- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Quantitative Assay to Measure Stress Granule Association of Proteins and Peptides in Semi-permeabilized Human Cells
Published: Vol 10, Iss 24, Dec 20, 2020 DOI: 10.21769/BioProtoc.3846 Views: 5924
Reviewed by: Steven BoeynaemsIndranil MalikAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2018

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Stress granules (SGs) are membrane-less organelles that form in the cytoplasm through phase separation, in response to diverse stressors. SGs contain translationally stalled mRNAs, proteins involved in translation, and various RNA-binding proteins (RBPs). Due to the high local concentration of aggregation-prone RBPs, SGs might act as condensation sites for aberrant phase transitions of RBPs and could favor formation of solid protein aggregates underlying the pathological cytoplasmic inclusions found in numerous neurodegenerative diseases. Most assays aiming at studying the recruitment of RBPs into SGs are based on overexpression and SG recruitment of RBPs in intact cells. These approaches are, however, often limited by the predominantly nuclear localization of many RBPs, which precludes cytoplasmic RBP concentrations sufficient for SG localization, and does not address RBP recruitment independent of SG formation. Here, we present a quantitative method to assess recruitment of recombinant RBPs into pre-formed SGs, independent of the RBP’s nuclear localization, using semi-permeabilized cells and fluorescence microscopy. In this assay, SGs are firstly induced by a stressor, and then the plasma membrane of the stressed cells is subsequently selectively permeabilized to provide access of the recombinant protein to SGs. Nuclear import of the protein-of-interest is prevented by blocking nuclear pores with wheat germ agglutinin. This assay allows one to study the molecular mechanisms underlying recruitment of RBPs into SGs quantitatively, in absence of their nuclear import and under controlled conditions. The method allows for a direct comparison of wildtype, mutant or posttranslationally modified RBPs, for addressing the influence of other proteins’ preventing or promoting SG association of RBPs, and is also applicable to synthetic peptides.
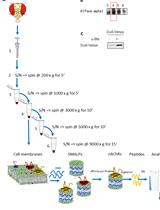
Graphic abstract
Workflow overview for analysis of SG recruitment of recombinant proteins or peptides in semi-permeabilized cells
Background
Stress granules (SGs), membrane-less organelles that contain translationally stalled mRNAs and several RNA-binding proteins (RBPs), form by phase separation as part of a cytoprotective mechanism in response to cellular stress (Protter and Parker, 2016; Gomes and Shorter, 2019). However, SGs have also been suggested to act as precursors for pathological protein aggregates in neurodegenerative diseases, in particular amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (Bentmann et al., 2013; Alberti and Dormann, 2019; Wolozin and Ivanov, 2019; Zhang et al., 2019). First, pathological inclusions containing the RBPs Fused in Sarcoma (FUS) or TAR DNA-binding protein 43 (TDP-43) in ALS/FTD post mortem tissue have been shown to harbor proteins commonly found in SGs, such as PABPC1 or TIA-1 (Dormann et al., 2010; Liu-Yesucevitz et al., 2010; Bentmann et al., 2012; Mackenzie et al., 2017). Second, ALS-associated mutations in RBPs, such as FUS, TDP-43, several hnRNPs and TIA-1, were demonstrated to promote SG localization of these proteins and/or to reduce SG dynamics (Bosco et al., 2010; Dewey et al., 2011; Dormann et al., 2010; Kim et al., 2013; Mackenzie et al., 2017). Third, optogenetic approaches have elegantly demonstrated that persistent or repetitive formation of SGs can lead to cytoplasmic assemblies resembling pathological inclusions (Zhang et al., 2019). Disease-linked RBPs have the tendency to phase separate at high concentration in vitro into liquid-like condensates that can solidify over time, a process promoted by distinct ALS-associated mutations (Murakami et al., 2012; Molliex et al., 2015; Patel et al., 2015; Mackenzie et al., 2017). As SGs contain a high local protein concentration, they could act as condensation sites for disease-linked RBPs and thus promote their pathological aggregation.
What are the molecular mechanisms that determine SG association of RBPs and other proteins? In the past years, the role of individual domains or mutations of RBPs for SG localization was mainly studied by cellular (over) expression of RBPs. However, this approach does not distinguish (i) whether the ectopically expressed protein might promote or suppress the process of SG formation per se, (ii) whether reduced nuclear import of the protein-of-interest might increase its cytoplasmic concentration, and thereby its partitioning into SGs, or (iii) whether a specific mutation directly promotes or suppresses SG association of the protein-of-interest.
Here, we report an assay to study association of recombinant proteins or peptides with pre-formed SGs in semi-permeabilized cells by quantitative fluorescence microscopy. It has been adapted from an original protocol by Adam et al. (1992) who first established selective permeabilization of the plasma membrane by the detergent digitonin to study mechanisms of nuclear import of proteins. Our assay can determine SG association of recombinant proteins or peptides irrespective of their nuclear import, as nuclear pore complexes are selectively blocked by wheat germ agglutinin (WGA), known to bind to nucleoporins (Finlay et al., 1987). By the use of recombinant proteins or peptides, either as fusion protein or untagged, SG association in dependence of the protein sequence or post-translational modifications can directly be correlated with further biochemical characterization in vitro (Hofweber and Dormann, 2018; Ukmar-Godec et al., 2019; Bourgeois et al., 2020). Furthermore, the influence of other factors, such as molecular chaperones or interacting proteins, can be studied under controlled conditions in a quantitative manner (Hofweber and Dormann, 2018; Bourgeois et al., 2020). This protocol was first published by Hofweber and Dormann (2018) to study how SG association of FUS is altered by its nuclear import receptor and chaperone Transportin (TNPO1) as well as arginine methylation.
Materials and Reagents
High precision coverslips, 12 mm No 1.5 (Marienfeld, catalog number: 0 117520 )
Microscopy slides, 76 x 26 mm (any commercial available, e.g., Marienfeld, catalog number: 1000200 )
6- or 10-cm cell culture dishes (any commercially available, e.g., Thermo Fisher Scientific, catalog number: 150288 or Greiner Bio-One, catalog number: 664160 , respectively)
Humid-chamber (e.g., wet Whatman paper covered by parafilm in a 10- or 15 cm cell culture dish) (sterile)
Custom made recombinant protein or peptide (either fluorescently labelled or tagged suitably for immunostaining)
Milli-Q water
HEPES (any commercially available, e.g., Sigma, catalog number: H3375 )
Potassium acetate (any commercially available, e.g., Roth, catalog number: T874.2 )
Magnesium acetate (any commercially available, e.g., MERCK, catalog number: 7463348 )
EGTA (any commercially available, e.g., GERBU, catalog number: 1310 )
Potassium phosphate monobasic (KH2PO4) (any commercially available, e.g., Roth, catalog number: 3904.1 )
Potassium phosphate dibasic (K2HPO4·3H2O) (any commercially available, e.g., Roth, catalog number: 6878.1 )
Magnesium chloride (MgCl2) (any commercially available, e.g., Sigma, catalog number: M2670 )
Sodium chloride (any commercially available, e.g., Roth, catalog number: 0 601.2 )
Potassium chloride (KCl) (any commercially available, e.g., Roth, catalog number: 6781.1 )
Disodiumhydrogen phosphate (Na2HPO4) (any commercially available, e.g., Roth, catalog number: P030.2 )
DMSO (Sigma, catalog number: D2438 )
MG-132 (10 mM in DMSO) (Sigma, catalog number: C2211 )
DMEM high glucose (Thermo Fisher Scientific, Invitogen, catalog number: 61995059 )
10x Trypsin-EDTA (Sigma, catalog number: 59418C )
Sterile PBS solution (see solutions)
Fetal bovine serum (FBS) (Thermo Fisher Scientific, Invitogen, catalog number: 10270-106 )
Gentamycin (e.g., Thermo Fisher Scientific, Invitogen, catalog number: 15710049 )
Digitonin, high purity (Milipore, CAS: 11024-24-1 )
Trypan Blue solution (Thermo Fisher Scientific, Invitogen, catalog number: 152 500 61 )
Wheat germ agglutinin (WGA) (lectin from Triticum vulgaris) solution (Sigma, catalog number: A2408 )
37% Formaldehyde (AppliChem, catalog number: 131328.1211 )
TX-100 (any commercially available, e.g., AppliChem, catalog number: 9002-93-1 )
Serum matching the species that the secondary antibodies was raised in, e.g., donkey serum (Milipore, catalog number: S30 )
Tween-20 (Roth, catalog number: 9127.2 )
DAPI (Sigma, catalog number: D9542 )
ProLong Diamond Antifade mountant (Thermo Fisher Scientific, Invitogen, catalog number: P36961 )
DL-Dithiothreitol (DTT, Applichem, catalog number: A1101.0025 )
Aprotinin (Roth, catalog number: A162.2 )
Leupepstin hemisulfate (Roth, catalog number: CN33.2 )
Pepstatin A (Roth, catalog number: 3083.2 )
0.1% (w/v) poly-L-lysine solution (Sigma, catalog number: P8920 )
Antibody to detect SG core components, e.g., Rabbit anti-G3BP1 antibody (ProteinTech, catalog number: 13057-2-AP )
Optional: Antibody to detect tag of recombinant protein, e.g., Mouse anti-MBP antibody (ProteinTech, catalog number: 66003-1-1g )
Optional: Protein labeling kit (any commercially available, e.g., Thermo Fisher Scientific, Invitogen, catalog number: 46403 )
Fluorescently labelled secondary antibodies (e.g., donkey anti-rabbit Alexa 555 or Alexa 647; Thermo, catalog number: A-31572 or A-31573 ; donkey anti-mouse A-21202)
DMEM/10% FBS/10 µg/ml gentamycin (see Recipes)
1x Trypsin/EDTA (see Recipes)
10 mM MG-132 in DMSO (see Recipes)
Reaction buffer, either transport buffer (TPB) or potassium-phosphate buffer (KPB) (see Recipes)
10% digitonin/reaction buffer (see Recipes)
2 mg/ml WGA in reaction buffer (see Recipes)
3.7% formaldehyde in PBS (see Recipes)
10% and 0.5% TX-100/PBS solution (see Recipes)
PBS/0.1% Tween-20 (see Recipes)
1% donkey serum in PBS/0-1% Tween or alternative blocking solution for immunostaining (see Recipes)
DAPI solution (see Recipes)
Equipment
P1000, P200, P20, P10 pipettes
Fine tip curved tweezers
Cell culture hood
CO2 incubator
pH meter
Vacuum pump
Optional: upright microscope for brightfield microscopy
Confocal scanning microscope (e.g., Leica SP8 with 63x/1.4 oil objective or equivalent) equipped with laser lines matching chosen secondary antibodies and protein/peptide labels. Detection by standard photomultiplier tube (PMT) has been sufficient for visualization and quantification of protein localization to SGs in our hands.
Software
LASX software (Leica)
Fiji/ImageJ (NIH, https://imagej.net/Fiji)
Excel (Microsoft)
GraphPad Prism (GrapPad)
Procedure
Prepare recombinant protein of choice by established purification protocols from bacteria (or equivalent expression system)
For subsequent detection, the protein should either be fused to a fluorescent protein (e.g., EGFP) or another tag suitable for subsequent immunostaining. We have had good experience using a MBP-tag, as it also serves as a solubility tag for aggregation prone proteins and does not interfere with SG recruitment. Alternatively, proteins can also be labeled using commercially available labeling kits. Peptides can already be purchased fluorescently labeled.
Notes:
The recombinant protein should be highly pure, i.e., free of RNA (A260/280 ratio ~0.6-0.7) and of contaminating proteins and/or degradation products. Endotoxin removal is not necessary.
In order to avoid major changes to the buffer composition of the assay, one needs to consider that depending on the storage buffer and concentration of the protein/ peptide stock, additional salt can be introduced into the reaction. If possible, dialyze the recombinant protein(s) into the assay buffer. Alternatively, preparing a highly concentrated stock (e.g., ≥ 100x) of protein/peptide can help to prevent unphysiological salt concentrations in the final assay.
Presence of high concentrations of detergent might interfere with cellular physiology and integrity of the nuclear membrane. The storage buffer should therefore not contain any detergents to improve protein/ peptide solubility.
Protein stabilizing agents in the protein storage buffer, such as glycerol or sucrose, have not interfered with the assay in our hands, provided the protein stock is sufficiently highly concentrated (see point 2 above).
Coat coverslips with poly-L-lysine (optional; 30-60 min)
Note: This step can be omitted if working with well adherent cell lines, such as HeLa cells. However, it can improve cell retention on coverslips and hence increase cell numbers for subsequent analysis.
Work under sterile conditions in cell culture hood.
Spread out coverslips either directly in appropriate cell culture dish (if subsequently used directly) or on parafilm in cell culture dish (if prepared in advance). If coverslips are not considered to be sterile, dip them once into 70% ethanol or absolute isopropanol before spreading them (on parafilm) in cell culture dish. Wait for EtOH/isopropanol to evaporate before proceeding.
Cover each coverslip with 0.1% poly-L-lysine solution using P1000 pipette and incubate for 20-30 min at room temperature.
Collect poly-L-Lysine solution using P1000 pipette (solution can be re-used up to 4 times) or aspirate using vacuum pump.
Wash coverslips once by covering them with sterile MilliQ water.
Aspirate water and let coverslips dry. Coverslips can be stored in a dry and dark place up to 2 weeks. Alternatively, rinse coverslips once with 3 ml medium (in 6 cm dish) if cells are to be seeded directly (Procedure C).
Seed cells onto coverslips (15-20 min)
Estimate confluency of HeLa cell culture.
Aspirate medium and gently wash cells once with sterile PBS. Aspirate PBS and dissociate cells off the dish using pre-warmed 1x trypsin and 2-5 min incubation at 37 °C. Resuspend cells thoroughly in appropriate medium (e.g., DMEM supplemented with 10% FBS and 10 µg/ml gentamicin) to inhibit trypsin activity and to achieve single cells in suspension.
Note: In order to ensure optimal and equal permeabilization of all cells, it is important to avoid cell clusters that would interfere with efficient permeabilization by digitonin. Also, cell density should be kept similar between individual replicates to minimize variability in cell permeabilization.
Calculate cell concentration following standard procedures (e.g., hemocytometer) or based on cell confluency. Seed cells onto (poly-L-lysine coated) coverslips so they reach ~60% confluency at the time of the assay (e.g., HeLa cells double every 20-23 h, so seed them at ~30% confluency). Ensure that coverslips do not float on top of the medium.
Let cells adhere onto coverslips (overnight [12-20 h]).
Stress granule (SG) association assay (~1.5 h)
Incubate cells with 10 µM MG-132 for 2-3 h at 37 °C and 5% CO2 to induce SGs.
Cool humidified chamber on ice.
Place coverslips with cells facing upwards well separated into the humidified chamber (e.g., draw a grid onto the parafilm to keep individual reactions in order).
Aspirate left over medium off the coverslips and wash cells once in 50-100 µl reaction buffer.
Notes:To cover a 12 mm coverslip completely, 40-50 µl solution are usually sufficient. For the washing steps this however should be increased to at least 100 µl per coverslip.
Choice of the reaction buffer depends largely on the nature of the protein or peptide used. We have made good experience with physiological buffers, such as transport buffer (TPB) or potassium phosphate buffer (KPB) that are also commonly used in nuclear import assays using semi-permeabilized cells (Adam et al., 1990; Arnold, 2006). Choose the buffer that results in the best signal-to-noise ratio, as judged by enrichment of your protein-of-interest in SGs compared to the surrounding cytoplasm.
Selectively permeabilize the plasma membrane of cells for 4-5 min on ice using 0.004-0.01% digitonin diluted in your reaction buffer. For this step, place a drop of the digitonin solution (50 µl per 12 mm coverslip) onto the coverslip. Renewing the digitonin solution after 2 min can improve permeabilization.
Note: The exact incubation time and final digitonin concentration largely depends on the digitonin batch, cell type and cell density and needs to be experimentally determined by trypan-blue staining. Insufficient permeabilization will interfere with protein access to SGs. Overpermeabilization can result in a leaky nuclear membrane and, particularly for RBPs, enrichment of proteins in nuclei, which correlates to an underrepresenation of the protein in the cytoplasm (see Notes 1-3 below).
Aspirate the digitonin solution, wash coverslips once briefly and then 4x for 4-5 min on ice in your reaction buffer.
Block nuclear pores by applying 40 µl of 0.2 mg/ml wheat germ agglutinin (WGA) diluted in your reaction buffer onto coverslips. Incubate on ice for 15 min.
Aspirate the WGA solution and replace with your protein(s) in reaction buffer (40 µl final volume/12 mm coverslip). For subsequent detection of recombinant proteins in SGs, ~100-200 nM final protein concentration in our hands is usually sufficient, but this might need to be optimized on an individual basis. In contrast to recombinant proteins, peptides can require higher concentrations, likely due to smaller molecular size and lack of signal amplification by a direct fluorescent label. If the influence of a second protein (such as a protein suspected to act as a molecular chaperone) on SG association is to be tested, a titration of the second protein with the first one held at a constant concentration is recommended.
Note: Include a “buffer only” control to determine bleed-through from the antibody staining for the SG marker (G3BP1) and/or unspecific binding of the antibody used to detect binding of the recombinant protein to SGs.
Incubate reactions for 30 min at room temperature by placing the humidified chamber from the ice onto the bench. Avoid evaporation, as this would result in changes in protein/peptide concentration, and/or drying out of coverslips.
Place humified chamber back on ice and aspirate reaction mix. Wash cells 3 x for 5 min (100 µl final volume/12 mm coverslip) each with reaction buffer (continue immediately with Procedure E).
Immunostaining of SGs (and recombinant protein) (~3 h or overnight)
Fix cells with 3.7% formaldehyde/PBS solution (50 µl final volume/12 mm coverslip) for 7-10 min at room temperature.
Note: Formaldehyde is toxic and should be handled and disposed of with necessary caution.
Wash cells 2 x in PBS (100 µl final volume/12 mm coverslip). (Possible break point; cells can be stored in PBS overnight and immunostaining continued on the following day, although we prefer to perform the primary antibody incubation step overnight, if the protocol cannot be completed on the same day.)
Permeabilize cells using 0.5% TX-100/PBS (50 µl final volume/12 mm coverslip) for 5 min at room temperature. This step ensures access of antibodies and efficient SG staining.
Wash cells 2 x in PBS (100 µl final volume/12 mm coverslip).
Block unspecific binding sites using 1% donkey serum (50 µl final volume/12 mm coverslip) in PBS/0.1% Tween-20 (or alternative blocking buffer, such as 5% BSA or similar) for 10 min at room temperature.
Stain for SGs using G3BP1 antibody (1:1,000 in blocking buffer) and, if required, recombinant protein (e.g., using anti-MBP antibody; 1:1,000 in blocking buffer) for 1-2 h at RT or overnight at 4 °C (50 µl final volume antibody dilution/12 mm coverslip).
Wash coverslips 3 x 5 min each in PBS/ 0.1% Tween-20 (100 µl final volume/12 mm coverslip).
Incubate with secondary antibodies for 30-60 min at room temperature (diluted 1:1,000 in blocking buffer; 100 µl final volume/12 mm coverslip). If bleed-through is a concern, use spectrally well separated dyes (e.g., Alexa 488 and Alexa 647). Staining for SGs in the visible range, however, allows for easier judgement of permeabilization efficiency (see Notes below).
Wash 3x 5 min in PBS/0.1% Tween-20 (100 µl final volume/12 mm coverslip).
Stain nuclei using 0.5 µg/ml DAPI freshly diluted in PBS for 5 min at room temperature (50 µl final volume/12 mm coverslip).
Wash cells twice in PBS (100 µl final volume/12 mm coverslip).
Place a drop (5-10 µl/coverslip) of ProLong Diamond mounting medium onto microscopy slide, avoid air bubbles.
Use fine tip curved tweezers to take individual coverslips, dip excess PBS off using a paper towel and invert coverslips into the mounting medium.
Allow mounting medium to polymerize and dry in a dark place in a horizontal position (overnight). Mounted coverslips can be stored at 4 °C for up to several months.
Imaging (1-4 h, depending on number of samples)
Note: Acquisition settings (laser power, gain, detector settings) need to be determined individually. It is important to avoid pixel saturation in order to allow for quantitative image analysis. Also, samples to be compared directly with each other need to be recorded using identical settings. Check for bleed-through and cross talk applying standard protocols (e.g., by including a “buffer only” control in the subsequent immunostaining or by applying secondary antibody in absence of primary antibody etc).
Establish acquisition settings, using the sample showing the strongest SG recruitment of your protein/peptide.
Avoid imaging of the coverslip edge, as cells here are prone to dry out more easily, resulting in unspecific signal or leaky nuclei.
Choose (equally) well-permeabilized cells. These cells usually display little or no diffuse G3BP1 staining in the cytoplasm and only strong SG enrichment of G3BP1 (Figure 1).
Note: If the experiment is performed the first time, it can be helpful to include one MG-132-treated, non-permeabilized coverslip in the first experiment, in order to distinguish well from poorly permeabilized cells (Figure 1). Staining for TIA-1, another commonly used SG marker, is not well suitable to appropriately judge cell permeabilization, due to its predominantly nuclear localization.
Find ideal z-position by focusing on SGs stained by G3BP1 (or alternative SG marker) antibody. Imaging an area of 73.81 x 73.81 µm (0.059 µm resolution) with a 63x objective results in sufficient pixel area per SG for subsequent analysis. Record several independent fields of view as single z-planes in all channels with a total of at least 10 cells per condition.

Figure 1. Immunostaining for the SG marker G3BP1 in permeabilized vs. unpermeabilized cells. Unstressed (untreated) cells or MG-132-treated cells were fixed and stained for G3BP1 either before (MG-132) or after the cells have been permeabilized using digitonin (MG-132 + perm). Note that well-permeabilized cells (MG-132 + perm) are characterized by strong SG formation in comparison to untreated cells, but display reduced cytoplasmic staining for G3BP1 in comparison to cells that have been fixed directly after MG-132 treatment. Scale bar, 20 µm.
Data analysis
Open image files in Fiji/ImageJ as hyperstack and select G3BP1 (SG) channel.

Choose the magic wand tool. Double-click will allow one to adjust the threshold in order for a SG to be recognized as a ROI (marked by solid line).
Pressing [t] will add this ROI to the ROI manager (alternatively, “Analyze → Tools → ROI Manager → Add [t]”).
Mark all SGs in the image and add them to the ROI manager. ROIs can be renamed as “SG1”, “SG2” etc. by choosing “Rename” in the ROI manager.
Finally draw a ROI outside the area of any cell to determine the background fluorescence intensity. Rename this ROI “background”.
Transfer ROIs into the channel to be quantified. Select each ROI, choose “Properties” in the ROI manager and enter the channel number corresponding to the channel you want to quantify (usually numbered in sequence of recording).
Set measurements by “Analyze” → “Set measurements”. Make sure “Area”, “Mean gray value”, “Min & max gray value” and “Stack position” and “Display label” is generated.
Mark all ROIs and press “Measure”. ROIs can be saved for documentation (“File” → “Save”).
Save generated measurements as an excel file.
Open Excel file. For each image, subtract the “mean” measured for the respective background from the “mean” measured for the individual SG ROIs.
Transfer background corrected measurements into GraphPad Prism and plot the values, e.g. as dot or violin plot, displaying the mean ± SEM in addition to the individual values. To combine several replicates (at least 3 independent replicates are recommended), calculate the mean of the individual experiments and display those as mean ± SEM.
Apply appropriate statistical tests. If two groups are to be compared with each other, apply Shapiro-Wilk normality test to check data distribution. Apply Paired t-test for samples with normal distribution, Wilcoxon-Mann-Whitney test for non-normal distribution. If >3 conditions are to be compared, apply 1-way ANOVA with appropriate post-hoc test. A typical outcome for the SG association of the RBP FUS in absence or presence of its import receptor and chaperone is shown in Figure 2.

Figure 2. Typical data set obtained by the SG association assay in semi-permeabilized cells. A. MBP-FUS-EGFP (green in merged image) associates with SGs (marked by G3BP1 stain, red in merged image) in the absence, but not presence, of its nuclear import receptor and chaperone TNPO1. DAPI is shown in turquoise in the merge. Scale bar: 10 µm. B. Quantification of FUS SG association of a single replicate shown in (B), ***P < 0.001 by Wilcoxon paired t-test.
Notes
The digitonin concentration and incubation time to achieve efficient permeabilization without causing nuclei to get leaky needs to be experimentally determined in advance. Successful permeabilization can be controlled by inverting the coverslip in trypan blue solution. Sufficient permeabilization will cause the nuclei to be stained blue. The incubation time should be kept to a minimum, aiming for the maximum number of cells being permeabilised. Longer digitonin incubation might result in a leaky nuclear membrane, preventing a successful block of nuclear pores by WGA and resulting in a strong nuclear localization of the recombinant protein (particularly if it has nucleic acid binding capacity).
We have observed that the 10% digitonin stock solution in DMSO loses permeabilization efficiency after several freeze-thaw cycles. Therefore, prepare small aliquots (10 µl) and use a fresh aliquot once permeabilization efficiency drops.
The protocol described here uses HeLa cells, as they are well-adherent and their plasma membrane can be selectively permeabilized without compromising the nuclear membrane. However, it is applicable to any other cell line meeting these criteria. Our own observations suggest that HEK293 (T) cells are too fragile and not suitable for selective permeabilization by digitonin.
Cells towards the edge of the coverslip might suffer from evaporation artefacts. Hence, be sure to image cells located in the center of the coverslip and do not allow the coverslip to dry out during the procedure.
We have successfully used both TPB and KPB for SG association of recombinant proteins and peptides. However, we have, on occasion, observed differences in the level of SG association over the surrounding cytoplasmic background signal and recommend a pilot experiment to determine ideal buffer conditions for each protein/peptide.
SGs are RNA-dependent MLOs and dissolve in presence of RNase, however in our experience it is not necessary to prepare RNase-free solutions.
We have made good experience using G3BP1 as a SG marker to verify SG localization of our protein-of-interest. G3BP1 represents a core SG protein and its signal is stable upon digitonin-mediated permeabilization of the plasma membrane. TIA-1, another common marker for SGs, might also be used, but we have observed significant reduction and stronger variability of its intensity in SGs, likely due to wash out after permeabilization. Additionally, unpermeabilized cells display a substantial level of diffuse G3BP1 in the cytoplasm surrounding SGs, allowing to distinguish unpermeablized/not well-permeabilized cells from those suitable for analysis. Unexperienced users can easily learn to judge the level of permeabilization by including a coverslip that had not been treated with digitonin in the G3BP1 immunostaining (Figure 1).
Antibodies against tags might cause considerable background staining, due to cross-reactivity with residual cellular proteins in SGs. Make sure to test for unspecific SG signal caused by your antibody of choice, by performing immunostaining on MG-132-treated cells in the absence of exogenously added recombinant protein.
SG composition has been described to vary depending on the stressor used for SG induction (Aulas et al., 2017). Hence, SG association of the protein/peptide-of-interest might vary depending on reagent/treatment used for SG induction. We have mainly used MG-132, as this treatment is efficient and does not interfere with cell morphology (e.g., rounding up of cells, which might hamper subsequent analysis).
When comparing different proteins that have been labelled using commercially available labeling kits, it is essential to check the degree-of-labeling (DOL) of each protein and to compare only proteins with similar DOL. As labelling efficiency can vary considerably between individual proteins/experiments, this can for example be achieved by doping in unlabelled protein until similar DOLs are reached.
Recipes
DMEM containing 10% FBS, 10 µg/ml gentamycin
Add 50 ml FBS to 500 ml of DMEM
Add 0.5 ml of 10 mg/ml gentamycin
Mix well and store at 4 °C (keep sterile)
1x Trypsin-EDTA
Dilute 5 ml 10x trypsin-EDTA in 45 ml 1x sterile PBS (keep sterile)
1x sterile PBS (can also be stored as 10x solution or purchased)
For 1 L, dissolve 8 g NaCl, 17.8 g Na2HPO4, 2 g KCl and 2.4 g KH2PO4 in 800 ml MilliQ H2O
Adjust pH to 7.4 and adjust volume to 1 L
Autoclave and keep sterile
10 mM MG-132 in DMSO
Prepare 10 mM MG-132 stock solution in DMSO
Aliquot and store at -20 °C
Prepare stocks of DTT and protease inhibitors
Prepare 1 M DTT in MilliQ H2O
Prepare 1 mg/ml each of aprotinin and leupeptin hemisulfate in MilliQ H2O
Prepare 1 mg/ml pepstatin in either DMSO or ethanol
Aliquot and store at -20 °C (can be thawed/frozen multiple times)
1x Transport Buffer (TPB) (can also be prepared and stored as 10x solution)
Prepare 20 mM HEPES pH 7.3-7.4, 110 mM KOAc, 2 mM Mg(OAC)2, 1 mM EGTA
Filter through 0.2 µm filter and store at 4 °C
Directly add 2 mM DTT and protease inhibitors right before use (1 µg/ml each of aprotinin, leupeptin and pepstatin)
1x KPB (can be stored at room temperature)
Prepare 1 M stock solution of KH2PO4
Prepare 1 M stock solution of K2HPO4
Mix 19 ml 1 M KH2PO4 with 81 ml 1 M K2HPO4 to achieve 1 M K-phosphate buffered solution at pH 7.4
Prepare 20 mM potassium phosphate buffer pH 7.4, 5 mM MgCl2, 200 mM KOAc, 1 mM EGTA
10% digitonin in DMSO
Prepare 10% stock solution in DMSO
Note: Digitonin is toxic, weigh in with care, e.g., by wearing a mask!
Prepare 10 µl aliquots and store at -20°C
If required, 1% DMSO stock solutions can be prepared and stored in small aliquots
Prepare digitonin working solution freshly at the digitonin concentration tested to be efficient (see Note 1 above)
2 mg/ml WGA solution
Prepare 2 mg/ml WGA solution in reaction buffer. Aliquot and store at -20 °C
Prepare working dilution freshly (final 0.2 mg/ml) in reaction buffer
3.7% Formaldehyde/PBS
Dilute 37% formaldehyde solution in PBS freshly before use
Note: Formaldehyde is toxic and needs to be handled and disposed of with care (e.g., under fume hood).
0.5% TX-100/PBS
Dissolve 10 ml TX-100 stock solution in 100 ml PBS. Solution might need extended stirring and can be stored at room temperature
Use 10% TX-100/PBS solution to prepare 0.5% TX-100/PBS working solution freshly
1x PBS/0.1% Tween
Dissolve 1 ml Tween-20 in 1 L PBS and stir thoroughly. Solution can be stored at room temperature
0.5 µg/ml DAPI working solution
Dissolve DAPI in MilliQ water (DAPI does not dissolve in PBS!) at 1 mg/ml final concentration
Aliquot and store at -20 °C
Prepare freshly: 0.5 µg/ml DAPI working solution in PBS
Acknowledgments
We thank Erin Sternburg for comments on the manuscript and acknowledge the BioImaging Facility of the BioMedical Center. This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) within Emmy Noether grants DO1804/1-1 and DO1804/1-2, research grant DO1804/3-1, the priority programme SPP2191 (ID 402723784), the Munich Cluster for Systems Neurology (EXC2145 SyNergy-ID 390857198), the Fritz Thyssen Foundation (Az. 10.19.1.001MN) and the Junior Researcher Fund of Ludwig-Maximilians-Universität München.
This protocol has been first published for FUS (Hofweber and Dormann, 2018), and subsequently for Tau and lysine-rich peptides (Ukmar- Godec et al., 2019), as well as for the RBP Cold-inducible RNA-binding protein (CIRBP) (Bourgeois et al., 2020). We acknowledge Adam et al. (1992), who first established the semi-permeabilized cell assay to study nuclear import of proteins, which served as basis for this protocol.
Competing interests
The authors declare no competing interests.
References
- Adam, S. A., Sterne-Marr, R. and Gerace, L. (1992). Nuclear protein import using digitonin-permeabilized cells. Methods Enzymol 219: 97-110.
- Alberti, S. and Dormann, D. (2019). Liquid-Liquid Phase Separation in Disease. Annu Rev Genet 53: 171-194.
- Aulas, A., Fay, M. M., Lyons, S. M., Achorn, C. A., Kedersha, N., Anderson, P. and Ivanov, P. (2017). Stress-specific differences in assembly and composition of stress granules and related foci. J Cell Sci 130(5): 927-937.
- Bentmann, E., Haass, C. and Dormann, D. (2013). Stress granules in neurodegeneration--lessons learnt from TAR DNA binding protein of 43 kDa and fused in sarcoma. FEBS J 280(18): 4348-4370.
- Bentmann, E., Neumann, M., Tahirovic, S., Rodde, R., Dormann, D. and Haass, C. (2012). Requirements for stress granule recruitment of fused in sarcoma(FUS) and TAR DNA-binding protein of 43 kDa(TDP-43). J Biol Chem 287(27): 23079-23094.
- Bosco, D. A., Lemay, N., Ko, H. K., Zhou, H., Burke, C., Kwiatkowski, T. J., Sapp, P., McKenna-Yasek, D., Brown, R. H. and Hayward, L. J. (2010). Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet 19(21): 4160-4175.
- Bourgeois, B., Hutten, S., Gottschalk, B., Hofweber, M., Richter, G., Sternat, J., Abou-Ajram, C., Gobl, C., Leitinger, G., Graier, W. F., Dormann, D. and Madl, T. (2020). Nonclassical nuclear localization signals mediate nuclear import of CIRBP. Proc Natl Acad Sci U S A 117(15): 8503-8514.
- Dewey, C. M., Cenik, B., Sephton, C. F., Dries, D. R., Mayer, P., Good, S. K., Johnson, B. A., Herz, J. and Yu, G. (2011). TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol Cell Biol 31(5): 1098-1108.
- Dormann, D., Rodde, R., Edbauer, D., Bentmann, E., Fischer, I., Hruscha, A., Than, M. E., Mackenzie, I. R., Capell, A., Schmid, B., Neumann, M. and Haass, C. (2010). ALS-associated fused in sarcoma(FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J 29(16): 2841-2857.
- Finlay, D. R., Newmeyer, D. D., Price, T. M. and Forbes, D. J. (1987). Inhibition of in vitro nuclear transport by a lectin that binds to nuclear pores. J Cell Biol 104(2): 189-200.
- Gomes, E. and Shorter, J. (2019). The molecular language of membraneless organelles. J Biol Chem 294(18): 7115-7127.
- Hofweber, M. and Dormann, D. (2019). Friend or foe-Post-translational modifications as regulators of phase separation and RNP granule dynamics. J Biol Chem 294(18): 7137-7150.
- Kim, H. J., Kim, N. C., Wang, Y. D., Scarborough, E. A., Moore, J., Diaz, Z., MacLea, K. S., Freibaum, B., Li, S. and Molliex, A. (2013). Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495(7442): 467-473.
- Liu-Yesucevitz, L., Bilgutay, A., Zhang, Y. J., Vanderweyde, T., Citro, A., Mehta, T., Zaarur, N., McKee, A., Bowser, R., Sherman, M., Petrucelli, L. and Wolozin, B. (2010). Tar DNA binding protein-43(TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One 5(10): e13250.
- Mackenzie, I. R., Nicholson, A. M., Sarkar, M., Messing, J., Purice, M. D., Pottier, C., Annu, K., Baker, M., Perkerson, R. B. and Kurti, A. (2017). TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics. Neuron 95(4): 808-816e809.
- Molliex, A., Temirov, J., Lee, J., Coughlin, M., Kanagaraj, A. P., Kim, H. J., Mittag, T. and Taylor, J. P. (2015). Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163(1): 123-133.
- Murakami, T., Yang, S. P., Xie, L., Kawano, T., Fu, D., Mukai, A., Bohm, C., Chen, F., Robertson, J., Suzuki, H., Tartaglia, G. G., Vendruscolo, M., Kaminski Schierle, G. S., Chan, F. T., Moloney, A., Crowther, D., Kaminski, C. F., Zhen, M. and St George-Hyslop, P. (2012). ALS mutations in FUS cause neuronal dysfunction and death in Caenorhabditis elegans by a dominant gain-of-function mechanism. Hum Mol Genet 21(1): 1-9.
- Patel, A., Lee, H. O., Jawerth, L., Maharana, S., Jahnel, M., Hein, M. Y., Stoynov, S., Mahamid, J., Saha, S., Franzmann, T. M., Pozniakovski, A., Poser, I., Maghelli, N., Royer, L. A., Weigert, M., Myers, E. W., Grill, S., Drechsel, D., Hyman, A. A. and Alberti, S. (2015). A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 162(5): 1066-1077.
- Protter, D. S. W. and Parker, R. (2016). Principles and Properties of Stress Granules. Trends Cell Biol 26(9): 668-679.
- Ukmar-Godec, T., Hutten, S., Grieshop, M. P., Rezaei-Ghaleh, N., Cima-Omori, M. S., Biernat, J., Mandelkow, E., Soding, J., Dormann, D. and Zweckstetter, M. (2019). Lysine/RNA-interactions drive and regulate biomolecular condensation. Nat Commun 10(1): 2909.
- Wolozin, B. and Ivanov, P. (2019). Stress granules and neurodegeneration. Nat Rev Neurosci 20(11): 649-666.
- Zhang, P., Fan, B., Yang, P., Temirov, J., Messing, J., Kim, H. J. and Taylor, J. P. (2019). Chronic optogenetic induction of stress granules is cytotoxic and reveals the evolution of ALS-FTD pathology. Elife 8: 39578.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Hutten, S. and Dormann, D. (2020). A Quantitative Assay to Measure Stress Granule Association of Proteins and Peptides in Semi-permeabilized Human Cells. Bio-protocol 10(24): e3846. DOI: 10.21769/BioProtoc.3846.
Category
Neuroscience > Cellular mechanisms
Cell Biology > Cell-based analysis > Protein interaction
Biochemistry > Protein > Interaction > Protein-ligand interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.