- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Quantitative Irreversible Tethering (qIT) for Target-directed Covalent Fragment Screening


Published: Vol 10, Iss 24, Dec 20, 2020 DOI: 10.21769/BioProtoc.3855 Views: 3476
Reviewed by: Joana Alexandra Costa ReisManish Kumar PatelSneha Ray
Original research article
The authors used this protocol in:

May 2018
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Small molecules that react to form covalent bonds with proteins are widely used as biological tools and therapeutic agents. Screening cysteine-reactive fragments against a protein target is an efficient way to identify chemical starting points for covalent probe development. Mass spectrometry is often used to identify the site and degree of covalent fragment binding. However, robust hit identification requires characterization of the kinetics of covalent binding that can be readily achieved using quantitative irreversible tethering. This screening platform uses a non-specific cysteine-reactive fluorogenic probe to monitor the rate of reaction between covalent fragments and cysteine containing biomolecules. Fragment libraries are simultaneously screened against the target protein and glutathione, which functions as a control, to identify hit fragments with kinetic selectivity for covalent modification of the target. Screening by quantitative irreversible tethering accounts for variations in the intrinsic reactivity of individual fragments enabling robust hit identification and ranking.
Keywords: Covalent fragmentsBackground
Site-directed ligand discovery was first reported in 2000 and utilizes surface-exposed cysteine residues to covalently trap disulfide-linked fragments that bind at an adjacent pocket (Erlanson et al., 2000). Since then, irreversible cysteine-targeting inhibitors have grown immensely in popularity and now represent front-line therapies in multiple oncology indications, with notable examples targeting BTK and EGFR as well as previously undruggable targets such as KRAS(G12C). Following these developments, covalent fragment-based ligand discovery has emerged as an efficient route towards the design of target-specific covalent inhibitors (Resnick et al., 2019). Libraries of cysteine-reactive fragments are now commercially available from a range of compound vendors and utilize a variety of reaction types for covalent bond formation such as conjugate addition and nucleophilic substitution. Protein mass spectrometry was adopted early on as a useful assay for conducting covalent fragment screening, providing a direct readout on the extent and the site of fragment binding. However, even the earliest reports of covalent fragment-based ligand discovery emphasized the challenge that fragment-dependent electrophilicity presents (Nonoo et al., 2012): the intrinsic reactivity of covalent fragments can vary by several orders of magnitude within a library. Fragments with high intrinsic reactivity will covalently bind to proteins regardless of their structure and this reactivity-driven binding does not constitute a useful starting point for fragment optimization. In pioneering work by Statsyuk, it was demonstrated that, through careful design of the reactive warhead architecture, it is possible to limit the variations in intrinsic reactivity, resulting in more robust screening outcomes (Kathman et al., 2014). However, in many cases it is desirable to screen a wide variety of warhead structures, maximizing the exploration of chemical space and enlarging the pool of accessible targets (Keserű et al., 2016).
Quantitative irreversible tethering (qIT) is a plaform for screening cysteine-reactive fragments that has been designed to overcome the intrinsic reactivity problem (Craven et al., 2018). The fluoregenic probe CPM (7-diethylamino-3-(4'-Maleimidylphenyl)-4-Methylcoumarin) is used to quantify the rate of reaction between covalent fragments and cysteine residues. Covalent fragments are simultaneously screened against glutathione (GSH) as well as the target protein and the rates of reactions (kobs) determined in high-throughput. GSH functions as a control cysteine containing biomolecule that lacks significant secondary structure. The rate of reaction between a fragment and GSH represents a measure of the covalent fragment’s intrinsic reactivity. Hit fragments are those that react significantly faster with the target protein than with GSH, as quantified by the rate enhancement factor (REF = kobs(target)/kobs(GSH). Screening by REF analysis ensures that qIT can be carried out in high-throughput and does not suffer from high false-positive/negative hit rates because it accounts for intrinsic fragment reactivity. The key advantage of screening by quantitative irreversible tethering is that, in accounting for intrinsic fragment reactivity, true hits can be readily distinguished from non-selective reactive molecules which otherwise lead to high levels of false positives. The platform is compatible with a broad range of cysteine-reactive fragments and is applicable to most cysteine-containing soluble protein domains. Additionally, the high-throughput nature and low cost of using a plate-based fluorescence readout means that quantitative irreversible tethering projects can be performed rapidly and with high efficiency.
Since the original publication of qIT (Craven et al., 2018), we have updated the screening protocol to make it more applicable to a wider variety of proteins by changing the buffer system, more high-throughput by reformatting from 96-well to 384-well format and easier to implement by changing the environment of operation from 4 °C to room temperature. In our lab we have successfully applied this updated protocol to a range of protein classes including kinases, GTPases, oxidoreductase and proteases.
Materials and Reagents
Black 384-well plates (Corning, 384-well low flange black flat bottom NBS, catalog number: 3575 )
Pippette tips for pipetting station (CyBio, SELMA 384/25 µl pipette tips (standard), catalog number: OL3800-25-513-N )
Plate sealing tape (Bio-Rad, optical sealing tape, catalog number: 2239444 )
384-Well compound storage plates (Nunc, catalog number: 269390 )
PD-10 column (GE Healthcare, catalog number: 52130800 )
15 ml Centrifuge tubes (Star Lab, Catalog number: E1415-0200 )
Reagent reservoirs (Thermo Scientific, catalog number: 8096-11 )
Immobilized TCEP agarose (Pierce, catalog number: 77712 ). Store at 4 °C (shelf-life ~1 month, soluble TCEP can be added to a final concentration of 50 µM to extend the shelf-life)
CPM: 7-diethylamino-3-(4'-maleimidylphenyl)-4-methylcoumarin (ThermoFisher, catalog number: D346 ). Store at 0.5 mM in DMSO at -20 °C in aliquots of 56 µl
L-Glutathione reduced (Sigma-Aldrich, catalog number: G4251 ). Store at 4 °C
Target protein–containing a single surface-exposed/reactive cysteine residue (cloning and expression by standard molecular biology protocols as required). See Note 1 for compatibility considerations
Control protein–this should be identical in sequence to the target protein but have the reactive cysteine knocked-out (cloning and expression by standard molecular biology protocols as required). Typically, the reactive cysteine is mutated to alanine or serine
Covalent fragment library (Compounds stored at 50 mM in DMSO in 384-well compound storage plates, 320 compounds per plate in columns 3-22, columns 1, 2, 23 and 24 are blank). See Note 2 for composition and storage considerations
Iodoacetamide (Sigma-Aldrich, catalog number: I1149 )
HEPES (Sigma-Aldrich, catalog number: H3375 )
Sodium chloride (Sigma-Aldrich, catalog number: S7653 )
(Optional) Argon gas (BOC, catalog number: 11Y )
DMSO (Sigma-Aldrich, catalog number: D8418 )
Reaction buffer (see Recipes)
Quench buffer (see Recipes)
CPM Quench solution (see Recipes)
3x TCEP agarose stock (1.5% suspension) (see Recipes)
3x GSH stock (15 µM) (see Recipes)
3x Target protein stock (15 µM) (see Recipes)
3x Control protein stock (15 µM) (see Recipes)
Equipment
Timer (Lab Alert, catalog number: HS24670 )
Single channel pipettes (Gilson)
12-Channel pipette (Sartorius, Picus electronic 10-300 µl, catalog number: 735461)
Plate reader (BMG LABTECH, model: CLARIOstar )
Semi-automated pipetting station (Analytik Jena, model: CyBio SELMA 384/25 µl)
Centrifuge with plate adapter (Ependorf, model: 5804R)
NanoDrop spectrophotometer (Thermo Scientific, catalog number: ND-2000)
Software
Microsoft Excel
GraphPad Prism 8
Procedure
Pilot assay to assess protein construct compatibility and robustness
Assemble the reaction plate (see Table 1 for layout).
Table 1. Reaction plate layout for the pilot assay
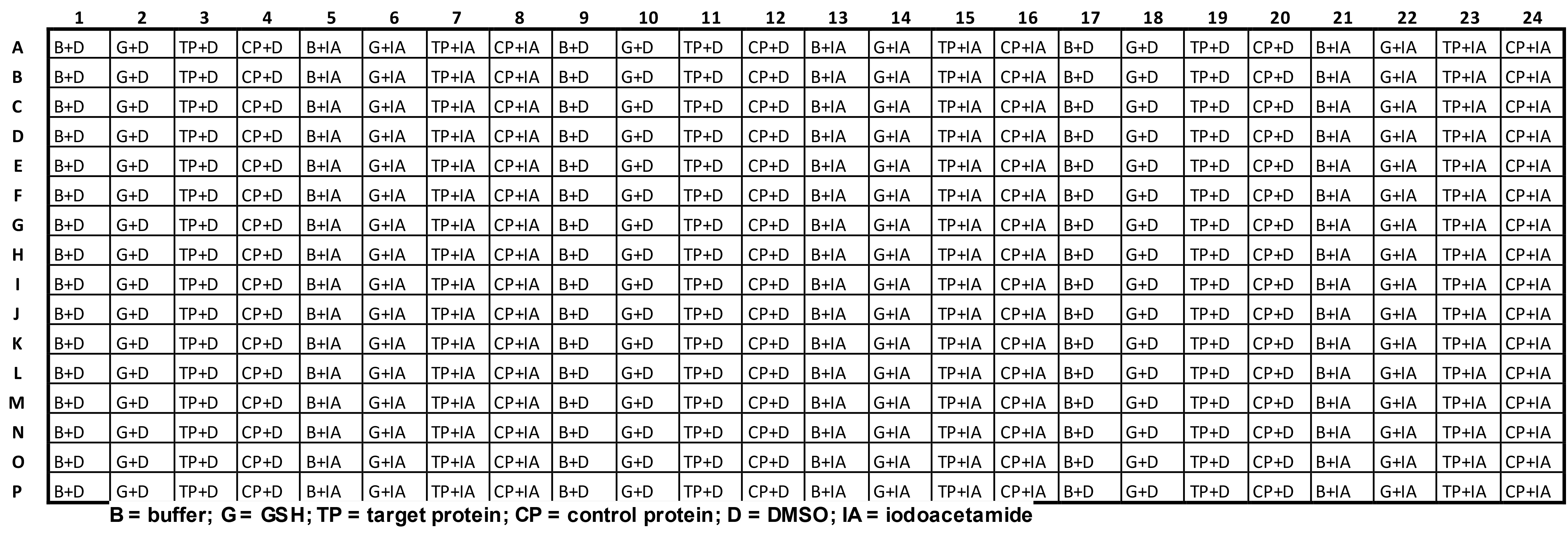
Take a black 384-well plate and label it “Reaction plate”.
Dispense 20 µl of freshly prepared 3x TCEP agarose stock from a reagent reservoir into each well of the reaction plate using a 12-channel pipette. During pipetting, the beads require regular agitation in the reservoir to maintain a homogeneous suspension and the bottoms of the pipette tips should be cut off to prevent them from blocking.
Dispense 20 µl of reaction buffer into columns 1, 5, 9, 13, 17 and 21 of the reaction plate from a reagent reservoir using a 12-channel pipette.
Dispense 20 µl of 3x GSH stock into columns 2, 6, 10, 14, 18 and 22 of the reaction plate from a reagent reservoir using a 12-channel pipette.
Dispense 20 µl of 3x target protein stock into columns 3, 7, 11, 15, 19 and 23 of the reaction plate from a reagent reservoir using a 12-channel pipette.
Dispense 20 µl of 3x control protein stock into columns 4, 8, 12, 16, 20 and 24 into the reaction plate from a reagent reservoir using a 12-channel pipette.
Incubate at room temperature (RT) for 2 h to ensure complete reduction of cysteine residues by TCEP agarose.
Initiate the iodoacetamide and DMSO control reactions.
Dispense 20 µl of reaction buffer containing 3% DMSO into columns 1, 2, 3, 4, 9, 10, 11, 12, 17, 18, 19 and 20 of the reaction plate from a reagent reservoir using a 12-channel pipette.
Dispense 20 µl of 1.5 mM iodoacetamide in reaction buffer containing 3% DMSO into columns 5, 6, 7, 8, 13, 14, 15, 16, 21, 22, 23 and 24 from a reagent reservoir using a 12-channel pipette.
Mix all wells by aspirating and dispensing 25 µl three-times using the 384 well semi-automated pipetting station (for details of how to operate the pipetting station please refer to www.analytik-jena.com/products/liquid-handling-automation/flexible-benchtop-automation/cybio-selma/).
Start a timer.
Centrifuge the plate at 200 x g for 3 min to pellet the TCEP agarose.
Perform the first CPM quench (Q1).
Take a new black 384-well plate and label it “Quench plate Q1”.
Prepare 20 ml of fresh CPM quench solution (see Recipes).
Dispense 27 µl of CPM quench solution into each well of quench plate Q1.
Using the 384 well semi-automated pipetting station aspirate 3 µl from the top of each well of the reaction plate and dispense into quench plate Q1 (aspirating from the top ensures that no TCEP agarose is transferred).
Mix all wells of the quench plate by aspirating and dispensing 25 µl three-times.
Record the time on the timer (this should be between 5 and 15 min).
Seal the reaction plate with plate sealing tape to prevent evaporation and store at RT until the next quench.
Incubate the quench plate Q1 for 1 h at RT.
Measure the fluorescence intensity of quench plate Q1 (excitation/emission: 384/470 nm).
Perform the second CPM quench (Q2) by repeating Step A3 when the timer is at 1 h.
Perform the third CPM quench (Q3) by repeating Step A3 when the timer is at 5 h.
Perform the fourth CPM quench (Q4) by repeating Step A3 when the timer is at 24 h.
After the final measurement export the data into Microsoft Excel for data processing.
Covalent fragment screening of 320 compounds (see Figure 1 for schematic workflow)
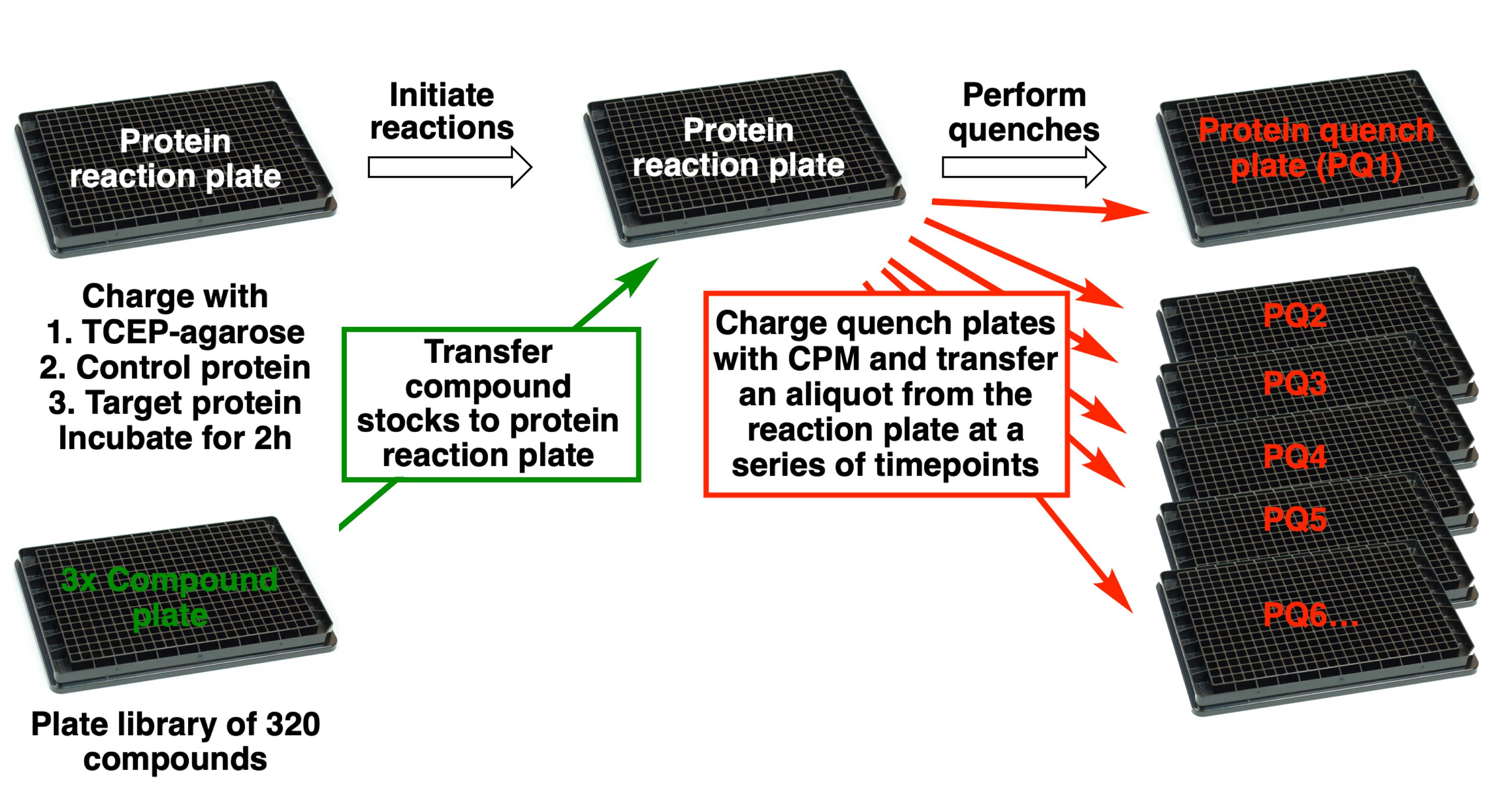
Figure 1. Schematic workflow for covalent fragment screening by qITAssemble the protein reaction plate.
Take a black 384-well plate and label it “Protein reaction plate”.
Dispense 20 µl of freshly prepared 3x TCEP agarose stock from a reagent reservoir into each well of the protein reaction plate using a 12-channel pipette. During pipetting, the beads require regular agitation in the reservoir to maintain a homogeneous suspension and the bottoms of the pipette tips should be cut off to prevent them from blocking.
Dispense 20 µl of 3x control protein stock into columns 1 and 2 of the protein reaction plate from a reagent reservoir using a 12-channel pipette.
Dispense 20 µl of 3x target protein stock into columns 3-24 of the protein reaction plate from a reagent reservoir using a 12-channel pipette.
Incubate at RT for 2 h to ensure complete reduction of cysteine residues by TCEP agarose.
Assemble the GSH reaction plate.
Take a new black 384-well plate and label it “GSH reaction plate”.
Dispense 20 µl of freshly prepared 3x TCEP agarose stock from a reagent reservoir into each well of the protein reaction plate using a 12-channel pipette. During pipetting, the beads require regular agitation in the reservoir to maintain a homogeneous suspension and the bottoms of the pipette tips should be cut off to prevent them from blocking.
Dispense 20 µl of reaction buffer into columns 1 and 2 of the GSH reaction plate from a reagent reservoir using a 12-channel pipette.
Dispense 20 µl of 3x GSH stock into columns 3-24 of the GSH reaction plate from a reagent reservoir using a 12-channel pipette.
Incubate at RT for 2 h to ensure complete reduction of GSH by TCEP agarose.
Assemble the 3x compound stock plate.
Take a new black 384-well plate and label it “3x compound plate”.
Dispense 58.2 µl of reaction buffer into all wells of the 3x compound plate from a reagent reservoir using a 12-channel pipette.
Dispense 1.8 µl of DMSO into columns 1, 2, 23, and 24 of the 3x compound plate using the semi-automated pipetting station.
Using the semi-automated pipetting station aspirate 1.8 µl from each well of the covalent fragment library storage plate (compounds are in columns 3-22, columns 1, 2, 23 and 24 are blank) and dispense into the 3x compound plate.
Mix all wells of the 3x compound plate by aspirating and dispensing 25 µl three-times.
Centrifuge the 3x compound plate at 200 x g for 3 min to pellet any precipitates.
Seal and store the covalent fragment library storage plate.
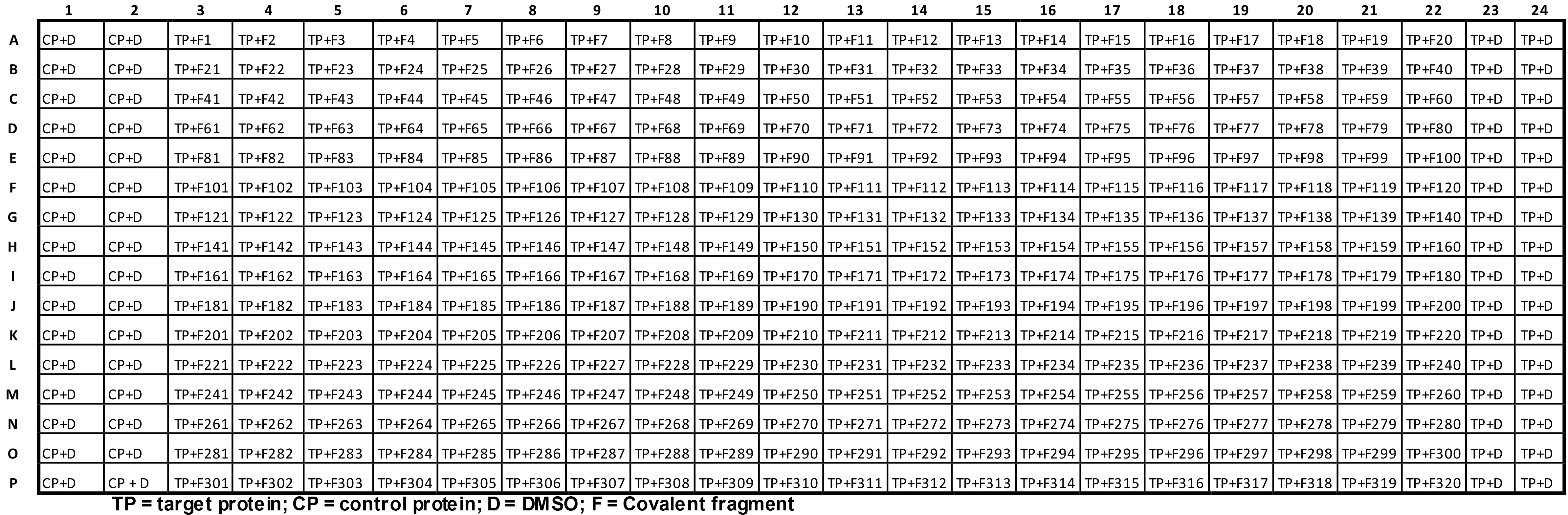
Initiate the protein reactions (see Table 2 for final layout).
Table 2. Protein reaction plate layout for the covalent fragment screen
Aspirate 20 µl from each well of the 3x compound plate and dispense into the protein reaction plate using the semi-automated pipetting station.
Mix all wells of the protein reaction plate by aspirating and dispensing 25 µl three-times.
Start a timer (protein reaction timer).
Centrifuge the protein reaction plate at 200 x g for 3 min to pellet the TCEP agarose.
Initiate the GSH reactions.
Aspirate 20 µl from each well of the 3x compound plate and dispense into the GSH reaction plate using the semi-automated pipetting station.
Mix all wells of the GSH reaction plate by aspirating and dispensing 25 µl three-times.
Start a timer (GSH reaction timer).
Centrifuge the GSH reaction plate at 200 x g for 3 min to pellet the TCEP agarose.
Perform the first protein CPM quench (PQ1) and GSH CPM quench (GQ1).
Take two new black 384-well plates and label one “Protein quench plate PQ1” and the other “GSH quench plate GQ1”.
Prepare 20 ml of fresh CPM quench solution (see Recipes).
Dispense 27 µl of CPM quench solution into each well of quench plates PQ1 and GQ1.
Using the 384 well semi-automated pipetting station aspirate 3 µl from the top of each well of the protein reaction plate and dispense into quench plate PQ1.
Mix all wells of the PQ1 by aspirating and dispensing 25 µl three-times.
Record the time on the protein timer (this should be between 5 and 15 min).
Seal the protein reaction plate with plate sealing tape to prevent evaporation and store at RT until the next quench.
Using the 384 well semi-automated pipetting station aspirate 3 µl from the top of each well of the GSH reaction plate and dispense into quench plate GQ1.
Mix all wells of the GQ1 plate by aspirating and dispensing 25 µl three-times.
Record the time on the GSH timer (this should be between 5 and 15 min).
Seal the GSH reaction plate with plate sealing tape to prevent evaporation and store at RT until the next quench.
Incubate the quench plates PQ1 and GQ1 for 1 h at RT.
Measure the fluorescence intensity of the Q1 quench plates (excitation/emission: 384/470 nm).
Perform subsequent CPM quenches (Q2-Q9) by repeating Step B6 when the timers are at 0.5 h, 1 h, 2 h, 4 h, 6 h, 18 h, 22 h and 26 h.
After the final measurement export the data into Microsoft Excel for data processing.
Data analysis
Analysis of pilot assay to assess protein construct compatibility and robustness
Analysis of quench Q1
In Microsoft Excel, calculate the mean and standard deviation for the fluorescence intensity of “Buffer + DMSO” (columns 1, 9 and 17), “Buffer + Iodoacetamide” (columns 5, 13 and 21), “GSH + DMSO” (columns 2, 10, and 18), “GSH + Iodoacetamide” (columns 6, 14, and 22), “Target protein + DMSO” (columns 3, 11, and 19), “Target protein + Iodoacetamide” (columns 7, 15, and 23), “Control protein + DMSO” (columns 4, 12, and 20) and “Control protein + Iodoacetamide” (columns 8, 16 and 24).
Calculate the Z-factor: Z = 1 - (3(σp + σn)/|µp - µn| (σp = standard deviation of positives, σn = standard deviation of negatives, µp = mean of positives, µn = mean of negatives) for the following combinations:
Positive = “GSH + DMSO”; negative = “Buffer + DMSO”
Positive = “GSH + DMSO”; negative = “GSH + Iodoacetamide”
Positive = “Target protein + DMSO”; negative = “Control protein + DMSO”
Positive = “Target protein + DMSO”; negative = “Target protein + Iodoacetamide”
To check for edge effects, confirm that the column 1 gives a similar mean fluorescence intensity to columns 9 and 17 and that column 24 gives a similar mean fluorescence intensity to columns 8 and 16. See Note 3 if edge effects are observed.
Repeat the above Z-factor analysis for Q2, Q3 and Q4.
Interpretation of pilot assay results.
In general, for application to small molecule screening a Z-factor > 0.5 means that the assay is considered suitable for screening (Zhang et al., 1999). Because this assay is time-dependent we apply the following criteria.
Z-factor (Positive = “GSH + DMSO”; negative = “Buffer + DMSO”) should be > 0.5 for all quenches, with a dynamic range ≥ 5. We often see a slight drop for this Z-factor over time but in our lab it is typically > 0.6 at Q4. If a dramatic drop in the dynamic range and Z-factor is observed over time, this indicates that the TCEP-agarose beads may have oxidized and need replacing.
Z-factor (Positive = “GSH + DMSO”; negative = “GSH + Iodoacetamide”) should increase over time and be > 0.5 for Q3 and Q4.
Z-factor (Positive = “Target protein + DMSO”; negative = “Control protein + DMSO”) should be > 0.5 for all quenches, with a dynamic range ≥ 10. This indicates that the cysteine of interest on the target protein is reacting successfully with CPM.
The mean fluorescence intensity of the “Control protein + DMSO” should also be compared against “Buffer + DMSO”. Here, a dynamic range of up to 2 is typical. But if it is substantially more than 2, this may indicate that there are other cysteines on the construct that are reacting with CPM. If this is the case, the construct should be reevaluated (see Note 1) and additional cysteine mutations may be required.
Z-factor (Positive = “Target protein + DMSO”; negative = “Target protein + Iodoacetamide”) should increase over time and be > 0.5 for Q3 and Q4.
Covalent fragment screening
Normalize the fluorescence intensity values for PQ1 in Microsoft Excel (see Supplementary Excel Sheet for a template including mock data).
Calculate the mean fluorescence intensity of “Control protein + DMSO” (columns 1 and 2).
Calculate the mean fluorescence intensity of “Target protein + DMSO” (columns 23 and 24).
Subtract the mean fluorescence intensity of “Control protein + DMSO” from the mean fluorescence intensity of “Target protein + DMSO” to give the “Baselined maximum signal”.
Subtract the mean fluorescence intensity of “Control protein + DMSO” from the fluorescence intensities of each of the reaction wells (columns 3-22) to give a “Baselined fluorescence intensity” for each reaction.
Divide each of the 320 “Baselined fluorescence intensity” values by the “Baselined maximum signal” to give a “Normalised fluorescence intensity” for each reaction at Time (of quench) = Q1.
Normalize the fluorescence intensity values for PQ2-9 as above for PQ1.
Create an XY data table titled “Protein kinetics” in GraphPad Prism with Time (of quench) in minutes = X and Normalized fluorescence intensity = Y for each covalent fragment.
Insert: New Analysis: Non-linear regression (curve fit): One Phase Decay with the following restraints: 0 > Y0 > 0.2; Plateau > 0.8.
Insert: New Graph for Existing Dataset: select Create a New Graph for Each Dataset.
Check the R squared values for each reaction and highlight any that are low. Note that in the case of very slow or fast reactions, the R squared value will be low and in this case the observed rate constant should not be taken at face value. Manually inspect each graph for outliers. Sometimes one of the individual quenches may exhibit outliers across the whole plate and these should be excluded.
Copy the K value for each reaction into a new Microsoft Excel table called “REF Analysis” under the column heading “kobs target protein”.
Normalize the fluorescence intensity values for GQ1 in Microsoft Excel
Calculate the mean fluorescence intensity of “Buffer + DMSO” (columns 1 and 2).
Calculate the mean fluorescence intensity of “GSH + DMSO” (columns 23 and 24).
Subtract the mean fluorescence intensity of “Buffer + DMSO” from the mean fluorescence intensity of “GSH + DMSO” to give the “Baselined maximum signal”.
Subtract the mean fluorescence intensity of “Buffer + DMSO” from the fluorescence intensities of each of the reaction wells (columns 3-22) to give a “Baselined fluorescence intensity” for each reaction.
Divide each of the 320 “Baselined fluorescence intensity” values by the “Baselined maximum signal” to give a “Normalised fluorescence intensity” for each reaction at Time (of quench) = Q1.
Normalize the fluorescence intensity values for GQ2-9 as above for GQ1.
Create an XY data table titled “GSH kinetics” in GraphPad Prism with Time (of quench) in minutes = X and Normalised fluorescence intensity = Y for each covalent fragment.
Insert: New Analysis: Non-linear regression (curve fit): One Phase Decay with the following restraints: 0 > Y0 > 0.2; Plateau > 0.8.
Insert: New Graph for Existing Dataset: select Create a New Graph for Each Dataset.
Check the R squared values for each reaction and note any that are low. Manually inspect each graph for outliers.
Copy the K value for each GSH reaction into the Microsoft Excel table called “REF Analysis” under the column heading “kobs GSH”.
Divide the kobs Target protein column by the kobs GSH column to generate a REF for each reaction.
Calculate the geometric mean and standard deviation of the REFs.
Hit fragments may be defined as those with a REF that is 3 standard deviations over the geometric mean or can be ordered by REF and an arbitrary hit rate defined (e.g., top 2%). The protein and GSH reaction graphs in Prism should then be manually inspected for all hit compounds to confirm that the curve fitting and data quality are suitable.
Follow up from primary covalent fragment screen: All hit compounds from a primary screen should be rescreened in triplicate by the same protocol to confirm reproducibility and for reliable quantitative hit ranking. It may also be desirable to rescreen hits at multiple concentrations (e.g., 500, 250, 100 and 50 µM) to gain further insights into the concentration dependency of the modifications. Where possible, hits should then be orthogonally validated by mass spectrometry, biochemical assays and/or X-ray crystallography. For an example of how to conduct hit-follow up please see our recent publication on qIT ( Craven et al., 2020 ).
Notes
Where possible, the structure of the target protein should be analyzed to identify any cysteine residues that are likely to be exposed to solvent. In cases where the target has more than one solvent-exposed cysteine residue, site-directed mutagenesis should be applied to generate a construct containing only the target cysteine residue on the surface. Point-mutations can affect the structure or biochemical activity of the target protein and new constructs should be compared against the wild-type to check this has not taken place. Purification tags should also be free from cysteine residues or cleaved off. We generally recommend including reducing agents (such as DTT) in protein purification and storage buffers; however, these reducing agents must be fully removed prior to assay initiation, which can be readily achieved by buffer exchange using a PD-10 column.
Commercial vendors of cysteine-targeted covalent fragments include Enamine, LifeChemicals and Cominnex. A review of considerations for covalent fragment library design can also be found (Keeley et al., 2020). Covalent fragment libraries should be subject to ongoing quality control. Storage in D6-DMSO enables periodic assessment of purity and degradation by NMR. If possible, freeze thaw cycles of DMSO stocks should be avoided to minimize the introduction of atmospheric water.
Edge effects typically arise from evaporation or temperature differences across the plate. We do not observe any edge effects in this protocol. But if they are observed, then care should be taken to minimize evaporation by sealing plates carefully when not in use and to ensure that all components are kept at room temperature. If edge effects are still observed then the plate layout can be redesigned to exclude the use of the outside wells of the plate.
Recipes
Reaction buffer
25 mM HEPES (pH 8.0)
150 mM NaCl
Quench buffer
25 mM HEPES (pH 7.5)
150 mM NaCl
Notes:
Additional buffer components may be added to either the reaction buffer and/or quench buffer if required by the target protein for stability e.g., salts and glycerol. However, soluble reducing agents such as DTT, βME or TCEP must not be added as they will react with CPM. The addition of any new buffer components (particularly detergents) should always be tested for compatibility with CPM.
(Optional) In cases where proteins are highly sensitive to aerobic oxidation, buffers should be degassed by bubbling argon for 30 min prior to use.
CPM Quench solution
To prepare 20 ml of 1.39 µM CPM quench solution, add 55.6 µl of 0.5 mM CPM in DMSO to 20 ml of quench buffer. A 56 µl aliquot of 0.5 mM CPM in DMSO should be thawed immediately prior to diluting into the quench buffer and the quench should be performed with 15 min of preparation. CPM has low stability at room temperature in water and the performance of the assay is maximized by adhering to this practice
3x TCEP agarose stock (1.5% suspension)
Pipette 300 µl of immobilized TCEP agarose (stored as a 50% suspension) into a 15 ml centrifuge tube and dilute to 15 ml with reaction buffer
Mix by manual inversion
Centrifuge at 600 x g for 1 min to pellet beads
Aspirate and discard the supernatant
Repeat the dilution, mixing, centrifugation and aspirating sequence an additional 4 times
Dilute to 10 ml with reaction buffer
3x GSH stock (15 µM)
Prepare fresh immediately before use
Dissolve 46 mg of reduced glutathione in 10 ml reaction buffer to give a 15 mM stock
Add 15 µl of the 15 mM stock into 15 ml of reaction buffer to give a 15 µM stock
3x Target protein stock (15 µM)
Buffer exchange target protein into reaction buffer using a PD-10 column
Determine the protein concentration by NanoDrop and dilute to 15 µM in reaction buffer
Centrifuge at 2,400 x g for 5 min to remove precipitants
3x Control protein stock (15 µM)
Buffer exchange the cysteine-knockout analogue of the target protein into reaction buffer using a PD-10 column
Determine the protein concentration by NanoDrop and dilute to 15 µM in reaction buffer
Centrifuge at 2,400 x g for 5 min to remove precipitants
Acknowledgments
This work was supported by grants from the Institute of Chemical Biology (Imperial College London), the UK Engineering and Physical Sciences Research Council (Studentship award EP/F500416/1) and the Cancer and Polio Research Fund. The protocol described here is adapted from a previous publication (Craven et al., 2018).
Competing interests
G.B.C., A.A., and D.J.M. are co-inventors on a patent application covering qIT: PCT/GB2017/052456.
References
- Craven, G. B., Affron, D. P., Allen, C. E., Matthies, S., Greener, J. G., Morgan, R. M. L., Tate, E. W., Armstrong, A. and Mann, D. J. (2018). High-throughput kinetic analysis for target-directed covalent ligand discovery. Angew Chem Int Ed Engl 57(19): 5257-5261.
- Craven, G. B., Affron, D. P., Kosel, T., Wong, T. L. M., Jukes, Z. H., Liu, C. T., Morgan, R. M. L., Armstrong, A. and Mann, D. J. (2020). Multiparameter Kinetic Analysis for Covalent Fragment Optimization by Using Quantitative Irreversible Tethering(qIT). Chembiochem. doi: 10.1002/cbic.202000457.
- Erlanson, D. A., Braisted, A. C., Raphael, D. R., Randal, M., Stroud, R. M., Gordon, E. M. and Wells, J. A. (2000). Site-directed ligand discovery. Proc Natl Acad Sci U S A 97(17): 9367-9372.
- Kathman, S. G., Xu, Z. and Statsyuk, A. V. (2014). A fragment-based method to discover irreversible covalent inhibitors of cysteine proteases. J Med Chem 57(11): 4969-4974.
- Keeley, A., Petri, L., Abranyi-Balogh, P. and Keseru, G. M. (2020). Covalent fragment libraries in drug discovery. Drug Discov Today.
- Keserű, G. M., Erlanson, D. A., Ferenczy, G. G., Hann, M. M., Murray, C. W. and Pickett, S. D. (2016). Design principles for fragment libraries: maximizing the value of learnings from pharma fragment-based drug discovery(FBDD) programs for use in academia. J Med Chem 59(18): 8189-8206.
- Nonoo, R. H., Armstrong, A. and Mann, D. J. (2012). Kinetic template-guided tethering of fragments. ChemMedChem 7(12): 2082-2086.
- Resnick, E., Bradley, A., Gan, J., Douangamath, A., Krojer, T., Sethi, R., Geurink, P. P., Aimon, A., Amitai, G., Bellini, D., Bennett, J., Fairhead, M., Fedorov, O., Gabizon, R., Gan, J., Guo, J., Plotnikov, A., Reznik, N., Ruda, G. F., Diaz-Saez, L., Straub, V. M., Szommer, T., Velupillai, S., Zaidman, D., Zhang, Y., Coker, A. R., Dowson, C. G., Barr, H. M., Wang, C., Huber, K. V. M., Brennan, P. E., Ovaa, H., von Delft, F. and London, N. (2019). Rapid covalent-probe discovery by electrophile-fragment screening. J Am Chem Soc 141(22): 8951-8968.
- Zhang, J. H., Chung, T. D. and Oldenburg, K. R. (1999). A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 4(2): 67-73.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Craven, G., Mann, D. J. and Armstrong, A. (2020). Quantitative Irreversible Tethering (qIT) for Target-directed Covalent Fragment Screening. Bio-protocol 10(24): e3855. DOI: 10.21769/BioProtoc.3855.
Category
Biochemistry > Protein > Modification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.