- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Attachment of a 32P-phosphate to the 3′ Terminus of a DNA Oligonucleotide
Published: Vol 10, Iss 20, Oct 20, 2020 DOI: 10.21769/BioProtoc.3787 Views: 5355
Reviewed by: Anna A. ZorinaEmilie ViennoisAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Biochemical investigations into DNA-binding and DNA-cutting proteins often benefit from the specific attachment of a radioactive label to one of the two DNA termini. In many cases, it is essential to perform two versions of the same experiment: one with the 5′ DNA end labeled and one with the 3′ DNA end labeled. While homogeneous 5′-radiolabeling can be accomplished using a single kinase-catalyzed phosphorylation step, existing procedures for 3′-radiolabeling often result in probe heterogeneity, prohibiting precise DNA fragment identification in downstream experiments. We present here a new protocol to efficiently attach a 32P-phosphate to the 3′ end of a DNA oligonucleotide of arbitrary sequence, relying on inexpensive DNA oligonucleotide modifications (2′-O-methylribonucleotide and ribonucleotide sugar substitutions), two enzymes (T4 polynucleotide kinase and T4 RNA ligase 2), and the differential susceptibility of DNA and RNA to hydroxide treatment. Radioactive probe molecules produced by this protocol are homogeneous and oxidant-compatible, and they can be used for precise cleavage-site mapping in the context of both DNase enzyme characterization and DNA footprinting assays.
Graphic abstract
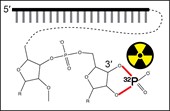
Background
A common goal in the study of DNA and DNA-binding proteins is to identify the position and frequency of DNA backbone cleavage events, an essential set of experiments if the investigator aims to determine the mechanism of a novel DNase enzyme or discern protection parameters from DNA footprinting assays. In the classic version of this experiment, the investigator first uses T4 polynucleotide kinase (PNK) to transfer a 32P-phosphate from [γ-32P]-adenosine triphosphate (ATP) to the 5′-OH of a DNA oligonucleotide of interest, yielding a 5′-radiolabeled DNA “probe”. After subjecting the probe to a cleavage process, the investigator then resolves the cleavage fragments by polyacrylamide gel electrophoresis (PAGE), visualizes them by autoradiography, and determines cleavage position and frequency from band location and intensity, respectively. However, if a DNA probe molecule is cleaved more than once, the described experiment only allows detection of the cleavage event closest to the 5′ end. To detect additional cleavage events with equivalent positional resolution, an analogous experiment must be performed with the 3′ DNA end radiolabeled.
Because there is currently no known kinase activity that can specifically transfer a 32P-phosphate to the 3′-OH of a DNA oligonucleotide, standard 3′-radiolabeling procedures instead employ either a polymerase or a terminal transferase to catalyze primer extension at the 3′ DNA end, using [α-32P]-nucleotide triphosphates (NTPs) as a reagent (in different strategies, the sugar moiety of the NTPs could be 2′-deoxyribose, ribose, or 3′-deoxyribose) (Wu et al., 1976). While the resulting probes are suitable for certain experiments, they cannot always be used for unambiguous DNA cleavage-site mapping (i.e., determining at exactly which phosphodiester a DNA molecule was cut, as measured from the 3′ DNA end). For example, the polymerase- or transferase-based methods suffer from heterogeneity in probe length and/or incorporation of radionuclides at internal backbone positions, both of which can destroy the unique band-to-fragment correspondence that is required for unambiguous cleavage-site mapping. When using probes generated by one of these methods, a single band on a PAGE autoradiograph could be attributed to any of several distinct radioactive cleavage fragments that migrate with indistinguishable electrophoretic mobility. Note that these problems arise even when the 32P-phosphate lies between the final two nucleotides, as certain nuclease enzymes and chemical processes (such as those associated with permanganate footprinting) can remove the final nucleoside without its phosphate. In one protocol that cleverly addresses this problem, the authors expose an internal radionuclide by sequentially treating a ribonucleotide-tailed radioprobe with alkali, phosphatase, and periodate. However, while this procedure does yield the desired outcome–a DNA probe homogeneously labeled with a terminal 3′-32P-phosphate–the initial tailing step requires high concentrations of expensive radioactive ribonucleotides to promote efficient labeling (Jay et al., 1974).
More recently, many synthesis companies have begun to offer custom DNA oligonucleotides with a fluorophore chemically conjugated to the 3′ end. If a laboratory has the optical instrumentation to directly image fluorophore-tagged DNA in a polyacrylamide gel, these reagents offer a viable alternative to 3′-radiolabeled DNA probes. Still, this detection technique is far less sensitive than autoradiography and will miss minor products in complex cleavage patterns. Additionally, common fluorophores may be degraded under the harsh chemical treatments associated with certain experiments, such as permanganate footprinting.
Considering the previously mentioned shortcomings, the ideal 3′-labeling strategy would: (1) take advantage of the high sensitivity and chemical compatibility of 32P-phosphate-based detection; (2) produce a homogeneous population of DNA probe with a single 3′-terminal radionuclide; and (3) employ catalytically efficient combinations of common enzymes and substrates to make the procedure inexpensive and fast. We developed a protocol to meet these needs in the context of our studies of DNA binding and cleavage by the RNA-guided DNase CRISPR-Cas12a (Cofsky et al., 2020). In our protocol, we begin with a DNA oligonucleotide in which the penultimate and final deoxyribonucleotides have been changed to a 2′-O-methylribonucleotide and a ribonucleotide, respectively, which are standard and cheap modifications at oligonucleotide synthesis companies (Figure 1A). To introduce the 32P-phosphate to the system, we first perform a standard 5′-radiolabeling procedure on a “phosphate shuttle” RNA oligonucleotide (Figure 1B). We then form a double-stranded junction by annealing the phosphate shuttle RNA oligonucleotide, the DNA oligonucleotide, and a bridging splint RNA oligonucleotide (Figure 1C). With the sugar modifications at the DNA oligonucleotide’s 3′ end, this bridge structure can be efficiently ligated by T4 RNA ligase 2, attaching the 32P-phosphate to the 3′-OH of the DNA oligonucleotide (Nandakumar and Shuman, 2004; Figure 1D). After ligation, we treat the probe precursor with hot hydroxide, leaving the DNA intact and dissociating the RNA into mononucleotides that can be removed with a spin column (Figures 1E, 1F). The final result is a DNA oligonucleotide with a penultimate 2′-O-methylribonucleotide and a 3′-terminal ribonucleotide bearing a 2′,3′-cyclic-32P-phosphate (Figure 2).
Figure 1. Protocol for 3′-radiolabeling, in brief
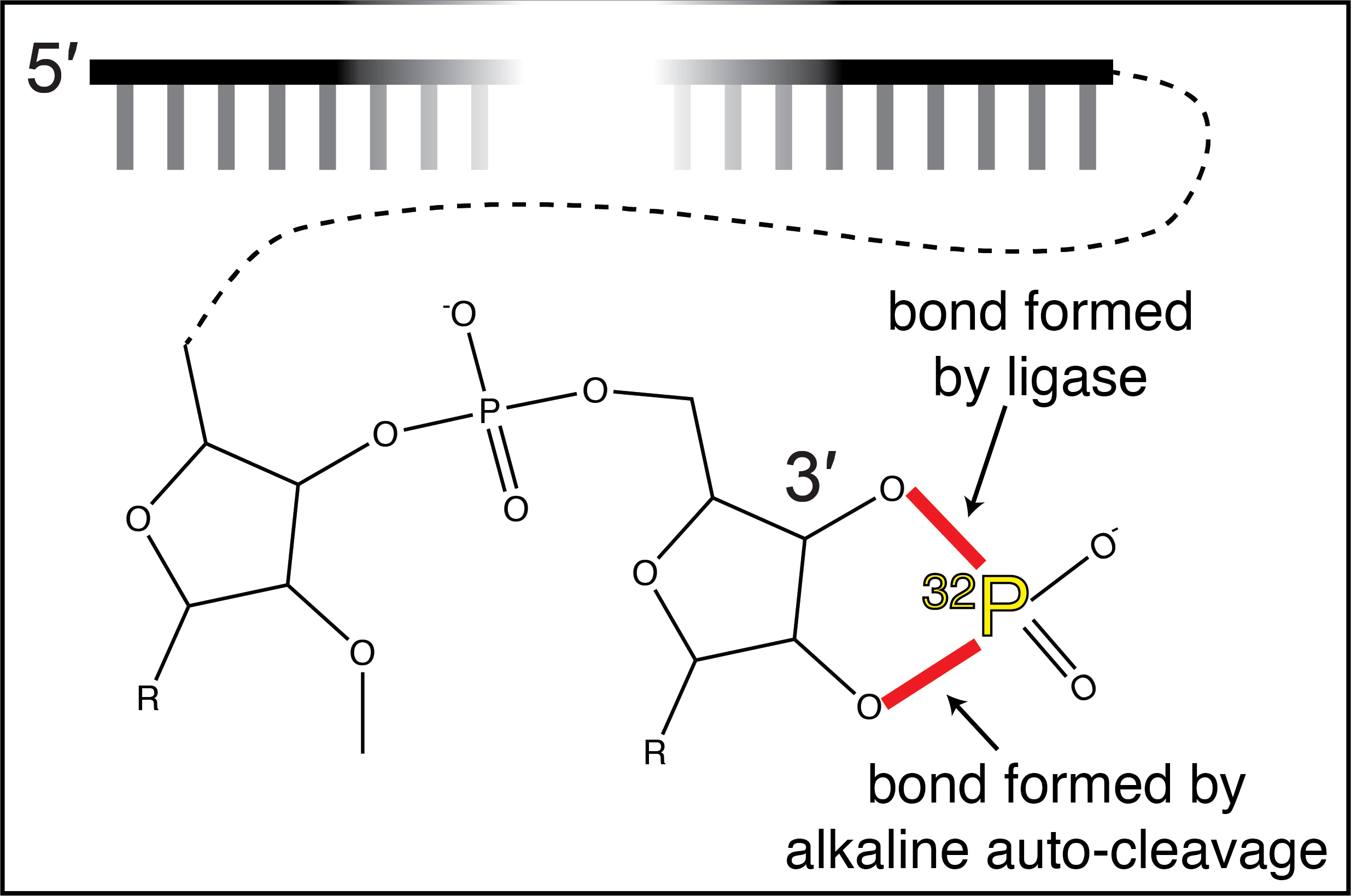
Figure 2. Chemical structure of final 3′-radiolabeled product
This method produces homogeneous 3′-terminally radiolabeled DNA probe with minimal reagent consumption and sample manipulation. Additionally, if both 5′-and 3′-radiolabeling experiments are being performed, the investigator only needs to purchase a single radionucleotide ([γ-32P]-ATP) to produce the required probes, as compared to polymerase-/transferase-based strategies that would also require expensive [α-32P]-NTPs. In our experiments, we used these probes to detect Cas12a-generated DNA cleavage products (including minor and transient products), leading to the unforeseen finding that Cas12a cleaves its DNA substrate multiple times to form a five-nucleotide gap. We also used these probes in permanganate footprinting experiments that yielded highly position-specific information about the conformation of Cas12a-bound DNA (Cofsky et al., 2020). Additionally, by using a 32P-phosphate label in these experiments, we avoided the problems associated with the use of fluorophores in the presence of strong oxidants. Finally, when using this protocol to generate many variant DNA probes, reagent costs were ~90% the cost of the cheapest commercially synthesized fluorophore-tagged DNA oligonucleotides, according to list prices at the time of writing. Therefore, this method may be used to prepare inexpensive DNA probes for any cleavage-site mapping experiment that requires high precision, sensitivity, and chemical compatibility.
Materials and Reagents
- Illustra MicroSpin G-25 columns (Cytiva, catalog number: 27532501 ), stored at room temperature
- 3′-modified DNA oligonucleotide (Integrated DNA Technologies) (see Procedure for details), stored at -20 °C
- Phosphate shuttle RNA oligonucleotide (in vitro transcribed or commercially synthesized, Integrated DNA Technologies), stored at -80 °C
- Splint RNA oligonucleotide (in vitro transcribed or commercially synthesized, Integrated DNA Technologies), stored at -80 °C
- T4 polynucleotide kinase, 10,000 U/ml (New England Biolabs, catalog number: M0201S ), stored at -20 °C
- T4 polynucleotide kinase reaction buffer, 10x (New England Biolabs, catalog number: B0201S ; included with product M0201), stored at -20 °C
- [γ-32P]-ATP EasyTide, 3000 Ci/mmol, 10 mCi/ml, 1 mCi (PerkinElmer, catalog number: BLU502A001MC ), stored at -20 °C
- T4 RNA ligase 2, 10,000 U/ml (New England Biolabs, catalog number: M0239S ), stored at -20 °C
- T4 RNA ligase 2 reaction buffer, 10x (New England Biolabs; included with product M0239S ), stored at -20 °C
- Common reagents:
NaOH (1.35 M stock)
HCl (1.35 M stock)
KCl
Tris-Cl
EDTA
Sodium citrate
MilliQ water (all instances of the word “water” in this protocol refer to MilliQ water) - 5x annealing buffer (see Recipes), stored at 4 °C
- 1x RNA storage buffer (see Recipes), stored at 4 °C
Equipment
- Standard shielding and other protective equipment for work with 32P
- Temperature-controlled block and/or thermocycler
- Vortexer and microcentrifuge (for use with MicroSpin G-25 columns)
- Optional: benchtop radiation counter (Bioscan QC-4000)
Procedure
- Order the DNA oligonucleotide with the appropriate modifications
- Order your DNA oligonucleotide with sequence of interest (Note 1). The 3′-most nucleotide should be modified to be a ribonucleotide. The penultimate nucleotide (immediately 5′ to the ribonucleotide) should be modified to be a 2′-O-methylribonucleotide. To specify these modifications in the IDT ordering interface, begin with an order for an RNA oligonucleotide at 100 nmol scale. If the sequence of your DNA oligonucleotide is NNNNNN-3′, your sequence input should be NNNNmNrN.
In the example presented in this protocol, the DNA oligonucleotide sequence is:
5′-GTCATAATGATTTTATCTTCTGGATTGTTGTAAGCAGCATTTGAGCAAAAATCTGTTmGrC-3′, where all letters indicate deoxynucleotides except “mG” (which indicates 2′-O-methylguanosine) and “rC” (which indicates cytidine). - Resuspend the DNA oligonucleotide to 10 µM in water (Note 2).
- Order your DNA oligonucleotide with sequence of interest (Note 1). The 3′-most nucleotide should be modified to be a ribonucleotide. The penultimate nucleotide (immediately 5′ to the ribonucleotide) should be modified to be a 2′-O-methylribonucleotide. To specify these modifications in the IDT ordering interface, begin with an order for an RNA oligonucleotide at 100 nmol scale. If the sequence of your DNA oligonucleotide is NNNNNN-3′, your sequence input should be NNNNmNrN.
- Order or transcribe the phosphate shuttle RNA oligonucleotide and the splint RNA oligonucleotide
- The phosphate shuttle RNA oligonucleotide can be ordered from an oligonucleotide synthesis company or produced by in vitro transcription. If producing by in vitro transcription (Notes 3 and 4), use a method whose final product contains a 5′-OH (either by treating a T7 RNA polymerase product with phosphatase or including a self-cleaving hammerhead ribozyme in the transcript). It is essential that the phosphate shuttle RNA contain a 5′-OH for the 5′-radiolabeling step.
- Phosphate shuttle RNA oligonucleotide sequence (Note 5):
5′-GGGUCGGCAUGGCAUCUC-3′ - We include below the sequence of an in vitro transcription DNA template for T7 RNA polymerase to generate the phosphate shuttle RNA oligonucleotide (Note 6). Transcripts from this template contain a 5′ hammerhead ribozyme that cleaves to yield a 5′-OH on the final phosphate shuttle oligonucleotide.
5′-GTCGAAATTAATACGACTCACTATAGGCGACCCCTGATGAGGCCTTCGGGCCGAAACGGTGAAAGCCGTAGGGTCGGCATGGCATCTC-3′
- Phosphate shuttle RNA oligonucleotide sequence (Note 5):
- The 5′ half of the splint RNA oligonucleotide should be complementary to the phosphate shuttle RNA oligonucleotide. The 3′ half of the splint RNA oligonucleotide should be complementary to the 3′ end of the DNA oligonucleotide to be labeled. The splint RNA oligonucleotide can be ordered from an oligonucleotide synthesis company or produced by in vitro transcription. The substituents on either end of this oligonucleotide are unimportant, as the ligase enzyme will bind to its center.
- Sequence of splint RNA oligonucleotide in this example:
5′-GGAGAUGCCAUGCCGACCCGCAACAGAUUUUUGCUCA-3′ - We include below the sequence of an in vitro transcription DNA template for T7 RNA polymerase to generate the splint RNA oligonucleotide:
5′-GTCGAAATTAATACGACTCACTATAGGAGATGCCATGCCGACCCGCAACAGATTTTTGCTCA-3′
- Sequence of splint RNA oligonucleotide in this example:
- Resuspend each RNA oligonucleotide to 10 µM in RNA storage buffer.
- The phosphate shuttle RNA oligonucleotide can be ordered from an oligonucleotide synthesis company or produced by in vitro transcription. If producing by in vitro transcription (Notes 3 and 4), use a method whose final product contains a 5′-OH (either by treating a T7 RNA polymerase product with phosphatase or including a self-cleaving hammerhead ribozyme in the transcript). It is essential that the phosphate shuttle RNA contain a 5′-OH for the 5′-radiolabeling step.
- 5′-radiolabel the phosphate shuttle RNA oligonucleotide
- Combine the following reagents (50 µl total reaction volume) (Note 7):
5 µl 10x T4 PNK buffer
30 µl water
5 µl phosphate shuttle RNA oligonucleotide (10 µM stock)
2.5 µl T4 PNK enzyme (10,000 U/ml stock)
7.5 µl [γ-32P]-ATP (3000 Ci/mmol, 10 mCi/ml stock) - Allow phosphorylation to progress at 37 °C for 2 h.
- Heat-inactivate the T4 PNK by incubating at 65 °C for 20 min.
- Add 50 µl water to the reaction.
- If interested in determining yield, use the benchtop radiation counter to measure the radioactivity in the reaction at this step.
- Spin the reaction through a MicroSpin G-25 column (no resin pre-equilibration necessary), following the manufacturer’s instructions. This step largely removes any free [γ-32P]-ATP that may remain after the reaction.
- Assuming complete recovery of the RNA oligonucleotide, you should have 100 µl of eluate containing 500 nM RNA oligonucleotide, some fraction of which has a 5′-32P-phosphate. Use the benchtop radiation counter to measure the radioactivity in the eluate, and determine the efficiency of 5′-radiolabeling by comparing to the radioactivity measurement recorded before free ATP removal.
- Combine the following reagents (50 µl total reaction volume) (Note 7):
- Anneal ligase substrates
- For each DNA oligonucleotide to be 3′-radiolabeled, combine the following (33 µl total reaction volume):
6.6 µl 5x annealing buffer
0.3 µl water
24 µl 5′-radiolabeled phosphate shuttle RNA oligonucleotide (500 nM stock, directly from eluate in previous step)
1 µl modified DNA oligonucleotide (10 µM stock)
1.1 µl splint RNA oligonucleotide (10 µM stock) - Place the annealing reaction on a thermocycler, and run the following temperature program (Note 8):
- 95 °C, 2 min.
- Cool to 25 °C over the course of 40 min.
- Hold at 25 °C.
- For each DNA oligonucleotide to be 3′-radiolabeled, combine the following (33 µl total reaction volume):
- Ligate the DNA oligonucleotide to the phosphate shuttle RNA oligonucleotid
- To the 33-µl annealing reaction, add:
4 µl 10x T4 RNA ligase 2 reaction buffer
1 µl 40 mM MgCl2
2 µl T4 RNA ligase 2 (10,000 U/ml stock) - Incubate at 37 °C overnight (~16 h). If performing quality control, keep a sample from immediately after this incubation to run on a gel and determine ligation efficiency (Figure 3).
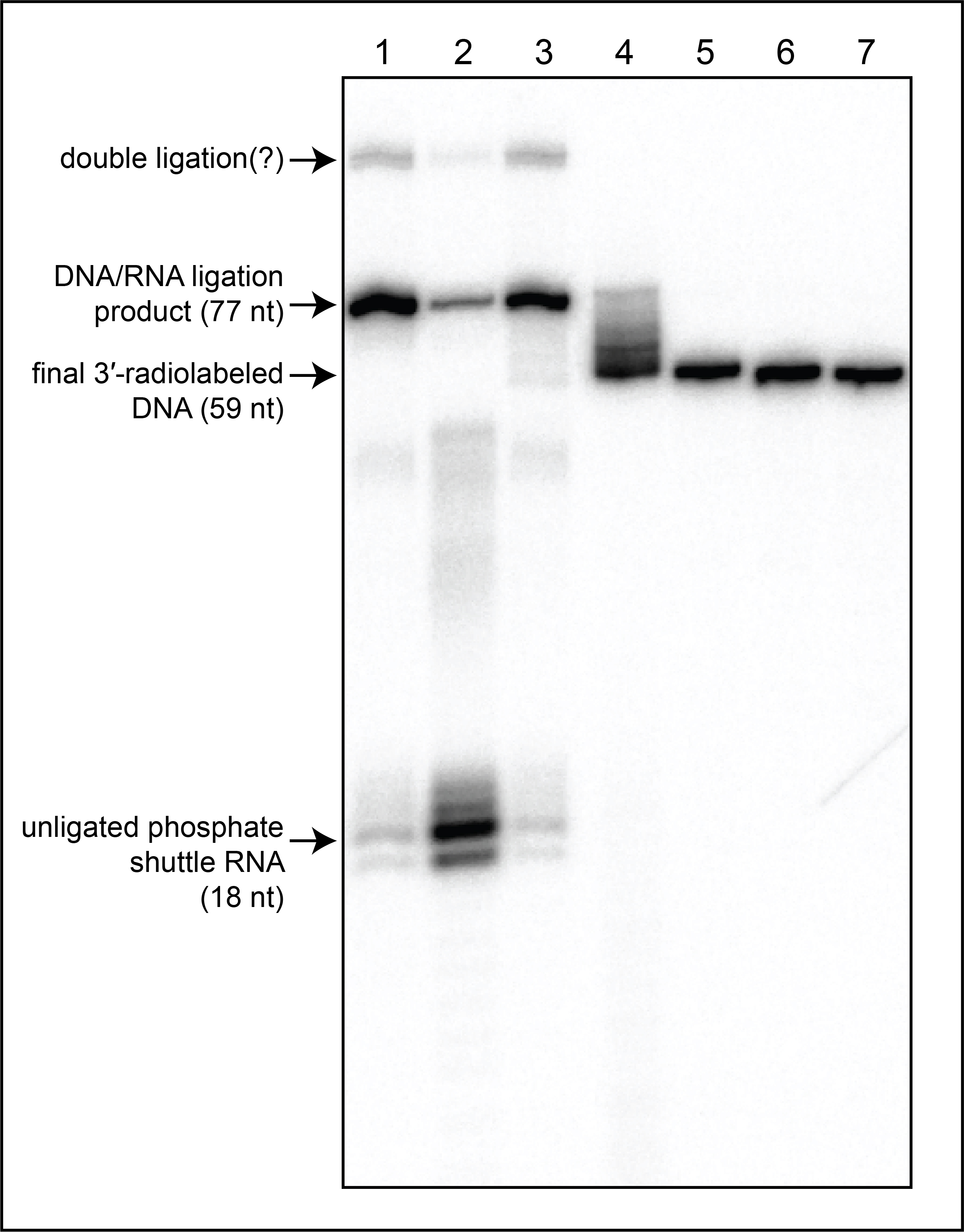
Figure 3. Example autoradiograph of denaturing PAGE analysis. Lane 1: products of ligation reaction, standard protocol. Lane 2: products of ligation reaction using a DNA oligonucleotide that omits the 2′-O-methyl modification at the penultimate position (but retains the ribonucleotide modification at the final position), demonstrating the importance of the 2′-O-methyl modification for ligation efficiency. Lanes 3-7: products after RNA degradation of the ligation products in lane 1 with [NaOH] at 20 mM (lane 3), 50 mM (lane 4), 100 mM (lane 5), 150 mM (lane 6), and 200 mM (lane 7). The band in lane 6 represents the product generated by the conditions recommended in this protocol.
- To the 33-µl annealing reaction, add:
- Degrade RNA
- To the 40-µl ligation reaction, add 5 µl 1.35 M NaOH. Mix thoroughly.
- Incubate at 95 °C for 10 min.
- Place on ice, and immediately add 5 µl 1.35 M HCl. Mix thoroughly.
- Remove salts and mononucleotides by buffer exchange
- Pre-equilibrate a MicroSpin G-25 column with 20 mM Tris-Cl, pH 7.9 (room temperature) according to the manufacturer’s instructions (5 x 400-µl equilibration spins).
- Apply the 50-µl degradation reaction to the column, and spin according to the manufacturer’s instructions. Add 50 µl water to the eluate.
- At this point, the sample should have 100 µl total volume, containing 10 mM Tris-Cl, pH 7.9 and 100 nM 3′-radiolabeled DNA oligonucleotide (as well as the heat-inactivated kinase and ligase enzymes).
- Yield and quality check
- Use the benchtop radiation counter to check the yield of transfer of radioactivity from the phosphate shuttle RNA oligonucleotide to the final DNA oligonucleotide. We commonly see recovery of ~75% of transferable phosphates (Note 9).
- Run a sample of the labeled DNA oligonucleotide on denaturing PAGE to verify that the sample contains a pure radiolabeled DNA fragment of the correct size (Notes 10 and 11, Figure 3).
- Store the radiolabeled DNA at -20 °C. The limiting factor for the expiration of the radiolabeled oligonucleotide is the radioactivity, as the chemical stability of the product (> years) long outlasts the radioactive decay lifetime of 32P (half-life 14 days). The product can continue to be used as long as it retains enough radioactivity for the researcher’s particular experimental purposes.
Notes
- For the purposes of mapping a DNA cleavage site with single-nucleotide resolution, the baseline purification option from oligonucleotide synthesis companies is often insufficient (the product contains significant concentrations of truncated synthesis products that may confound experimentally important cleavage products). To achieve adequate DNA oligonucleotide purity before 3′-radiolabeling, either perform a PAGE-purification in house or opt for more extensive purification in your commercial order.
- Standard precautions for working with RNA should be followed. Keep gloves and surfaces clean, and when possible, use filter-containing pipette tips. We do not detect any RNase activity when using MilliQ water in reactions containing RNA, but the experimentalist can purchase commercially certified RNase-free water if there are suspicions of RNase contamination. RNA samples should be stored at -80 °C (ideal) or -20 °C when not in use, but they can be briefly handled on a room-temperature bench between the steps described in this protocol.
- Consult any standard protocol for the optional steps involving in vitro transcription, PAGE purification, analytical PAGE, and autoradiography.
- If RNA oligonucleotides are produced by in vitro transcription, the resulting transcript should then be PAGE-purified for optimal results.
- The phosphate shuttle RNA oligonucleotide sequence is arbitrary, and we simply included here a sequence that worked well in our experiments. This sequence could presumably be changed provided that compensatory substitutions are also made in the splint RNA oligonucleotide.
- For the in vitro transcription template sequences included in this protocol, only the top strand is shown. Double-stranded DNA templates were assembled and amplified by polymerase chain reaction.
- This protocol yields enough 5′-radiolabeled phosphate shuttle RNA oligonucleotide to 3′-radiolabel 4 different DNA oligonucleotides (with a final yield of 100 µl at 100 nM for each 3′-radiolabeled DNA oligonucleotide). Scale the volumes up or down to suit the needs of your particular application, taking care to spread the sample across multiple G-25 MicroSpin columns if the volume limitation of an individual column is exceeded.
- In the absence of a thermocycler, an alternative annealing procedure is as follows: incubate samples at 95 °C for 2 min on a heat block, turn off the heat block, and allow the system to gradually equilibrate to room temperature.
- The ~75% yield is after accounting for the stoichiometric excess of phosphate shuttle RNA oligonucleotide in the annealing reaction, which results in a 17% subpopulation whose 5′-32P-phosphate is unavailable for transfer. The absolute fraction of recovered radioactivity is ~60%.
- The most common contaminants are depurination products and n+1 products (some fraction of the DNA molecules contain two ribonucleotides on the 3′ end instead of one, as the n+1 phosphodiester was not cleaved in the RNA degradation reaction). In our preparations, these products were minor or not detected at all (there was variation between different probe preparations), and they did not interfere with our experiments. If the contaminant concentration is too high for your particular oligonucleotide preparation, try quenching the RNA degradation reaction with HCl at a variety of temperatures or time points. Identify a combination of temperature and quenching time that yields an acceptable balance of depurination products (which accumulate over time) and n+1 products (which disappear over time due to cleavage and release of the terminal ribonucleotide). Alternatively, PAGE-purify the 3′-radiolabeled DNA oligonucleotide for maximal purity.
- The identity of the final species can also be confirmed by treating with T4 PNK and checking by PAGE/autoradiography that all radioactivity has moved from the DNA oligonucleotide to an inorganic phosphate (due to the phosphatase activity of the T4 PNK enzyme that removes 2′,3′-cyclic phosphates).
Recipes
- 5x annealing buffer
50 mM Tris-Cl, pH 7.9 at 25 °C
250 mM KCl
5 mM EDTA - 1x RNA storage buffer
2 mM sodium citrate, pH 6.4 at 25 °C
0.1 mM EDTA, pH 8.0
Acknowledgments
This protocol was developed with support from a National Science Foundation Graduate Research Fellowship (J.C.C.) and National Science Foundation Award # MCB-1817593 (J.A.D.). The protocol was originally developed for use in Cofsky et al., 2020. We thank David Colognori for a critical reading of the protocol draft.
Competing interests
The Regents of the University of California have patents issued and pending for CRISPR technologies on which J.A.D. is an inventor. J.A.D. is a cofounder of Caribou Biosciences, Editas Medicine, Scribe Therapeutics, and Mammoth Biosciences. J.A.D. is a scientific advisory board member of Caribou Biosciences, Intellia Therapeutics, eFFECTOR Therapeutics, Scribe Therapeutics, Mammoth Biosciences, Synthego, Felix Biosciences, and Inari. J.A.D. is a Director at Johnson & Johnson and has research projects sponsored by Biogen, Pfizer, AppleTree Partners, and Roche. J.C.C. has no competing interests to report.
Ethics
This protocol does not include the use of human or animal subjects.
References
- Cofsky, J. C., Karandur, D., Huang, C. J., Witte, I. P., Kuriyan, J. and Doudna, J. A. (2020). CRISPR-Cas12a exploits R-loop asymmetry to form double-strand breaks. Elife 9: e55143.
- Jay, E., Bambara, R., Padmanabhan, R. and Wu, R. (1974). DNA sequence analysis: a general, simple and rapid method for sequencing large oligodeoxyribonucleotide fragments by mapping. Nucleic Acids Res 1(3): 331-353.
- Nandakumar, J. and Shuman, S. (2004). How an RNA ligase discriminates RNA versus DNA damage. Mol Cell 16(2): 211-221.
- Wu, R., Jay, E., and Roychoudhury, R. (1976). Nucleotide Sequence Analysis of DNA. In: Methods in Cancer Research. Vol. 12. Busch, H. (Ed.). Academic Press, 87-176.
Article Information
Copyright
![]() Cofsky and Doudna. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Cofsky and Doudna. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Cofsky, J. C. and Doudna, J. A. (2020). Attachment of a 32P-phosphate to the 3′ Terminus of a DNA Oligonucleotide. Bio-protocol 10(20): e3787. DOI: 10.21769/BioProtoc.3787.
- Cofsky, J. C., Karandur, D., Huang, C. J., Witte, I. P., Kuriyan, J. and Doudna, J. A. (2020). CRISPR-Cas12a exploits R-loop asymmetry to form double-strand breaks. Elife 9: e55143.
Category
Molecular Biology > DNA > DNA modification
Molecular Biology > DNA > DNA labeling
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.