- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Confocal Microscopy of Reovirus Transport in Living Dorsal Root Ganglion Neurons
Published: Vol 10, Iss 22, Nov 20, 2020 DOI: 10.21769/BioProtoc.3825 Views: 4457
Reviewed by: John SL ParkerAbraam YakoubKristin L. Shingler
Original research article
The authors used this protocol in:
Feb 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Neurotropic reoviruses repurpose host machinery to traffic over long distances in neuronal processes and access distal replication sites. Understanding mechanisms of neuronal transmission is facilitated by using simplified in vitro primary neuronal culture models. Advances in the design of compartmentalized microfluidic devices lend robustness to neuronal culture models by enabling compartmentalization and manipulation of distinct neuronal processes. Here, we describe a streamlined methodology to culture sensory neurons dissociated from dorsal root ganglia of embryonic rats in microfluidic devices. We further describe protocols to exogenously label reovirus and image, track, and analyze transport of single reovirus particles in living neurons. These techniques can be adapted to study directed axonal transport of other neurotropic viruses and neuronal factors involved in signaling and pathology.
Background
Viruses from diverse families including the Flaviviridae, Herpesviridae, Picornaviridae, and Rhabdoviridae, breach protective barriers of the nervous system inflicting severe disease and economic burden (Koyuncu et al., 2013; Bohmwald et al., 2018; Tyler, 2018). Mammalian orthoreovirus (reovirus), belonging to the Reoviridae, causes serotype-dependent neuronal infection in a wide variety of young mammals that can result in lethal encephalitis (Tyler et al., 1986; Dermody et al., 2013]. Reovirus is nonenveloped with a segmented dsRNA genome encased by two concentric protein shells and serves as a pliable tool to study viral infections of the nervous system (Dermody et al., 2013). Although cellular and molecular mechanisms of reovirus infection have been widely studied using transformed cell lines, these systems do not capture the complexities of polarized neuronal cells. Viruses infecting neurons must travel over long distances in axons to reach distal sites of replication and egress. To understand mechanisms of reovirus entry and long-distance transport in neurons, we recently adapted techniques to culture primary neurons and image fluorescently labeled reovirus in living cells (Aravamudhan et al., 2020).
In vitro culture of primary neurons from embryonic and adult rodents provide simplified yet robust tools to study neuronal function and pathology. Dissociated sensory neurons from dorsal root ganglia (DRG) can be propagated in vitro and are used widely to study axonal regeneration and peripheral neuropathies (Haberberger et al., 2019; Melli and Hoke, 2009). DRGs are clusters of neuronal cell bodies located proximal to the spinal cord and relay sensory information from the periphery to the central nervous system. DRG neurons are pseudo-unipolar with no dendrites and one axon that bifurcates into two processes, one synapsing with peripheral organs and another connecting to the spinal cord. DRGs are infected during neuroinvasion by several viruses and have been used to study infection mechanisms (Flamand et al., 1991; Smith et al., 2001; Volpi et al., 2018). We describe procedures to cultivate dissociated DRG neurons from rat embryos. We also have used this protocol to cultivate DRG neurons from embryonic mice, which provide a more robust system amenable to genetic manipulation. However, we found that reovirus infects rat neurons more efficiently than mouse neurons in culture (unpublished observations). Therefore, the system must be chosen carefully depending on the application.
In vitro neuron culture in combination with microfluidic devices offers a powerful system to isolate neuronal soma from axons and distinguish axonal transport in the anterograde (soma to axonal termini) and retrograde (axonal termini to soma) directions (Neto et al., 2016). Microfluidic devices allow use of small quantities of reagents and provide access to neuronal microenvironments for studies of axonal growth and injury, biochemical investigations, and high-resolution imaging. The commercial availability of microfluidic devices makes it accessible to everyone without need of sophisticated fabrication instruments. We describe use of a commercially available two-compartment system for culturing DRG neurons and imaging reovirus transport (Nagendran et al., 2018). This device was developed by Xona microfluidics and comes preassembled on optically transparent plastic suitable for high-resolution imaging.
We share an integrated set of protocols to isolate and culture DRG neurons in microfluidic devices to study reovirus transport. In addition, we describe image analysis tools to track single reovirus particles and share programs to obtain biophysical parameters describing reovirus transport. These methods can be adapted to investigate molecular mechanisms of viral neuronal infection. Individual aspects of this procedure can be applied to fluorescently label other nonenveloped viruses, study transport of other neurotropic pathogens, and dissect functions of local RNA and protein metabolism in axonal signaling. Extension of co-culture techniques can be used to study communication between neurons and glia and peripheral tissues. Culture of neurons in microfluidic devices embedded with electrode arrays can be used to simultaneously study electrical activity and biochemical signaling, further enhancing the utility of protocols described here.
Materials and Reagents
Sterilization pouches (VWR, catalog number: 58753-194 )
Transfer pipette (VWR, catalog number: 414004-002 )
Glass Pasteur pipette (VWR, catalog number: 14673-043 )
Plug the wide end with of the glass pipette with cotton. Polish the capillary end using a Bunsen burner by holding the tip in the flame and rotating it rapidly. Pause every few seconds and check the size of the opening to ensure that the edge is smooth and round and the opening is not too narrow. A narrow opening will disrupt cells during the trituration. Autoclave the fire polished Pasteur pipette to sterilize before use.
35 mm Petri dishes (VWR, Greiner Bio-One, catalog number: 82050-540 )
100 mm Petri dishes (VWR, catalog number: 25384-342 )
150 mm Petri dishes (Midland Scientific Inc., Kord Valmark, catalog number: VM 2902 )
Microfluidic devices (Xona microfluidics, catalog number: XC450 )
Glass bottom dishes (MatTek, catalog number: P35G-1.5-14-C )
Closures for dialysis membrane tubing (VWR, Spectrum Laboratories [132735], catalog number: 25224-075 )
Glass vial (Qorpak, catalog number: GLC-00987 )
Dialysis membrane tubing, MWCO 12000-14000 (Fisher Scientific, SpectrumTM S432697, catalog number: 08-667A )
Syringe filter 0.22 μm (VWR, catalog number: 28145-477 )
Conical tubes (VWR, Corning, catalog number: 21008-656 )
Eppendorf tubes (FisherbrandTM, catalog number: 0 5408129 )
E14.5 pregnant Sprague Dawley rat
Immersion oil for microscopy (Cargille, catalog number: 16484 )
Papain (Worthington Biochemical, catalog number: LS003126 )
L-cysteine (Sigma-Aldrich, catalog number: C7352 )
Ovomucoid protease inhibitor (Worthington, catalog number: LK003182 ). Reconstitute in 32 ml of HBSS and store at 4 °C
Collagenase A (Roche, catalog number: 10103578001 )
DNase I (Sigma-Aldrich, Catalog number: D5025-15KU )
Trypan blue (Sigma-Aldrich, catalog number: T8154 )
Hanks’ balanced salt solution without calcium and magnesium, HBSS (CellGro-Corning, catalog number: 21-022-CV )
Poly-D-lysine hydrobromide, PDL (Sigma-Aldrich, catalog number: P0899 ). Reconstitute in HBSS at a concentration of 0.5 mg/ml, filter sterilize, make 5 ml aliquots in sterile conical tubes and store at -20 °C
Laminin (Corning, catalog number: 354232 )
Thaw slowly on ice, make aliquots of different volumes (10 to 50 μl) in sterile tubes, and store at -20 °C. Laminin will form a gel following rapid changes in temperature. For coating, combine multiple aliquots of different volumes to arrive at the final required volume. Do not re-freeze after thawing.
Alexa FluorTM 647 NHS Ester (Thermo Fisher Scientific, catalog number: A37573 ). Reconstitute at 10 mM concentration in DMSO using sterile conditions and make 2 μl aliquots. Store at -20 °C and use within 6 months. The dye solution becomes hydrolyzed over time, resulting in poor labeling efficiency. Do not refreeze after thawing.
Sodium bicarbonate, NaHCO3 (Fisher chemicalTM, catalog number: S233-500 )
NeurobasalTM medium (Gibco, catalog number: 21103049 )
UltraCULTURETM serum-free (Lonza, catalog number: BE12-725F )
GlutaMAXTM supplement (Gibco, catalog number: 35050079 )
Penicillin-streptomycin (Gibco, catalog number: 15070-063 )
HyCloneTM fetal bovine serum (GE Healthcare/Cytiva, catalog number: SH30088.03 ). Make 50 ml aliquots in sterile conical tubes and store at -20 °C. Store thawed aliquots at 4 °C up to 6 months
N-2 supplement (Gibco, catalog number: 17502048 ). Thaw on ice, make 500 μl aliquots in sterile tubes, and store at -20 °C
B-27TM supplement (Gibco, catalog number: 17504044 ). Thaw on ice, make 1 ml aliquots in sterile tubes, and store at -20 °C
Nerve growth factor 2.5S subunit, NGF (Gibco, catalog number: 13257019 ). Thaw on ice, make 15 μl aliquots in sterile tubes, and store at -20 °C. Supplied at 100 µg/ml
Hibernate-E medium (Gibco, catalog number: A1247601 )
Cytosine β-D-arabinofuranoside, AraC (Sigma-Aldrich, catalog number: C1768 ). Reconstitute in water at a concentration of 10 mM, filter sterilize, make 100 μl aliquots in sterile tubes, and store at -20 °C. To limit repeated freeze-thaws, each aliquot can be re-aliquoted in 10 μl upon thawing and stored at -20 °C
NaCl (Fisher ChemicalTM, catalog number: S671-3 )
KCl (Fisher ChemicalTM, catalog number: P217-500 )
Na2HPO4 (Sigma-Aldrich, catalog number: S-7907 )
KH2PO4 (Fisher ChemicalTM, catalog number: P285-500 )
Collagenase-DNase I mixture (see Recipes)
Base medium (see Recipes)
Complete medium (see Recipes)
Low NGF medium (see Recipes)
Papain solution (see Recipes)
10x PBS (see Recipes)
10x sodium bicarbonate buffer (see Recipes)
Equipment
Scissors (Fine Science Tools, catalog number: 91460-11 )
Forceps (2) (Fine Science Tools, catalog number: 11253-20 )
Spring scissors (Fine Science Tools, catalog number: 15023-10 )
Sharp forceps (Fine Science Tools, Dumont #5, catalog number: 11252-20 )
Dissection microscope (CarlZeissTM, StemiTM DV4 )
Spectrophotometer (Thermo Fisher Scientific, model: NanoDropTM 1000 )
Tube rotator (VWR, catalog number: 10136-084 )
Tabletop centrifuge (Thermo ScientificTM, model: SorvallTM LegendTM Micro 17R )
pH meter (VWR, model: SympHonyTM B10P )
Tissue culture (TC) hood (BSL-2 certified)
TC microscope (ZeissTM, model: InvertoskopTM 40C )
Hemacytometer (Bright-LineTM, Millipore sigma, catalog number: Z359629 )
Software
Fiji ImageJ (https://fiji.sc/; Rasband, 1997-2018; Schneider et al., 2012 )
MATLAB (version 9.07.0 [R2019b]; Natick, Massachusetts: The MathWorks Inc. 2019; https://www.mathworks.com/products/matlab.html)
Procedure
Preparation of microfluidic devices
Prepare microfluidic devices 1 to 2 days before plating DRGs. Work in a TC hood and employ sterile handling techniques, maintaining sterility of all reagents.
Arrange up to four pre-assembled Xona microfluidic devices in a 150 mm Petri dish. Place a 35 mm Petri dish without the lid within the larger dish and add 3 ml of sterile water (Figure 1A).
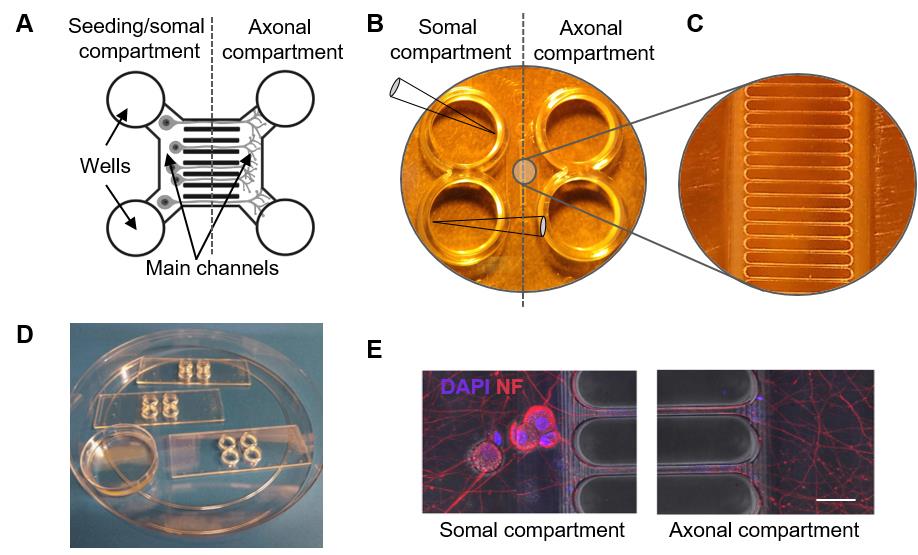
Figure 1. Microfluidic device configuration. A. Schematic depicts a microfluidic device with microgrooves connecting the two compartments. B-C. Each microfluidic device has two compartments (B) connected by thin grooves (enlarged in C). The left and right compartments are arbitrarily designated as somal and axonal compartments. Each compartment has two wells connected by the main channel. During sterilization and coating steps, volumes can be added by pointing the pipette tip away from the entrance to the main channel as depicted in the bottom left well. To seed dissociated neurons, introduce cells at the entrance to the main channel by pointing the pipette as depicted in the top left well. D. Set-up of microfluidic devices for experiments. Three XonaTM microfluidic devices are arranged within a 150 mm Petri dish and covered with a lid to maintain sterility. A smaller 35 mm Petri dish without a lid is filled with water and placed within the larger dish to create a humidified environment. E. Representative micrographs (adapted from Aravamudhan et al., 2020 ) show extension of axons into the axonal compartment 7 days post-plating of dissociated DRG neurons in the somal compartment. Neurons were stained with markers for nuclei (DAPI) and axons (non-phosphorylated neurofilament H, NF). Scale bar, 50 µm.This set up will provide a humidified environment and is essential to minimize evaporation of liquid from microfluidic devices.
Note: For large-scale experiments, it may be more cost-effective to engineer microfluidic devices in-house. Devices at a lower price are also available for in-house assembly on glass coverslips (Example: Xona Microfluidics, SND450). However, we find the latter approach to be cumbersome, often resulting in leaky devices, and we prefer preassembled devices.
Follow these general instructions during handling and preparation of microfluidic devices in subsequent steps (protocol adapted from the manufacturer’s instructions).
Work with four or less devices at a time to maintain sufficient moisture.
While adding liquid, always keep the pipette tip pointing away from the entrance to the main channel (Figures 1B-1C) and dispense slowly, taking care not to introduce bubbles into the main channels or microgrooves. When removing liquid, leave some behind and never allow the main channels to dry out. Bring all liquids to room temperature before adding to devices to avoid formation of bubbles.
To sterilize, coat, or wash devices:
- First remove any liquid from the device and slowly dispense volume into the top left well of the device. Allow liquid to flow through the main channel into the bottom well for 1 min. The right and left compartments of each device should be identical, and the designation is only for the sake of consistency in handling. We mark the top left corner of each device with a permanent marker.
- Fill the bottom left well with liquid.
- Repeat Steps A3ci and A3cii with other devices and wait 5 min to allow liquid to flow through the microgrooves into the right compartment.
- Add volume to the top right well and allow it to flow to the bottom right well for 1 min, add volume to the bottom right well, and wait for 5 min before proceeding to the next step
Following steps outlined in 3, sterilize and wet the surface of devices using 90% ethanol, adding 100 μl to each well. Use of 90-95% ethanol allows uniform wetting of the device with minimal introduction of air bubbles.
Wash devices twice using HBSS, adding 150 μl to each well.
Coat devices with 0.5 mg/ml PDL (100 μl to each well). Cover the Petri dish holding the microfluidic devices with a lid, incubate the devices at 37 °C overnight (minimum 4 h), and then wash the devices twice using HBSS. Do not allow coating to dry out.
Coat devices with laminin solution (100 μl per well; 1:50 dilution of 2.67 mg/ml laminin stock in HBSS) for a minimum of 16 h and up to 48 h.
Before dissociating DRGs for plating, remove laminin, wash devices twice with HBSS, and equilibrate devices in complete medium (100 μl per well) in a cell-culture incubator (37 °C with 5% CO2).
Note: For applications that do not require isolated treatment of neuronal soma and axons or observation of directional transport, DRGs also can be cultivated in glass-bottom MatTek dishes with the same procedure and used for live-cell imaging. For these applications, apply PDL-laminin coatings only to the depressed glass-covered portion of the dish and plate neurons in a small volume of medium only in this area for 4 hours before flooding the entire dish with medium. This strategy allows efficient use of DRGs by culturing neurons only in the imaging area of the dish.
Isolation of embryonic rat DRG neurons
Set up the following before starting dissection.
Autoclave dissection tools (scissors, forceps) ahead of time in sterilization pouches.
Tip: Cover the tips of tools with 10 µl pipette tips to prevent damage.
Before dissection, immerse instruments in a beaker with 70% ethanol. Place Kim wipes at the bottom of the beaker to prevent damage to the tools.
Place a 15 ml conical tube with sterile HBSS on ice and have a sterile transfer pipette handy.
Chill two 100 mm dishes half-filled with HBSS on ice.
Have sterile 100 mm Petri dishes ready for dissection (1 dish for 2 embryos).
Place a 1.5 ml Eppendorf tube with 0.5 ml each of complete medium and HBSS on ice. DRGs will be collected into this solution.
Note: We usually collect DRGs from 4 to 5 embryos into each Eppendorf tube. If you are working with more embryos, set up additional tubes to collect DRGs.
Set up dissecting microscope in a laminar flow hood or a work bench and disinfect work area with 70% ethanol.
Chill tabletop centrifuge to 4 °C.
Prepare papain solution (1 ml to digest DRGs from 4-5 embryos; scale up as required).
Euthanize a E14.5 pregnant rat using isoflurane overdose as described. Move the rat to a sealed anesthetic vaporizer chamber. Set the isoflurane concentration in the vaporizer to 3-4% to induce anesthesia. Once rat is deeply anesthetized, increase the vaporization to 5% to induce death. Leave the rat in the chamber for 3 min after the breathing ceases. Remove the rat from the chamber and proceed to secondary euthanasia by bilateral thoracotomy. Make an incision in the abdominal wall below the sternum, and cut bilaterally to expose the diaphragm. Lacerate the diaphragm using scissors or a scalpel blade.
Remove the amniotic sac containing embryos and place them in a Petri dish with HBSS on ice.
Carefully cut open each amniotic sac, remove the placenta and membranes, isolate the embryo, and immediately decapitate using scissors (Figure 2A). Move the decapitated embryo to a fresh Petri dish containing HBSS placed on ice. Repeat until all embryos are collected.
Note: It is best to isolate embryos and dissect DRGs using sterile conditions in a laminar flow hood. However, we have had success isolating DRGs on a laboratory bench and conducting further enzymatic dissociation and plating using sterile conditions in a TC hood.
To begin DRG dissection, place one embryo ventral side up on a clean Petri dish under a dissection microscope and add a few drops of cold HBSS with a transfer pipette to keep the tissue moist. Complete dissecting DRGs from this embryo before moving to the next one.
Note: Do not let tissues dry or warm up during dissection. Keep buffers and tissue chilled on ice and minimize the time from euthanasia to plating DRGs in order to maximize cell viability. Add a few more drops of HBSS as necessary to keep the tissue cold and moist.
Gently slit the ventral side open and remove viscera with forceps (Figure 2B).
With the ventral side facing up and rostral side close to you, carefully insert spring scissors into the spinal column through the opening at the neck and slit the column open by making superficial longitudinal cuts without disturbing the spinal cord.
Use fine forceps to open the spinal column wide and gently loosen the tissue surrounding the spinal cord leaving the cord intact (this step will make it easier to pull the DRGs along with the spinal cord in the next step) (Figure 2C).
Hold the spinal column down (at the neck) with one pair of fine forceps and slowly remove the spinal cord along with attached DRGs with another pair, working your way from neck to tail (Figure 2D). This process should result in an intact spinal cord fully detached from the surrounding tissue with DRGs attached to its entire length (Figure 2E).
Note: This is a tricky step. You must be gentle not to strip DRGs from the spinal cord. Isolating the maximal number of DRGs will require carefully loosening the tissue and removing small portions of spinal cord with attached DRGs at a time from the embryo.
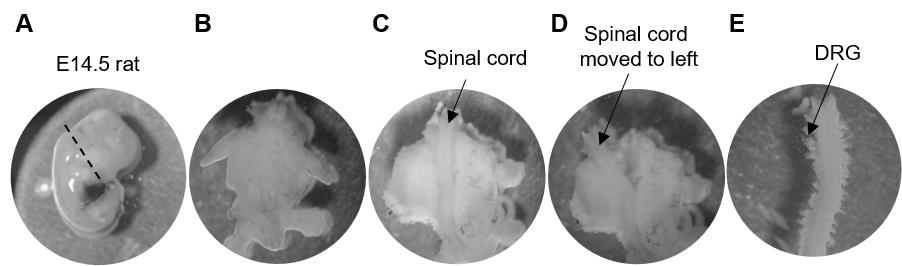
Figure 2. Dissection of DRGs from an embryonic rat. A. Decapitate isolated embryo along the dotted line. B. Ventral view of the decapitated embryo after removal of viscera. C. The spinal cord can be visualized after opening the spinal column. D. The spinal cord along with attached DRGs separated from the surrounding tissue and moved to the side. E. Isolated spinal cord with DRGs to be isolated attached all along the length.Move the spinal cord with attached DRGs to a clean spot on the dish and add a few drops of cold HBSS. With one pair of fine forceps, pin the spinal cord down, and with another pair, isolate DRGs by pinching at their bases or connections to the spinal cord. Collect DRGs into tubes containing a 1:1 mixture of complete medium and HBSS and place on ice, working along the length of the cord. There are 31 pairs of DRGs in rats, but it will not be possible to isolate all of them.
Repeat the procedure to collect DRGs from all embryos. Proceed with dissociation if you are plating on the same day.
Note: DRGs also can be stored up to one week in Hibernate E medium containing 50 units/ml penicillin and 50 μg/ml streptomycin at 4 °C before dissociation and plating. We usually plate DRGs from half the embryos (~4-5) from a rat on the day of dissection and store and plate the rest a week later. This strategy allows enough time to plan and optimize between two sets of experiments using embryos from the same rat.
Note: We find that the exact day of gestation is critical for the ease of isolation of DRGs.
We use Sprague-Dawley rat embryos for our experiments and find E14.5 to be optimal. Even one day later, innervations from DRGs connect them to the surrounding tissue, which makes it harder to remove the DRGs with the spinal cord.
Note: This protocol also works well for isolation and culture of DRGs from E12.5 mouse embryos.
Enzymatic dissociation of DRG
Allow DRGs to settle to the bottom of the tube (~ 5 min).
Notes:
Allow DRGs to settle before every centrifugation step. Not doing so will result in loss of DRGs that stick to the side of the tube while removing supernatant after centrifugation.
Employ sterile handling techniques and work in a TC hood whenever possible from the next step.
Centrifuge at 300 x g at 4 °C for 3 min to pellet DRGs. Using sterile conditions in a TC hood, remove supernatant without getting too close to the pellet, and rinse with 1 ml HBSS. Tap the tube gently to resuspend. Do not pipette DRGs up and down because they will stick to the pipette tip. Centrifuge again and remove the HBSS.
Add 1 ml of papain solution, tap on the tube to resuspend DRGs, and incubate at 37 °C for 17 min.
Centrifuge at 350 x g for 5 min to pellet DRGs and remove papain solution. Do not get too close to the pellet, as stringy material extending from the pellet (collagen and DNA released from lysed cells) will make it easy to dislodge.
Resuspend the DRG pellet by tapping into 100 μl ovomucoid and 1 ml collagenase-DNase I solutions. Incubate at 37 °C for 10 min.
Wash dissociated DRGs twice with 1 ml of base medium. Centrifuge at 350 x g and tap to resuspend during each wash step. Finally, add 1 ml of complete medium to the DRG pellet.
Note: The base and complete media differ only in NGF content. The complete medium contains NGF, while the base medium does not.
Coat a fire-polished pipette with complete medium by pipetting 10 times. This will prevent neurons from sticking to the walls of the pipette during the next dissociation step. Pipette DRG suspension in complete medium ~15 times to dissociate until there are no more visible clusters and the cells are uniformly suspended. Do not introduce bubbles and avoid excess pipetting to limit damage to cells and maintain viability.
Mix 10 μl each of dissociated DRG suspension and trypan blue. Count viable cells that exclude trypan blue using a hemocytometer and determine cell concentration. DRGs isolated from 4 embryos usually yield 1.7 x 106 dissociated viable cells.
Plating and maintenance of dissociated DRG neurons in microfluidic devices
Adjust the concentration of cells as required for plating. We usually plate 0.5-2 x 105 viable cells suspended in 250 μl per microfluidic device.
Remove medium from the left compartment of the microfluidic device (this will be the seeding or somal compartment; Figure 1B) just before seeding cells. Work with one device at a time, and do not allow wells to dry out.
Slowly add 140 μl of cell suspension into the top well placing the pipette tip close to the main channel entrance (Figure 1B). Cells will start to flow through the main channel and reach the bottom well. To counter the flow and allow cells to settle in the main channel, pipette 110 μl of cell suspension into the bottom well near the main channel entrance.
Note: As cells flow through the main channel of the seeded (somal) compartment, a fraction will settle down and extend axons into the opposing (axonal) compartment. It is normal for a majority of cells to settle in the seeded wells.
Return all seeded devices to the 37 °C incubator and allow the cells to adhere overnight.
At ~16 to 18 h post-plating, change to medium containing 2 μM AraC to discourage growth of non-neuronal cells and introduce an NGF gradient to promote axon growth into the axonal compartment. Cells should have adhered and started to extend out axonal processes by this time. To change medium, remove medium from the somal compartment (without drying out) and replace with 300 μl of low-NGF medium containing AraC. Remove medium from the axonal compartment and replace with 200 μl of complete medium containing AraC. Keep pipette tip pointing away from the main channel while changing medium.
Notes:
To maintain fluidic isolation between the somal and axonal compartments, introduce hydrostatic pressure by establishing a difference in volume between the two compartments. In the above case, we introduced a 100 μl volume difference between compartments to maintain a higher concentration of NGF in the axonal compartment. Solvent molecules slowly diffuse from the compartment with higher volume to the one with lower volume. Small molecules or virus added to the compartment with the lower volume remain isolated from the opposing compartment for at least 24 h.
Never completely remove medium, dry out the main channels, or expose neurons to air.
Periodically check the composition of cells in the dishes under a bright-field microscope. Remove AraC and replace with complete medium maintaining the NGF gradient when most of the non-neuronal cells appear dead. We find that treatment with 2 μM AraC for 48 h efficiently kills non-neuronal cells. However, the concentration of AraC (up to 4 μM) and length of treatment should be empirically established.
Change medium every 3 days thereafter. Extension of processes into the axonal compartment can be observed 4 to 5 days post-plating and develop into a dense network by 8 days (Figure 1D). We usually conduct experiments between 8 to 12 days and have maintained DRG neurons in microfluidic devices up to 14 days post-plating.
Note: Synapse formation and maturity of DRG neurons in culture can be assessed using procedures described elsewhere (Joseph et al., 2010; Newberry et al., 2016). DRG neurons cultivated using similar procedures exhibit peak spontaneous electrical activity around 11-14 DIV (Newberry et al., 2016).
Fluorescent labeling of reovirus (adapted from Fecek et al., 2006 )
Propagate and purify reovirus virions as described ( Berard et al., 2009 ; Kobayashi et al., 2010 ). Determine optical density of the purified virus stock at 260 nm using a NanoDrop spectrophotometer. Determine particle concentration using the relationship, 1 OD260 = 2.1 x 1012 particles/ml ( Smith et al., 1969 ). Calculate the volume of virus preparation required to obtain 3 x 1012 particles for labeling.
Note: Reovirus is a BSL2 infectious agent, and standard safety measures approved by an institutional biosafety committee should be employed while handling the virus. We recommend use of gloves and a lab coat and handling the virus only inside a biosafety cabinet.
Freshly prepare and filter-sterilize 50 mM NaHCO3 buffer.
Combine (in a biosafety hood) 50 μl of NaHCO3 buffer, 3 x 1012 reovirus particles, and sterile deionized water to obtain a total volume of 499 μl in a sterile tube. Add virus particles after diluting NaHCO3 with the required volume of water.
Add 1 μl of Alexa FluorTM 647 NHS ester dye (stock 10 mM) to virions diluted in bicarbonate buffer. Thaw an aliquot of the dye just before use, and do not leave for prolonged periods at room temperature.
Set the tube on a rocker or rotator and allow the labeling reaction to proceed at room temperature for 90 min in the dark. The tube containing the labeling mixture can be covered with aluminum foil to protect from light.
While the labeling reaction proceeds, dilute 10x PBS to 1x using sterile deionized water (filter-sterilize in a TC hood, if the water is not sterile) and pre-chill 3 L of 1x PBS at 4 °C.
Dialyze the labeled virus against 1 L of 1x PBS at 4 °C (in the cold room) to remove excess unlabeled dye. We usually change the buffer thrice (at 2 h, 18 h, and 20 h).
Transfer the labeled virus to a glass vial and store protected from light at 4 °C. We usually use the labeled virus in experiments up to 6 months.
Note: Labeled virus preparation can be used even after 6 months if the virions do not degrade. Lack of degradation can be verified by the electrophoretic mobility of viral proteins after subjecting 2 x 1010 particles to electrophoresis in a 10% SDS-polyacrylamide gel, followed by Coomassie brilliant blue staining.
Imaging reovirus transport in living neurons
- Inoculate axonal compartment with fluorescently labeled reovirus 30 min to 1 h before imaging.
- Prepare inoculum containing 109 particles of fluorescently labeled reovirus in 200 μl of complete medium.
Note: The inoculum can be decreased depending on the virus studied and the application. - Remove medium from the axonal compartment without drying out the wells. Ensure that the somal compartment is filled with medium (~300 μl)
- Add 120 μl of virus inoculum into the top well, allow the inoculum to flow through the main channel into the bottom well for 1 min, and add 80 μl of inoculum into the bottom well.
Note: The higher volume in the opposing somal compartment (300 μl) will prevent active diffusion of the virus from the axonal compartment (200 μl).
- Prepare inoculum containing 109 particles of fluorescently labeled reovirus in 200 μl of complete medium.
- Incubate cells with the inoculum at 37 °C for 30 min to 1 h.
- In the meantime:
- Warm 1 ml each of HBSS and complete medium to 37 °C.
- Warm up the imaging chamber, stage, and objective of the microscope to 37 °C. We use a Leica TCS-SP8 laser scanning confocal microscope equipped with a 63x (NA = 1.4; HC PL APO) oil immersion objective and an HyD detector for imaging. The microscope also is equipped with an environmental chamber (GSI TOKAI HIT Standard Heating Stage Top Incubator) and a lens heating collar, which allow maintenance of living cells at 37 °C during imaging. The procedure can be adapted for use with virtually any confocal imaging setup.
Notes:
i.For long-term imaging of living cells, it is essential to equilibrate the imaging chamber to contain 5% CO2 to maintain the pH of medium. However, we image neurons up to 30 min without CO2 equilibration and have not observed any adverse effects on axonal transport.
ii.Any confocal microscope, either spinning disc or line scan microscopes (LSM), can be used to image the transport of fluorescent virus in axons. The microscope must have a fast scanning option to obtain images at a rate of at least 4 frames per second. We have conducted similar analyses of images obtained using a different LSM microscope (Zeiss 710) and a spinning-disc confocal microscope from PerkinElmer. While epifluorescence microscopes also can be used for these analyses, confocal microscopes offer better z-resolution and can distinguish virus motion in different planes containing axons.
- Remove the excess inoculum from the axonal compartment of the microfluidic device, gently wash twice with pre-warmed HBSS, and add 200 μl of complete medium. Note that virions will stick to the coating in the axonal compartment, which makes it difficult to image virus transport in this area.
- Add a drop of oil to the objective. Transfer the microfluidic device to the pre-warmed microscope stage. Move the stage and position the objective roughly in line with the grooves in the microfluidic device. Raise the objective until the oil just starts to contact the bottom of the device. Let the device and medium re-equilibrate to 37 °C for 10 min.
- Focus using the eye piece and brightfield light to find a groove containing axons.
- Establish the imaging parameters. We use Leica Application Suite X (LAS X) software and the following parameters to image reovirus transport in living neurons:
- Bit-depth: 12
- Format: 512 x 512 pixels (82 x 82 μM area with two grooves imaged at once)
- Optical zoom: 2.25
- Bidirectional scanning: ON
- Pinhole: 1 AU
- Frame interval: 0.26 s
- Imaging interval: 30 s (We do not experience much drift over a 1 min span of imaging. You may need to correct for drift during longer intervals of imaging.)
- Set up laser intensity and gain. Turn PMT on.
Note: Exposure to a high laser intensity over prolonged periods can result in photobleaching, while a high gain setting will lead to amplification of background, resulting in a poor signal-to-noise ratio. A good balance between laser intensity and gain should be established for each setup, while achieving a high frame rate to track moving particles. - Acquire time-lapse images and save as “file_name.lif” files. These files can be analyzed further using ImageJ.
- For convenience, we image from the top groove of the device moving to the bottom groove when recording successive movies. Do not image the same groove multiple times to minimize phototoxicity. Restrict imaging time per device to 30 min if the imaging chamber is not equilibrated with CO2.
- Inoculate axonal compartment with fluorescently labeled reovirus 30 min to 1 h before imaging.
Data analysis
- Manual particle tracking and kymograph generation
- Open the file containing time-lapse images of virus transport using Fiji ImageJ.
- From the Fiji task bar, choose Image > Stacks > Tools > Grouped Z Project and max intensity for the projection method. This action will open a new image window with maximum intensity of each pixel projected on to a single frame from the time-lapse image series. Trajectories of individual fluorescent reovirus particles can be clearly visualized in this image (Figure 3A).
- Using the segmented line tool, trace a trajectory from the projected image (Figure 3B) and add the line trace to the ROI manager (Analyze > Tools > ROI Manager > Add). Repeat the process to add more trajectories to the ROI manager. The ROIs can be saved for future reference.
- To generate a kymograph, select the original image window and an ROI of interest from the ROI manager window. From the Fiji taskbar, select Analyze > Multi Kymograph > Multi Kymograph and a Linewidth of 3. This action will open a new image window displaying the kymograph of the selected trajectory (Figure 3C; kymographs display positions along the ROI on X-axis and time along Y-axis).
Note: If the image has multiple color channels, split the individual channels into separate images using Image > Color > Split Channels, and choose the image containing fluorescent reovirus to produce a kymograph. - The slope of individual line segments (distance divided by the interval) in the kymograph provides the velocity of the virus punctum during the assessed interval.
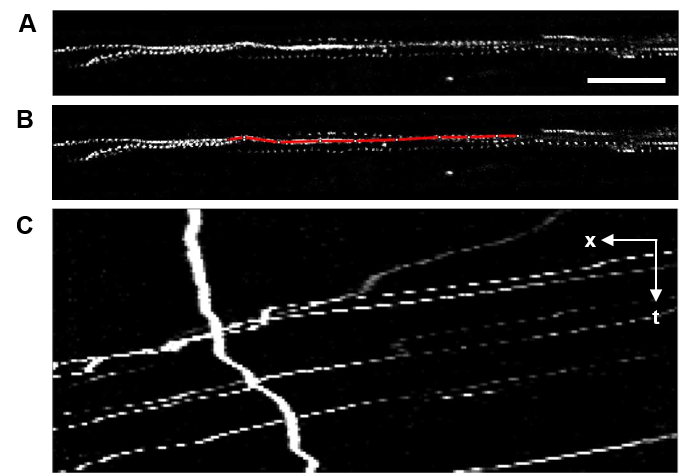
Figure 3. Particle tracking and kymograph generation using ImageJ. A. Representative image shows trajectories of multiple reovirus particles in axons within a groove of a microfluidic device. Trajectories were obtained by projecting maximum intensities from each frame of a time-lapse image series spanning 26 seconds onto a single frame using the “grouped Z-project” function in ImageJ. Scale bar, 10 µm. B. A single trajectory was manually traced (red line). C. Kymograph was generated along the traced line in (B) using the “Multi kymograph” function in ImageJ. Each line in the kymograph shows positions of a virus punctum along an axon (X-axis) over time (Y-axis). - Automated tracking of single reovirus particles
- To track the motion of individual reovirus particles in a field of view and record trajectories, open the file (file_name.lif) containing time-lapse images using Fiji ImageJ.
- Load TrackMate plugin (Plugins > Tracking > TrackMate). Algorithms and parameters used within this plugin have been described in detail (Tinevez et al., 2017).
- TrackMate should automatically extract and display the imaging parameters from the metadata associated with the image file (Figure 4A). Verify that these values are correct and proceed to the next step. Use arrows at the bottom-right of the screen to move to the previous or the next step.
- Establish parameters to identify individual reovirus puncta based on size and intensity.
- Choose the DoG detector.
- The “Estimated blob diameter” is a rough estimate of the size of the particle. A diameter of 0.75 μm works optimally to identify particles using our experimental conditions. Threshold values are dependent on the image quality (signal intensity over background) and can be empirically determined as described below. Check to use “Median filter” and “Sub-pixel localization”.
- Choose preview and employ the Zoom tool to ensure that the threshold used results in optimal identification of virus puncta. Particles that are selected will be marked with a magenta circle. Refer to Figures 4B-4C for differences in particle selection between low and apt threshold values.
- If the images have a high signal-to-background ratio, no further filtering of the spots is required.
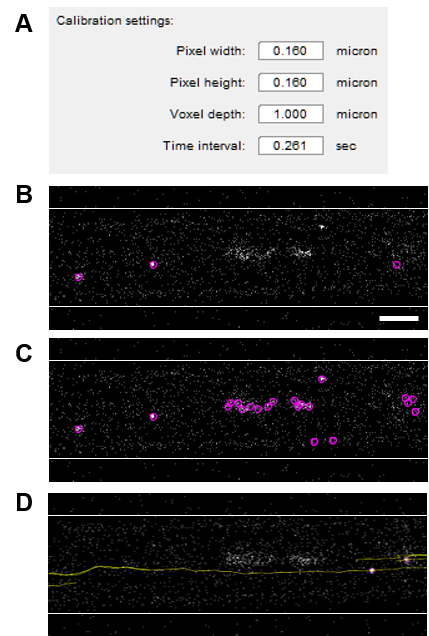
Figure 4. Tracking reovirus motion using TrackMate. A. An initial TrackMate window shows parameters automatically extracted from the image metadata. B-C. A representative example showing selection of fluorescent reovirus particles or background puncta within DRG axons in a groove while using an optimum (B) or a low (C) threshold value, respectively. Scale bar, 5 µm. D. Identified tracks (yellow) superimposed on a single image from the time-lapse series. Refer to Video 1 for the corresponding movie showing motions of individual reovirus puncta tracked.Video 1. Tracking reovirus motion in axons. A representative output movie from TrackMate showing motion (tracked in yellow lines) of fluorescent reovirus virions over 26 seconds within DRG axons in a microfluidic device groove. The device is oriented with the somal compartment to the left and the axonal compartment to the right of the imaged groove.
- Navigate forward and set up parameters to track particle trajectories.
- Choose “simple LAP tracker”. This tracker is optimal to identify trajectories of particle transport in axons, which follow a relatively linear track.
- We recommend the following initial parameters: Linking max distance: 2.5 μm, Gap-closing max distance: 3 μm, Gap closing max frame gap: 3 frames. These parameters may need to be refined empirically for optimal tracking.
- Identified trajectories will be displayed superimposed on the image as shown in Figure 4D and Video 1.
- Filter and record trajectories of interest in the next few steps.
- Filter out tracks with displacements less than 6.5 μm.
- Trajectories fitting this criterion will be displayed superimposed on the image screen.
Note: Manually review the time-lapse images or use the projected image to determine whether the chosen parameters resulted in the selection of most trajectories. Also, determine whether individual tracks are linked correctly. For example, the motion of a single virus particle can be split into multiple trajectories if parameters in step 5b are not optimal. Iteratively go to previous steps and alter parameters to achieve optimal trajectory selection. Given the nature of single-particle tracking, it may not be possible to perfectly identify the trajectories of all particles in a field-of-view. Therefore, choose parameters that result in consistently reproducible results across different images and experiments. We recommend reading “Configuring the simple LAP tracker” section of the TrackMate documentation for more detailed information. - When satisfied with the parameters, use the “Analysis” button to compile the results. Save the results under the window “Spots in tracks statistics” with a file name of choice (Example: Virus_Trajectory_1).
- Extraction of kinetic parameters from particle trajectories
- We use a homebuilt MATLAB script for analyzing trajectories of individual reovirus particles obtained using steps described in the previous section. The script was written for MATLAB 2019(b) executed in Windows 10 and provided as a Supplement with this protocol (Trajectory_analysis_MATLAB).
- The script has three parts: (a) user definition, (b) main program, and (c) exporting files.
- User definition: The user needs to define 5 parameters before running the program.
- Filename: Provide the complete path including the name of the file to be analyzed with in quotations (Example: “C:\Users\Name\Desktop\Virus_Trajectory_1”).
- no_fr: Number of frames captured in 1 s. We set the default value to four, as images were acquired every ~250 ms.
- threshold_time: Sets the minimum duration threshold for including trajectories. The default value is set to 5 s (trajectories of particles that were tracked for less than 5 s will be discarded).
- thresh_dist: Defines the minimum distance that a particle should move within 1 s for it to be considered “in motion”. The default value is set at 0.15 µm (if a particle moves < 150 nm in a 1 s interval, it will be considered “paused” during that interval).
- threshold_vel: Threshold to obtain instantaneous velocity of particles while “in motion.” The default value is set at 0.15 µm/s.
- Main program: This section forms the bulk of the program, computes biophysical parameters based on input values from the previous section, and need not be modified by the user.
- Export Files: Executing the MATLAB program by clicking the “Run” button will generate five Microsoft Excel files with the following computed values:
- 'inst_vel.xlsx': This file provides a list of instantaneous velocities for all trajectories from the input file. Instantaneous velocities are calculated as the displacement of individual punctum in consecutive ~1 s intervals. Instantaneous velocities from all trajectories from the input file are concatenated into a single column.
- 'msd.xlsx': This file provides the information required for plotting mean square deviation. The columns correspond to time, average mean square deviation and the corresponding standard deviation, and the number of data points.
- 'track_vel.xlsx': This file provides the average velocity of particles in individual trajectories and computed as the total distance traveled by a particle divided by the trajectory duration.
- 'frac_duration.xlsx': Individual values in this file provide the fraction of time in each trajectory that a particle spends in a “paused” state.
- 'pause_duration.xlsx': This file provides a list of pause durations from all trajectories in the input file compiled into a single column. Pause duration is defined as the amount of time spent by a particle in each “pause” (instantaneous velocity < 0.15 µm/s)
- User definition: The user needs to define 5 parameters before running the program.
Notes
Cortical and hippocampal neurons also can be used to study axonal transport of virus. Although other groups have reported success in culturing cortical neurons in microfluidic devices, in our experience, these neurons do not survive using the same growth conditions in these devices.
Fluorescent labeling of capsid proteins using reactive Alexa Fluor dyes provides a quickly adaptable strategy for labeling and imaging viruses not amenable to genetic tagging with fluorescent proteins. However, loss of labels from virions following uncoating limits use of this strategy to study early steps in virus entry and trafficking.
We have successfully labeled reovirus with other Alexa fluorophores (Alexa FluorTM 488 TFP ester and Alexa FluorTM 546 NHS ester) using the protocol described here. However, Alexa FluorTM 488 TFP ester tends to be less stable and hydrolyzes faster after dissolving compared with Alexa FluorTM 647 NHS ester, resulting in large differences in labeling efficiency between batches.
Recipes
Collagenase-DNase I mixture
Dissolve 15 KU DNase I in 1.5 ml of HBSS
Dissolve 12 mg solid Collagenase A in 10.5 ml HBSS
Combine Collagenase A and DNase I solutions
Add 120 μl of 100x penicillin-streptomycin
Filter sterilize in the TC hood with 0.22 μm filter
Make 1 ml aliquots in sterile tubes and store at -20 °C
Base medium (50 ml)
24 ml NeurobasalTM medium
24 ml UltraCULTURETM medium
1.5 ml HyClone serum
0.5 ml N-2
1 ml B-27
0.5 ml GlutaMAX
0.5 ml Penicillin-Streptomycin
Note: Prepare medium freshly as required and store at 4 °C. We do not store medium longer than a week.
Complete medium
Add NGF to base medium to a final concentration of 50 ng/ml
Low NGF medium
Add NGF to base medium to a final concentration of 20 ng/ml
Papain solution (prepare freshly before DRG dissection)
Freshly prepare a saturated solution of NaHCO3: 0.24 g in 2.4 ml water
Dissolve 16 mg L-cysteine in 6 ml HBSS
Mix 1.5 ml HBSS + 4 µl saturated NaHCO3 solution + 0.5 ml L-cysteine solution
Sterilize the solution using a 0.22 μm filter
Add papain to final concentration of 20 units/ml
Activate papain at 37 °C for 20 min, and then keep on bench while preparing DRGs for dissociation.
10x PBS (1 L; pH 7.4)
80 g NaCl
2 g KCl
14.4 g Na2HPO4
2.4 g KH2PO4
Dissolve in 900 ml of deionized water, adjust pH to 7.4, and bring volume to 1 L. Autoclave and store at room temperature
10x sodium bicarbonate buffer (50 mM, pH 8.5)
Dissolve 0.21 g of NaHCO3 in 5 ml of sterile deionized water. Prepare this solution freshly before labeling reovirus
Acknowledgments
We thank members of the Dermody laboratory, Bruce Carter, and Amrita Pathak for technical advice and helpful discussions in the development of protocols described here. This work was supported by U. S. Public Health Service (https://www.usphs.gov/) awards R01 AI038296 (PA and TSD). Additional support was provided by UPMC Children’s Hospital of Pittsburgh (PA and KR) and the Heinz Endowments (TSD). Protocols were adapted from previous work “Reovirus uses macropinocytosis-mediated entry and fast axonal transport to infect neurons” ( Aravamudhan et al., 2020 ).
Competing interests
The authors declare no competing interests.
Ethics
All animal work was conducted in accordance with the Public Health Service policy and approved by the University of Pittsburgh Institutional Animal Care and Use Committee. All personnel adhered to applicable federal, state, local, and institutional laws and policies governing ethical animal research including the Animal Welfare Act (AWA), the Public Health Service (PHS) Policy, the Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research and Training, and the Health Research Extension Act of 1985.
References
- Aravamudhan, P., Raghunathan, K., Konopka-Anstadt, J., Pathak, A., Sutherland, D. M., Carter, B. D. and Dermody, T. S. (2020). Reovirus uses macropinocytosis-mediated entry and fast axonal transport to infect neurons. PLoS Pathog 16(2): e1008380.
- Berard, A. and Coombs, K. M. (2009). Mammalian reoviruses: propagation, quantification, and storage. Curr Protoc Microbiol Chapter 15: Unit15C 11.
- Bohmwald, K., Galvez, N. M. S., Rios, M. and Kalergis, A. M. (2018). Neurologic alterations due to respiratory virus infections. Front Cell Neurosci 12: 386.
- Dermody, T. S., Parker, J. S. and Sherry, B. (2013). Orthoreoviruses. In. Fields Virology. Knipe, D. M. and Howley, P. M. (Eds.). Philadelphia, Lippincott Williams & Wilkins. 2: 1304-1346.
- Fecek, R. J., Busch, R., Lin, H., Pal, K., Cunningham, C. A. and Cuff, C. F. (2006). Production of Alexa Fluor 488-labeled reovirus and characterization of target cell binding, competence, and immunogenicity of labeled virions. J Immunol Methods 314(1-2): 30-37.
- Flamand, A., Gagner, J. P., Morrison, L. A. and Fields, B. N. (1991). Penetration of the nervous systems of suckling mice by mammalian reoviruses. J Virol 65(1): 123-131.
- Haberberger, R. V., Barry, C., Dominguez, N. and Matusica, D. (2019). Human dorsal root ganglia. Front Cell Neurosci 13: 271.
- Joseph, D. J., Choudhury, P. and Macdermott, A. B. (2010). An in vitro assay system for studying synapse formation between nociceptive dorsal root ganglion and dorsal horn neurons. J Neurosci Methods 189(2): 197-204.
- Kobayashi, T., Ooms, L. S., Ikizler, M., Chappell, J. D. and Dermody, T. S. (2010). An improved reverse genetics system for mammalian orthoreoviruses. Virology 398(2): 194-200.
- Koyuncu, O. O., Hogue, I. B. and Enquist, L. W. (2013). Virus infections in the nervous system. Cell Host Microbe 13(4): 379-393.
- Melli, G. and Hoke, A. (2009). Dorsal root ganglia sensory neuronal cultures: a tool for drug discovery for peripheral neuropathies. Expert Opin Drug Discov 4(10): 1035-1045.
- Nagendran, T., Poole, V., Harris, J. and Taylor, A. M. (2018). Use of pre-assembled plastic microfluidic chips for compartmentalizing primary murine neurons. J Visualized Exp (141).
- Newberry, K., Wang, S., Hoque, N., Kiss, L., Ahlijanian, M. K., Herrington, J. and Graef, J. D. (2016). Development of a spontaneously active dorsal root ganglia assay using multiwell multielectrode arrays. J Neurophysiol 115(6): 3217-3228.
- Neto, E., Leitao, L., Sousa, D. M., Alves, C. J., Alencastre, I. S., Aguiar, P. and Lamghari, M. (2016). Compartmentalized microfluidic platforms: The unrivaled breakthrough of in vitro tools for neurobiological research. J Neurosci 36(46): 11573-11584.
- Rasband, W. S.(1997-2018). ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA.
- Schneider, C. A., Rasband, W. S. and Eliceiri, K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9(7): 671-675.
- Smith, G. A., Gross, S. P. and Enquist, L. W. (2001). Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc Natl Acad Sci U S A 98(6): 3466-3470.
- Smith, R. E., Zweerink, H. J. and Joklik, W. K. (1969). Polypeptide components of virions, top component and cores of reovirus type. Virology 39(4): 791-810.
- Tinevez, J. Y., Perry, N., Schindelin, J., Hoopes, G. M., Reynolds, G. D., Laplantine, E., Bednarek, S. Y., Shorte, S. L. and Eliceiri, K. W. (2017). TrackMate: An open and extensible platform for single-particle tracking. Methods 115: 80-90.
- Tyler, K. L., (2018). Acute viral encephalitis. N Engl J Med 379(6): 557-566.
- Tyler, K. L., McPhee, D. A. and Fields, B. N. (1986). Distinct pathways of viral spread in the host determined by reovirus S1 gene segment. Science 233(4765): 770-774.
- Volpi, V. G., Pagani, I., Ghezzi, S., Iannacone, M., D'Antonio, M. and Vicenzi, E. (2018). Zika virus replication in dorsal root ganglia explants from interferon receptor1 knockout mice causes myelin degeneration. Sci Rep 8(1): 10166.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Aravamudhan, P., Raghunathan, K. and Dermody, T. S. (2020). Confocal Microscopy of Reovirus Transport in Living Dorsal Root Ganglion Neurons. Bio-protocol 10(22): e3825. DOI: 10.21769/BioProtoc.3825.
Category
Neuroscience > Peripheral nervous system
Microbiology > Microbial cell biology
Cell Biology > Cell-based analysis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.