture is recommended prior to adding 5KC-TCR cells to allow APCs to uptake and process antigens for presentation. - Prepare 5KC-TCR cell mixes
Collect eight 5KC-TCR cell lines (containing eight different fluorescent color combinations) in separate tubes and centrifuge.
- Remove medium above cell pellets and resuspend each line at 1.6 x 106 cells/ml.
- Combine equal volumes of each 5KC-TCR line (mixture is 1.6 x 106 cells per ml, equivalent to 2 x 104 of each line per 100 μl).
- Add 100 μl 5KC-TCR cell mixture per well to the stimulation plate.
- Return plate to incubator overnight.
- Flow cytometry
- Settings described below are based on a Beckman-Coulter Cytoflex with 405, 488, 638 nm laser beams, but will differ based on available flow cytometers.
- Collect flow cytometry data for stimulation plates between 18 and 32 h after the addition of 5KC-TCR cells.
- Set compensation for all fluorescent colors used before running stimulation plate.Each fluorescent protein appears in the channel as in Table 10.
Table 10. Fluorescent proteins and corresponding flow cytometer channels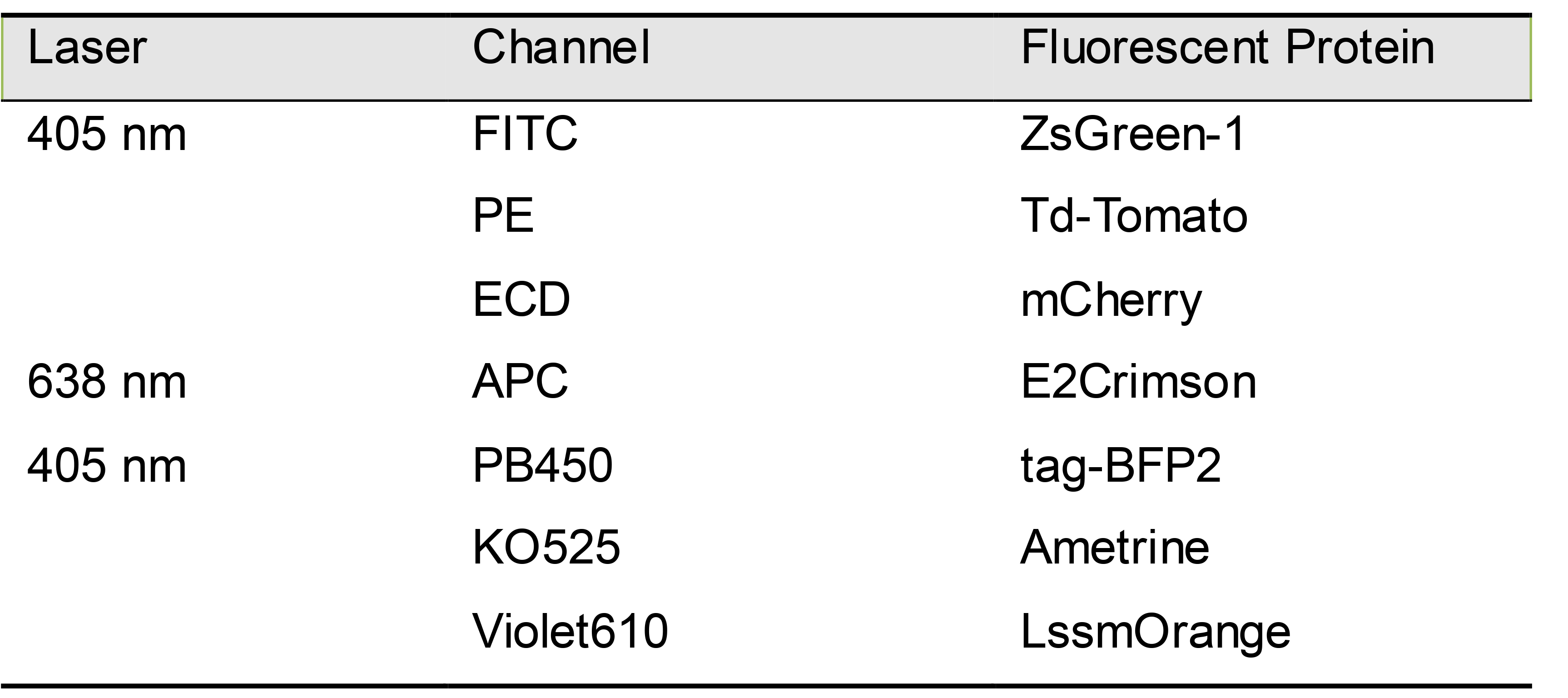
Centrifuge plate at 671 rcf at room temperature for 3 min. Dump media from plate in one smooth motion; pelleted cells should remain in the bottoms of the wells. Resuspend cells in 150 μl Phenol red-free RPMI using a multichannel pipettor.
Acquire data for 75,000 cells per well (sufficient to acquire at least 3,000 5KC-TCR cells expressing each color combination) using a flow cytometer equipped with a plate-loading function.
Flow rate can be set between 100 and 120 μl/min on Beckman-Coulter Cytoflex.
Set flow cytometer to stop acquisition after 150 μl.
- Set compensation for all fluorescent colors used before running stimulation plate.Each fluorescent protein appears in the channel as in Table 10.
Data analysis
Import data into FlowJo for analysis.
Gate cells according to the gating strategy shown in Figure 19.
Determine percent positivity of ZsGreen-1 from each well and import into an Excel file. Cells that exhibit higher intensity signals in the FITC channel than signals of the majority (i.e., >97-99%) of non-activated control cells are determined as positive for ZsGreen-1 expression.
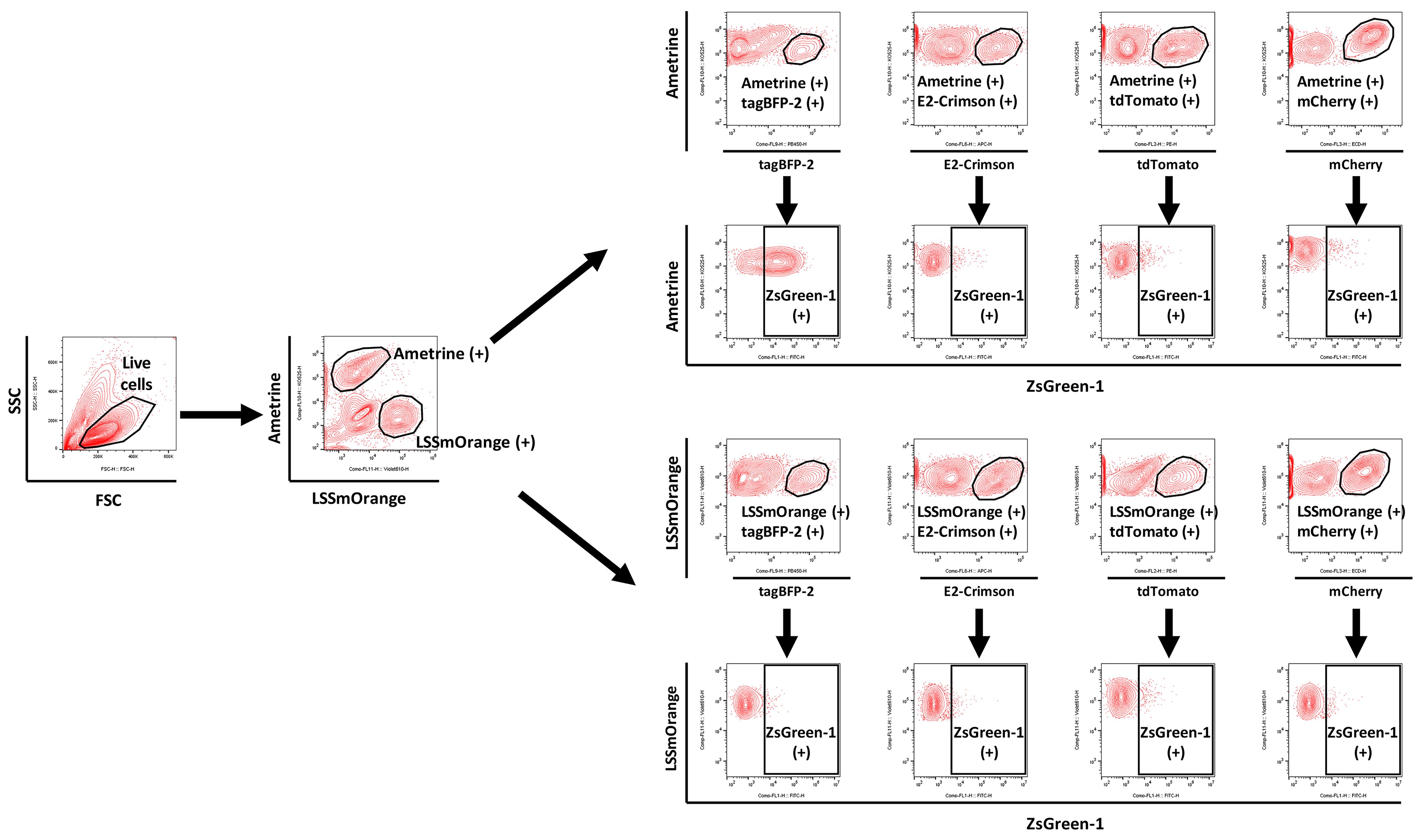
Figure 19. Gating strategy for determining response levels of each 5KC-TCR line in a mixture. After gating for live cells using the FSC vs. SSC scatter plot, K562-APCs can be excluded from 5KC-TCR cells by their lack of color. 5KC-TCR cells are gated on AM-positive and LO-positive cells. Within both AM and LO gating, cells are further gated for secondary color positivity (e.g., AM+ and BFP+), and ZsGreen-1 positivity is assessed based on gating determined by non-activated cell control. This figure is adapted from the article published by Mann et al. (2020) in Frontiers in Immunology (doi 10.3389/fimmu.2020.00633), which is licensed under by CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/). In this example, the TCR cell line identified by AM+/BFP+ is responding positively to stimulation, as seen by the expression of ZsGreen-1 along the X axis.Interpreting controls
The non-activated cell control is used to determine the base ZsGreen-1 signal level for each 5KC-TCR line without stimulation.
The no antigen control is used to determine the responsive level of non-antigen specific stimulation for each 5KC-TCR line.
ZsGreen-1 positive cells without antigen stimulation is typically < 3% and should not exceed 10%.
Promiscuous TCRs may respond to non-target antigens present in APCs expressing specific MHC molecules, resulting in high percentage of ZsGreen-1 positive cells.
Some fluorescent identifier combinations in conjunction with certain TCRs may result in a greater response in the absence of antigen stimulation. If > 10% of cells are positive for ZsGreen-1 expression, consider transducing the TCR onto a reporter cell line with different identifying fluorescent proteins.
The positive control is used to confirm that the TCR/CD3 complex is expressed and functional.
Normal response from CD3-stimulated cells is ≥ 80%.
Response level of 80%-60% indicates that TCRs are not well-expressed and may not be fully functional.
Response level of ≤ 60% indicates that the TCRs are unlikely to be functional.
Confirm that the non-activated cell control and no antigen control have sufficiently low levels of response before determining if a positive response has occurred in antigen stimulation wells.
Interpreting stimulation well response
Calculate stimulation index (SI) according to the following formula:
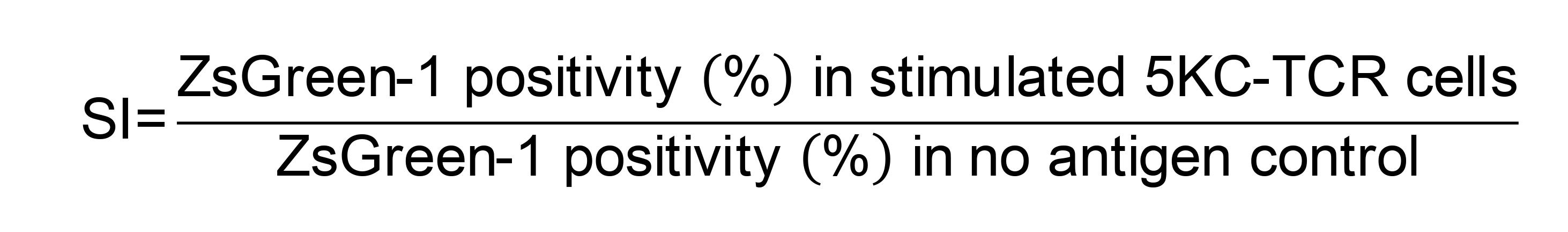
By using the SI, the effect of an non-antigen specific stimulation is mitigated.
Wells with SI ≥ 2 are considered positive
Higher SI indicates stronger response
For this application (screening large numbers of reactions), further statistical analysis is not typically performed. Instead, retest positive 5KC-TCR cells individually in three independent experiments to confirm reactivity to different concentrations of the antigens that induced ZsGreen-1 expression in the screening test. A T-test or ANOVA is used to compare ZsGreen-1 positivity between these three replicate experiments.
Notes
Vector construction, once synthetic DNA fragments are received, can be accomplished in four days by the following schedule:
Day 1: Digest vector backbone (and CaP2AIII for TCR vectors) and purify DNA fragment(s). [Optional break point: purified DNA fragments can be stored for several weeks at -20 °C.] Perform Gibson assembly reaction. [Optional break point: Gibson assembly reaction can be stored at 4 °C for up to 5 days.]
Day 2: Transform NEB 5-alpha Competent E. coli. [Optional break point: after incubating overnight, plates may be wrapped and stored at 4 °C for up to 3 days.]
Day 3: Grow mini-cultures and perform PCR check. [Optional break point, mini-cultures may be tightly covered and stored at 4 °C for up to 2 days.] Start large-scale cultures for Qiagen Midi-prep.
Day 4: Conduct Qiagen Midi-prep and vector sequencing.
The porcine teschovirus-1 2A cleaving peptide sequence (P2A) to join subunit gene sequences includes an N-terminal Gly-Ser-Gly linker for the improvement of self-cleaving efficiency.
When using DNA fragments and vectors purified by QIAQuick Gel Extraction Kit (Qiagen), we recommend performing a second purification using QIAQuick PCR Clean-up Kit (Qiagen) or Qiagen Minelute Gel Extraction Kit (Qiagen) before using the DNA fragments for Gibson assembly. A second purification removes residual contaminants that interfere with Gibson assembly reaction, and also enables further concentration of the DNA fragment, if needed.
For best transformation efficiencies, NEB 5-alpha Competent E. coli (NEB) are recommended for transformation when NEBuilder HiFi DNA Assembly Master Mix (NEB) is used for Gibson assembly.
When verifying vector sequence by Sanger sequencing, ensure vector sequences align perfectly with the designed, in-frame nucleotide sequence, without mutations, which can occur due to ligation error, PCR-introduced mutation, or DNA fragment synthesis errors. If a mutation is found, select another colony for Qiagen Midi-prep and sequencing. Vectors that are confirmed to have the correct sequence can be used directly for cell transfection.
For the construction of TCR vectors, backbone vector choice is determined by the TRBJ gene. If it is TRBJ1-# use MBC1-null; if it is TRBJ2-#, use MBC2-null.
When freezing and thawing cells, we recommend using wide-bore P1000 pipette tips to minimize mechanical stress on cells (see Equipment list).
After thawing, save a small portion of cells (<100 μl), make a 1:1 dilution with Trypan Blue, and count cells on a Countess II Automated cell counter. Divide the number of live cells by total number of cells to attain cell viability.
Expected viability is ≥ 90%
Phoenix-ECO cells often require 1-2 days to achieve normal cell growth rate and conditions after thawing. This can be assisted by:
Using an earlier passage number of cells.
Increase FBS in thawing media to 20%.
Treating culture dish with PDL prior to thawing to assist cells in adhering.
5KC α-β- and K562 culture conditions should be optimized prior to starting transfection and transduction procedures. Optimization should include:
Manually checking and calibrating CO2 levels of 37 °C incubator to 5% CO2 to prevent acidification of medium.
Testing multiple (2 or 3) lots of FBS for effect on cell growth rate and condition. Atlanta Biologicals is a recommended source of FBS for 5KC cells.
Determining sub-culturing conditions that maintain healthy 5KC α-β-growth rate and condition. 5KC α-β- cells prefer sparse conditions rather than dense. In general, this requires splitting cell cultures every day.
When cells are seeded at 5 x 104 cells/ml, they achieve a doubling rate of ≤12 h.
Cell condition is assessed using an inverted light microscope. Cells should appear bright with smooth edges and a roundish shape with occasional small blebs or points.
K562 growth conditions should be optimized based on manufacturer’s instructions. In general, they grow more slowly than 5KC α-β- cells and are therefore seeded at a higher density. They appear larger and more rounded than 5KC cells when visualized with an inverted light microscope.
The CD3-containing vectors (CD3-AM and CD3-LO) contain fluorescent protein genes because CD3 will not be expressed by 5KC α-β- cells before TCR genes have been added, and therefore fluorescence is used as a marker for cells that are transduced with the vector.
While AM and LO are expressed in the CD3-transduced 5KC α-β- cells, the AM or LO gene is behind the 2.3 kb of CD3 gene cassette, resulting in dim fluorescent expression. To ensure sufficient fluorescent expression, AM and LO are additionally introduced as FP identifiers during the second round of transfection and transduction (Step B2c, or Figure 12, step 2).
This results in two AM- or LO-positive populations during sorting after the fluorescent protein identifiers are added. Cells should be sorted on AM-high or LO-high to ensure that they have received both AM or LO FP vectors (Figure 16).
The cells generated in this step should be handled in the best manner possible and sufficiently expanded to allow many (> 16) vials to be frozen for future use. Avoid prolonged passaging of these cell lines before using to generate 5KC-TCR cell lines or freezing down additional vials.
When TCR is not expressed well:
Some primary T-cells contain two α chains, often one functional and one non-functional. When two TCRs of a set share the same β chains with two α chains, the non-functional TCR may not be reconstituted.
Some human V-α and V-β regions may be incompatible with the mouse signal peptide regions to be cleaved correctly. Replacing the mouse signal peptide region with an intrinsic human signal peptide region sequence may improve TCR expression.
We do not use codon-optimized nucleotides to express chimeric TCR genes in murine T-hybridoma cells, 5KC α-β- cells, since it is our experience that natural nucleotide sequences typically result in higher TCR/CD3 expression than the codon-optimized sequences. However, codon-optimization may occasionally improve TCR/CD3 expression for some TCRs.
Both 5KC and K562 cells must be treated as if they are shedding virus particles (non-replicating) for three complete media exchanges after transduction. Carry out all cell handling from transfection up to this point in BS level 2 conditions.
PBS is added to empty wells on culture plates to prevent excessive evaporation of media in dry climates. In humid climates this may not be necessary.
Various types of cells other than K562-APCs can be used as APCs in stimulation assays. Examples include autologous B cell lines transformed with Epstein-Barr virus, irradiated autologous peripheral blood mononuclear cells, and dendritic cells isolated/propagated/differentiated from autologous peripheral blood cells. The APC concentration needs to be optimized for each type of APCs.
For flow analysis, FSC vs. SSC scatter plots are used to gate living cells. We have tested live/dead staining under our assay conditions and it did not significantly influence the percentage of cells expressing ZsGreen-1 compared to using FSC vs. SSC gating. If sorting ZsGreen-1-positive cells for subsequent experiments is planned, we recommend using live/dead staining such as with Zombie NIR dye to improve the purity of sorted cells.
Recipes
Transfection medium
Rapid-Flow Sterile Disposable Filter Units (Nalgene)
DMEM (Gibco) 500 ml
HEPES buffer, 1 M (Gibco) 5 ml
Sodium pyruvate, 100 mM (Gibco) 5 ml
MEM-NEAA, 100x (Gibco) 5 ml
2-mercaptoethanol, 14.3 M (Sigma-Aldrich) 5 μl
Combine all ingredients, then filter to sterilize
Store at 4 °C for up to 1 week
5KC medium
IMDM (Gibco) 1,000 ml
FBS (Atlanta Biologicals) 100 ml
Pen/strep (Sigma-Aldrich) 10 ml
2-mercaptoethanol, 14.3 M (Sigma-Aldrich) 10 μl
Store at 4 °C for up to 2 weeks
Phoenix medium
DMEM (Gibco) 500 ml
FBS (Atlanta Biologicals) 50 ml
Pen/strep (Sigma-Aldrich) 5 ml
HEPES buffer, 1 M (Gibco) 5 ml
Sodium pyruvate, 100 mM (Gibco) 5 ml
MEM-NEAA, 100x (Gibco) 5 ml
2-mercaptoethanol, 14.3 M (Sigm-Aldrich) 5 μl
Store at 4 °C for up to 2 weeks
5KC Freezing Medium
Prepared 5KC medium 70%
FBS (Atlanta Biologicals) 20%
DMSO (Fisher Bioreagents) 10%
Combine enough reagents for 1 ml per each vial to be frozen
Filter through 0.2 μm syringe filter and use immediately
Phenol red-free RPMI
RPMI 1640, no phenol red (Gibco) 500 ml
FBS (Atlanta Biologicals) 10 ml
Pen/strep (Sigma-Aldrich) 5 ml
Sodium pyruvate, 100 mM (Gibco) 5 ml
MEM-NEAA, 100x (Gibco) 5 ml
2-mercaptoethanol, 14.3 M, (Sigma-Aldrich) 5 μl
Store at 4 °C for up to 2 weeks
Protamine sulfate, 5 mg/ml solution
Protamine sulfate powder (MP Biomedicals) 10 mg
Molecular biology grade water (HyClone) 2 ml
Dissolve powder and filter through 0.2 μm filter
Aliquot and store at -20 °C for up to one year
PDL, 0.1 mg/ml
PDL lyophilized powder (Sigma-Aldrich) 5 mg
Molecular biology grade water (HyClone) 50 ml
Add sterile cell culture grade water to PDL powder bottle and shake until powder is dissolved. Transfer to 50-ml conical and store at 4 °C for up to 2 months.
LB-ampicillin Broth
LB Broth (Gibco)
Ampicillin Ready Made Solution (Sigma-Aldrich)
1 μl of ampicillin is added per 1 ml of LB broth immediately before use
Store at 4 °C for up to 2 months
LB-ampicillin plates
LB Agar (BD Difco) 20 g
Milli-Q Ultrapure water (Millipore-Sigma) 500 ml
10-cm Petri dishes (Fisherbrand) ~25
Fully dissolve LB agar in water and autoclave at 121 °C for 20 min
Once agar has cooled to around 55 °C, add 500 μl ampicillin and pour into plates
Store at 4 °C for up to 1 month
Acknowledgments
This work was supported by the National Institutes of Health (R01DK099317, R01DK032083, P30CA046934, P30DK116073), JDRF (2-SRA-2018-480-S-B, 1-SRA-2020-911-A-N), and the Culshaw Family Junior Investigator Award. Development and validation of this protocol were described in a previous publication (Mann et al., 2020). A lentiviral vector pMH0001, which was used to generate a lentiviral vector pUS, is a gift from Jonathan Weissman (Addgene plasmid # 85969; http://n2t.net/addgene:85969; RRID:Addgene_85969). A retroviral vector Murine CD3 WTdelta-F2A-gamma-T2A-epsilon-P2A-zeta pMIA II, which was used to generate a retroviral vector CD3-LO, is a gift from Dario Vignali (Addgene plasmid # 52093 ; 52093 ">http://n2t.net/addgene: 52093 ; RRID:Addgene_ 52093 ).
Competing interests
The authors declare no conflict of interest or competing interests.
References
- Adamson, B., Norman, T. M., Jost, M., Cho, M. Y., Nuñez, J. K., Chen, Y., Villalta, J. E., Gilbert, L. A., Horlbeck, M. A., Hein, M. Y., Pak, R. A., Gray, A. N., Gross, C. A., Dixit, A., Parnas, O., Regev, A. and Weissman, J. S. (2016). A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 167(7): 1867-1882e1821.
- Holst, J., Wang, H., Eder, K. D., Workman, C. J., Boyd, K. L., Baquet, Z., Singh, H., Forbes, K., Chruscinski, A., Smeyne, R., van Oers, N. S., Utz, P. J. and Vignali, D. A. (2008). Scalable signaling mediated by T cell antigen receptor-CD3 ITAMs ensures effective negative selection and prevents autoimmunity. Nat Immunol 9(6): 658-666.
- Mann, S. E., Zhou, Z., Landry, L. G., Anderson, A. M., Alkanani, A. K., Fischer, J., Peakman, M., Mallone, R., Campbell, K., Michels, A. W. and Nakayama, M. (2020). Multiplex T cell stimulation assay utilizing a T cell activation reporter-based detection system. Front Immunol 11: 633.
- Michels, A. W., Landry, L. G., McDaniel, K. A., Yu, L., Campbell-Thompson, M., Kwok, W. W., Jones, K. L., Gottlieb, P. A., Kappler, J. W., Tang, Q., Roep, B. O., Atkinson, M. A., Mathews, C. E. and Nakayama, M. (2017). Islet-derived CD4 T cells targeting proinsulin in human autoimmune diabetes. Diabetes 66(3): 722-734.
- White, J., Pullen, A., Choi, K., Marrack, P. and Kappler, J. W. (1993). Antigen recognition properties of mutant V beta 3+ T cell receptors are consistent with an immunoglobulin-like structure for the receptor. J Exp Med 177(1): 119-125.
- Williams, T., Krovi, H. S., Landry, L. G., Crawford, F., Jin, N., Hohenstein, A., DeNicola, M. E., Michels, A. W., Davidson, H. W., Kent, S. C., Gapin, L., Kappler, J. W. and Nakayama, M. (2018). Development of T cell lines sensitive to antigen stimulation. J Immunol Methods 46265-73.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Landry, L. G., Mann, S. E., Anderson, A. M. and Nakayama, M. (2021). Multiplex T-cell Stimulation Assay Utilizing a T-cell Activation Reporter-based Detection System. Bio-protocol 11(2): e3883. DOI: 10.21769/BioProtoc.3883.
Category
Immunology > Immune cell function > Lymphocyte
Cell Biology > Cell-based analysis > Protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.