- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Live-cell Imaging and Quantitative Analysis of Meiotic Divisions in Caenorhabditis elegans Males
Published: Vol 10, Iss 20, Oct 20, 2020 DOI: 10.21769/BioProtoc.3785 Views: 4948
Reviewed by: Mara SchvarzsteinAnand Ramesh PatwardhanAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Live-imaging of meiotic cell division has been performed in extracted spermatocytes of a number of species using phase-contrast microscopy. For the nematode Caenorhabditis elegans, removal of spermatocytes from gonads has damaging effects, as most of the extracted spermatocytes show a high variability in the timing of meiotic divisions or simply arrest during the experiment. Therefore, we developed a live-cell imaging approach for in situ filming of spermatocyte meiosis in whole immobilized C. elegans males, thus allowing an observation of male germ cells within an unperturbed environment. For this, we make use of strains with fluorescently labeled chromosomes and centrosomes. Here we describe how to immobilize male worms for live-imaging. Further, we describe the workflow for the acquisition and processing of data to obtain quantitative information about the dynamics of chromosome segregation in spermatocyte meiosis I and II. In addition, our newly developed approach allows us to re-orient filmed spindles in silico, regardless of the initial 3D orientation in the worm, and analyze spindle dynamics in living worms in a statistically robust manner. Our live-imaging approach is also applicable to C. elegans hermaphrodites and should be expandable to other fluorescently labelled nematodes or other fully transparent small model organisms.
Keywords: Male meiosisBackground
In the past, studies on male meiosis have been performed on isolated and stained specimens to image meiotic spindles, with obvious limitations of using fixed specimens (Boring, 1909; Boveri, 1909; Goldstein, 1977). Live-imaging was applied to analyze the dynamics of meiosis in extracted spermatocytes of a number of species, including insects, such as grasshoppers (Nicklas and Staehly, 1967; Nicklas and Koch, 1969; Ault, 1986), the crane-fly Nephrotoma suturalis (Janicke and LaFountain, 1982; LaFountain et al., 2002 and 2012; Forer et al., 2018), the flatworm Mesostoma ehrenbergii (Fegaras and Forer, 2018), the fruit fly Drosophila melanogaster (Bonaccorsi and Gatti, 2017) and spiders (Golding and Paliulis, 2011; Felt et al., 2017). Extracted insect spermatocytes were observed with phase-contrast and polarized light microscopy and very elegant experiments were performed on spindles or single chromosomes using micromanipulators and laser microsurgery (King and Nicklas, 2000; Nicklas et al., 2001; Forer et al., 2013; Sheykhani et al., 2017). In general, a limitation of this approach is that cells have to be removed from the gonad and elaborate protocols have to be developed for each experiment using a specific organism to account for the physiological requirements for the culture media (i.e., pH, osmolarity and temperature).
In the nematode Caenorhabditis elegans, microscopic investigation of spermatocyte meiosis was previously achieved by labelling of isolated and fixed gonads with antibodies (Howe et al., 2001; Shakes et al., 2009; Peters et al., 2010; Schvarzstein et al., 2013). This method, while powerful for analyzing the molecular composition of male meiotic spindles, cannot be used to study spindle dynamics. Alternatively, male gonads can be dissected, and the extracted germ cells can be filmed using Normarski optics. Applying this approach (Figures 1A-1D), it was possible to film cell divisions but the components of the meiotic spindle such as chromosomes and spindle poles were impossible to detect reliably without appropriate fluorescent labels (Shakes et al., 2009; Gleason et al., 2012; Vielle et al., 2016; Hu et al., 2019). Moreover, the imaging time was limited to only a few minutes as the cells regularly arrested before completion of the meiotic divisions. In our hands, about 90% of the spermatocytes arrested prior to the end of the meiotic divisions due to unknown reasons.
We developed a live-imaging approach that enabled us to study spindle dynamics in 3D in living immobilized males (Figures 1E-1G). With this newly developed approach it is now possible to achieve a parallel imaging of multiple spindles over about 90 min without any damaging effects on spindle dynamics (Fang-Yen et al., 2009; Kim et al., 2013). Another method applies patterned agarose pads created by using Vinyl Records as a mold (Rivera Gomez and Schvarzstein, 2018). However, this method relies on the use of anesthetics to completely immobilize the worm for imaging. Here we intended to avoid the use of anesthetics for animal immobilization, thus excluding any possible side effects of drugs on the dynamics of cell division. A technical problem of this approach that had to be solved was the bleaching of the fluorescent markers during the course of imaging, i.e., a new sample had to be prepared about 1.5 h after the recording started. However, the high number of dividing cells within a single animal enables to collect sufficient data to determine the characteristics of dividing spindles in spermatocyte meiosis.
The challenge was to calculate the properties of dividing spindles over time. Due to the 3D live imaging spindles were not perfectly aligned in the imaging plane but rather randomly oriented in 3D with varying spindle axis angles in each frame. We solved this problem by computationally rotating each spindle in 3D to have a standard orientation for data analysis and then calculating kymographs for each fluorescent channel. By analyzing the kymographs, we could determine the dynamics of the chromosomes and the spindle poles over time with high accuracy. Further, this workflow can be used to image dividing mitotic germ cells in the distal gonad of males and hermaphrodites, which is also a cell type very difficult to image in situ. Our pipeline of imaging acquisition and data processing of cell division in C. elegans males can also be applied for imaging dividing embryos in hermaphrodites of this species and should also be applicable to other small model organisms with fluorescently labeled chromosomes and centrosomes.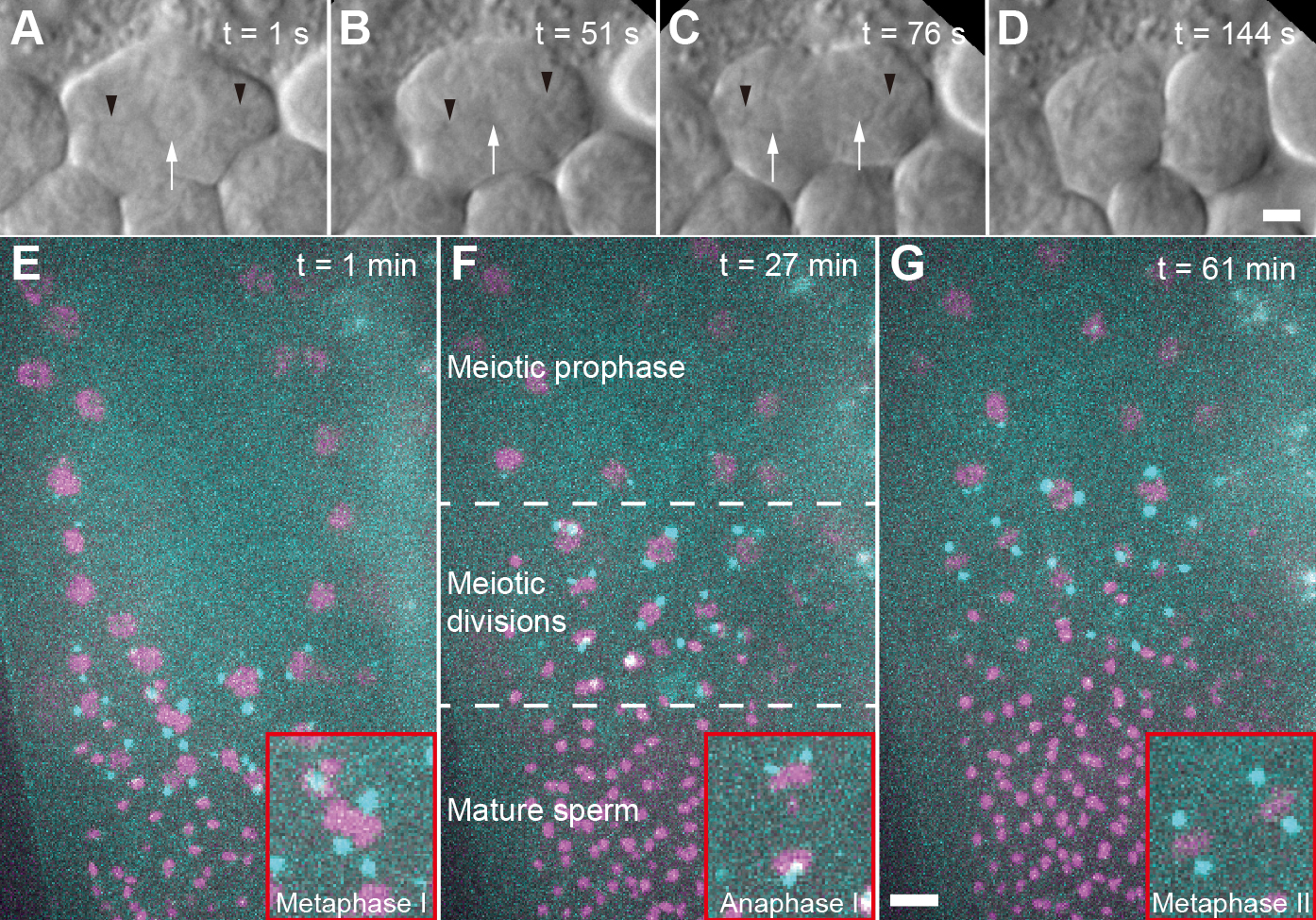
Figure 1. Imaging of C. elegans male germ cells. A. DIC imaging of a primary spermatocyte dissected from a wild-type (Bristol N2) male. The cell is in prometaphase with chromosomes visible in the center of the cell. Black arrow heads indicating the approx. position of the centrosomes, white arrow indicates the chromosomes. B. Cell in metaphase I. C. Cell in anaphase I. D. Cell finishing first meiotic division. Scale bar for (A-D), 2 µm. E. Fluorescence image of an immobilized male (strain TMR17) after bleach correction. This image is a projection of 21 z-slices taken one minute after starting the data acquisition. Chromosomes labelled with histone H2B::mCherry are shown in magenta, spindle poles are labelled with γ-tubulin::GFP and shown in cyan. The inset (red frame) shows a metaphase I spindle. F. Projection of 24 z-slices, 27 min after starting the image acquisition. In this field-of-view, different regions of a gonad are shown. These regions include meiotic prophase, various meiotic divisions of in meiosis I and II, and mature sperm. The inset (red frame) shows an anaphase I spindle. G. Projection of 21 z-slices, 61 min after starting the image acquisition. The inset (red frame) shows two spindles in metaphase II, Scale bar for (E-G), 5 µm.
Materials and Reagents
- Worm pick (flattened platinum wire mounted on a glass handle). Platinum wire with a diameter of 0.2 mm (Agar Scientific, catalog number: AGE404-2 )
- Parafilm
- Falcon tubes (50 ml, Greiner Bio-One, Germany)
- Microscope slides (76 x 26 mm x 1 mm, Engelbrecht, catalog number: 11102 )
- Empty plate or small container
- Filter paper Whatman Grade 589/2 (diameter: 90 mm, Merck, catalog number: WHA10300109 ). The filter paper was cut radially into 12 pieces
- Polystyrene beads (Polybead® Microspheres 0.10 μm; Polysciences, catalog number: 00876-15 )
- High-precision cover slips (18 mm x 18 mm x 0.17 µm, Carl Roth, catalog number: LH22.1 )
- Candle wax
- Glass Pasteur pipettes
- C. elegans strain N2 (reference strain; Brenner, 1974)
- C. elegans strain TMR17 (genotype: unc-119(ed3) III; ddIs6[tbg-1::GFP + unc-119(+)]; ltIs37[pAA64; pie-1::mCherry::HIS-58 + unc-119(+)] IV; strain maintained in the Müller-Reichert lab)
- E. coli strain OP50 (Brenner, 1974)
- Agarose (Carl Roth, catalog number: 3810.2 )
- Peptone (BD Biosciences, catalog number: 4197928 )
- NaCl (Carl Roth, catalog number: 9265.1 )
- Cholesterol (Carl Roth, catalog number: 8866.1 )
- CaCl2 (Carl Roth, catalog number: CN93.1 )
- MgCl2 (Carl Roth, catalog number: 2189.1 )
- NaOCl (Carl Roth, catalog number: 9062.3 )
- KH2PO4 (Carl Roth, catalog number: P749.2 )
- Na2HPO4 (Carl Roth, catalog number: P030.1 )
- MgSO4 (Carl Roth, catalog number: P027.1 )
- NGM-agar plates (see Recipes)
- KPO4 buffer (see Recipes)
- Bleaching solution (see Recipes)
- M9-buffer (see Recipes)
Equipment
- Fine tweezers
- Incubator set to 20 °C (Thermo Fisher, model: Heraeus BK 6160 )
- Incubator set to 30 °C to perform a heat-shock to animals (IKA, model: KS 4000 i control )
- Stereo microscope with transmitted light (Motic, model: SMZ-168 BL )
- Microwave
- Spoon/spatula
- Spinning disc confocal microscope (Olympus, model: IX83 )
- EMCCD camera (Andor, model: iXon Ultra 897 )
- Spinning disk unit (Yokogawa, model: CSU-X1 )
- 60x water immersion objective (Olympus, model: UPlanSApo 60x W , N.A. 1.2)
Software
- Fiji software package (www.fiji.sc, Schindelin et al., 2012)
- Arivis Vision4D (arivis AG, Munich)
- Microsoft Excel
- Python (https://www.python.org/)
Procedure
- Culture of C. elegans males
- Obtain male worms by applying the heat-shock method (Sulston and Hodgkin, 1988).
- Place 10 young L4 hermaphrodites on a fresh NGM-agar plate containing E. coli OP50.
- Seal the plate with Parafilm.
- Expose the plate to 30 °C for 6 h.
- Remove the parafilm and place the plate in an incubator at 20 °C for 2-3 days.
- Check then regularly for the presence of males on the plate. There should be 5-30 male worms on the plate.
- Transfer as many males as possible to a fresh NGM-agar plate and place 5 L4 hermaphrodites on this plate. Aim to have for 4-5 times more male individuals than hermaphrodites per plate. The progeny of the hermaphrodites should produce ~50% males.
- Maintain a culture of C. elegans males by transferring ~30 males and 5 L4 hermaphrodites to a fresh plate every three days.
- Synchronization of male worms
- Pipette 50 µl of bleaching solution on a fresh NGM-agar plate containing E. coli OP50.
- Add 10-20 adult hermaphrodites from a male culture plate into the bleaching solution.
- Incubate the plate for three days at 20 °C.
- Preparation of agarose pads
- Prepare about 30 clean glass slides and place them next to each other on the bench (Figure 2A).
- Prepare 10% (w/v) agarose powder in M9 buffer in a 50 ml Falcon tube (e.g., 0.5 g in 5 ml).
- Melt the agarose in a microwave with short heating intervals. Open the lid shortly in between the intervals.
Caution: Melted agarose is hot; protective gloves should be worn. - Use a small spoon or spatula to place a drop of the melted 10% agarose on a glass slide (Figure 2B).
- Immediately add a second glass slide on top and gently apply pressure for 5 s (Figure 2C).
- Repeat this procedure until all glass slides have agarose in between.
- Remove the top slide.
- Use a round object (e.g., cap of a permanent marker) to cut out agarose pads of a defined diameter of 10 mm. For this, select areas that do not contain any or only a few trapped air bubbles. Such bubbles might impair worm immobilization or the image quality later. Multiple agarose pads can be cut out of a single agar preparation (Figure 2D).
- Grab each agarose pad with fine tweezers and put it to a small container (e.g., empty plate) filled with M9 buffer (Figure 2E).

Figure 2. Immobilization of worms for live-imaging using agarose pads and Polystyrene beads. A. Arrangement of glass slides on the laboratory bench. This way, agarose pads can be produced quickly before the agarose starts to solidify. B. Placing of melted agarose on one glass slide using a small spoon. C. Placing of a second glass slide on top of the agarose droplet and simultaneously application of pressure until the agarose is solid (~5 s). D. Excision of an agarose pad (diameter of about 10 mm) using a cap of a permanent marker. E. Transfer of the agarose pad with a fine tweezer to M9 buffer until further use. F. Placing of the agarose pad on a glass slide to start a live-cell experiment and removal of excess of liquid from the side with a piece of filter paper. G. Application of a Polystyrene-bead solution (bead diameter 0.1 µm) to the top of the agarose pad. H. After addition of worms, a cover slip was placed on top of the agarose pad. Afterwards, a few microliters of M9 buffer were added to avoid dehydration of the animals. I. Sealing of the cover slip with candle wax and imaging of the slide using fluorescence microscopy. - Store agarose pads for several days in M9 buffer at 4 °C until further use. However, best results for worm immobilization are achieved with freshly prepared agarose pads.
- Live-imaging of meiotic spindles in male worms using fluorescent markers
- Select an agarose pad, place it with tweezers in the middle of a new glass slide. Be careful not to damage the surface of the agarose pad as this will reduce the immobilization effect on individual worms.
- Aspirate excess of liquid from the side of the agarose pad with a piece of filter paper (Figure 2F). Be careful not to touch the surface of the agarose pad as also cellulose fibers on the surface of the pad will reduce the immobilization of worms.
- Add 1 µl of a polystyrene bead solution on the agarose pad (Figure 2G).
- Place 5-10 three-day old male worms within the bead solution.
- Gently add a cover slip (Figure 2H). It is important to lower the covers slip gently as a dropping of the glass might damage the worms or activate spermatids within the males. A sliding of the mounted cover glass should also be avoided as this might impair the worms as well. If worms are still moving after the assembly procedure, discard the sample and start again with point no. 1.
- Add a few microliters of M9 buffer from the side under the cover slip. Be careful not to add too much as excess liquid will make the cover slip float and decrease the amount of immobilization.
- Seal the edges of the cover slip with melted candle wax (Figure 2I).
- Check again under a dissection microscope if the worms are immobile. If worms are perfectly immobilized at this point, they will not become mobile again, thus long-term imaging will be possible by using the settings described below.
- Place the prepared sample on a fluorescence microscope.
- Select a fully immobilized worm for imaging. Start image acquisition. Samples can be imaged for about 90 min without any detectable effects on cell division. Typically, z-stacks are recorded on a confocal spinning disk microscope in two fluorescence channels with an image stack every 30 s containing about 60 planes (0.33 µm z-spacing). For the analysis of spindle dynamics, use a worm strain expressing γ-tubulin::GFP and histone H2B::mCherry in the germ line. With a 60x water immersion objective (N.A.: 1.2), we generated images with a voxel size of 0.222 x 0.222 x 0.33 µm. As the specifications among microscopes and lasers can vary, it is not possible here to give absolute values for the laser power to be used. It is advisable to assess the degree of photobleaching after one hour by comparing the signal intensities in the recorded images at the beginning and at the end of data acquisition. If the signal is bleached very fast or cells stop dividing, reduce the laser power or the exposure time per frame. Then check, if the signal/noise ratio in a new recording allows to detect specific signals. This process needs to be repeated iteratively until the optimal imaging settings for the specific microscope have been determined.
- Image pre-processing
- Import the Z-stacks to ImageJ/Fiji. A user manual for ImageJ/Fiji can be found here.
- Convert them to a hyperstack.
- Correct for bleaching using the method “Exponential fit”.
- Save images.
- Image processing
- Import images into arivis Vision4D (Figure 3A).
- Crop out and export individual meiotic cells from the meiotic region of the male gonad (Figure 3B).
- Make a note in which frame anaphase starts.
- Use a 3D median filter with a kernel size of 1 pixel (Figure 3C) to reduce the noise.
- Segment individual centrosomes in each meiotic cell with the tool “Blob finder” (Figure 3D). The settings have to be chosen in accordance to the used imaging setup. As the image quality varied among the live recordings the segmentation settings had to be adjusted within a narrow range. Typically, we used the following settings for the “Blob Finder” tool: diameter: 1 µm, threshold: 10-20, split sensitivity: 85-95.
- Track the segments over time using the tool “Track segments”. Typically, we used the following settings: type: “Brownian motion”, max. distance: 2-4 µm.
- Edit and combine tracks in order to have one track connecting the individual centrosomes over time using the tool “Track editor”.
- Export a Microsoft Excel file containing the x, y and z coordinates of the geometric center of mass of each of the segmented centrosomes.
- Create a semicolon-delimited CSV-file for the first centrosome track, containing in each line the x, y, and z coordinate of the center of mass each time point.
- Create analogously a second CSV-file for the second centrosome track.
- Use a custom-made Python script to reorient (Watt, 1999) and resample the image data in order to create a kymograph along the spindle axis with a defined radius for each fluorescence channel (Figure 3F, https://cdn.elifesciences.org/articles/50988/elife-50988-code1-v4.py).
- Give the path to both CSV-files containing the coordinates of the center of the centrosomes (PATH).
- Indicate which fluorescence channel should be processed (CHANNEL).
- Indicate the frame of the data set that should be used as a start (START_FRAME; typically, this was ten time points before the frame of anaphase onset).
- Indicate the pixel size in the resampled output data set (LINE_PIXEL_SIZE; typically 0.1 µm was used).
- Indicate the radius around the spindle axis that is used to calculate the fluorescence pixel values within the kymograph (LINE_RADIUS; typically a ‘9’ was used, which means a radius of 0.9 µm around the spindle axis, given that the pixel size was set to 0.1 µm).
- Indicate the length of extrapolation along the spindle axis after the start and end coordinate (LINE_EXTRAPOLATE, typically 1.0 µm before and after the centrosome centers was added to each kymograph).
- Define an output filename for the kymograph CSV-file (OUTPUT_FILENAME).
- Calculate the Gaussian weighted sum of fluorescence (Burger and Burge, 2013) in each plane in 0.1 µm steps along the spindle axis with a radius of 0.9 µm (Figure 3G). This was repeated for all time points (Figure 3H) and the kymograph was saved as an image (Figure 4A) and a csv-file.
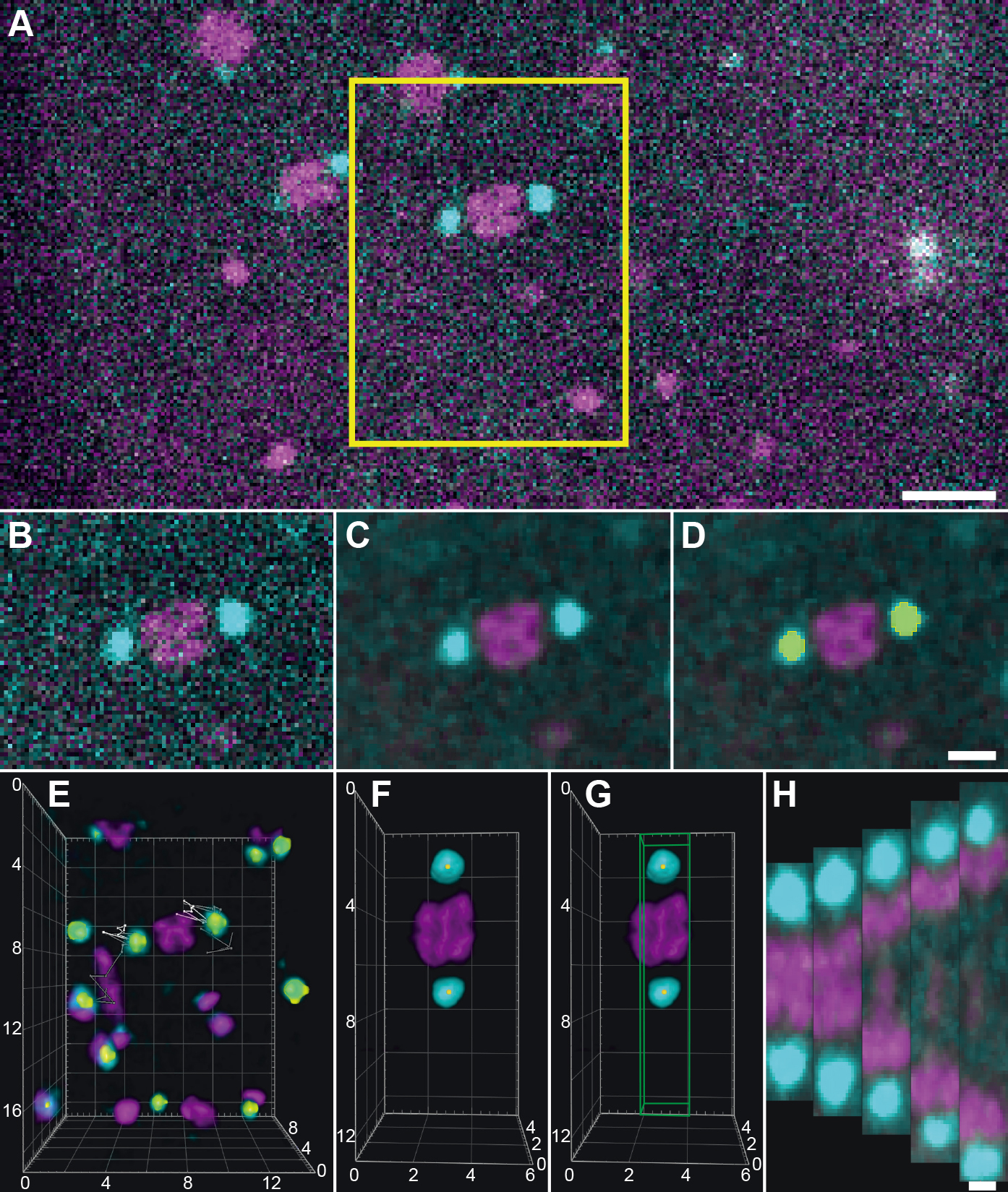
Figure 3. Workflow for image processing using arivis Vision4D. A. Data after pre-processing showing a meiosis I spindle in dual color fluorescence within one image plane. A selected area (indicated by a yellow box) is exported to a new file. Scale bar, 5 µm. B. Exported spindle as indicated in (A). C. Exported data after application of a 3D median filter (kernel size: 1 pixel). This algorithm removes the noise in the image while preserving the edges of structures. D. Segmentation of spindle poles (GFP-channel) using the “Blob finder” tool. The segmented volume is shown in yellow. Scale bar for B-D, 2 µm. E. 3D visualization of the exported image volume. The individual segmented spindle poles were traced over time as represented by lines on each pole. For 16 time points of the recording, white lines show the 3D position of the poles in the ‘past’ and gray lines the ‘future’ positions. F. Image showing a rotated and resampled spindle. Yellow dots indicate the center of mass of the segmented spindle poles. G. The volume that is used to assemble a kymograph is indicated by a box (green). Numbers in the coordinate system in (E-G) show distances in µm. H. Projections of the exported volumes at different time points that were used for the calculation of kymographs. Scale bar, 1 µm. - Analyze the kymograph for each fluorescence channel to determine the position of the peak intensity from both sides of the spindle over time (Figures 4B-4C).
- Calculate the distance of the peak intensities for each channel over time. This way, the autosome-to-autosome distance (Figure 4D) and the pole-to-pole distance (Figure 4E) can be calculated. In addition, the pole-to-autosome distance (Figure 4F) can also be determined by calculating the distance of the peak intensities from both fluorescence channels on each side of the spindle.
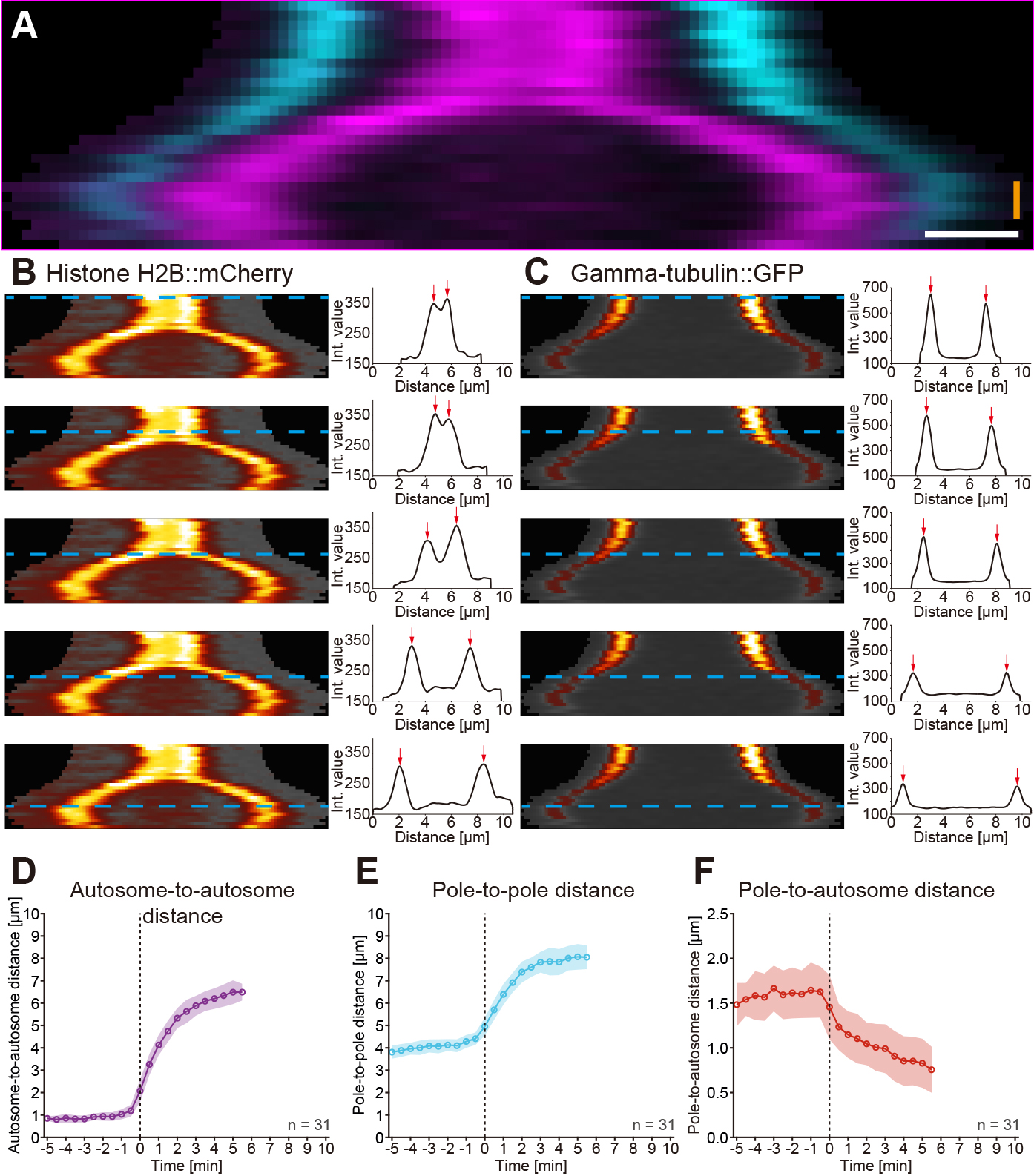
Figure 4. Kymographic analysis of spindle dynamics. A. Dual-color kymograph for the spindle as presented and processed in Figure 3. Autosomes are shown in magenta and spindle poles in cyan. Scale bar (white), 1 µm, time bar (orange), 2 min. B. Analysis of autosome dynamics. The kymograph of the histone::mCherry channel with the individual time points indicated by blue dashed lines is shown on the left, the fluorescence intensity profiles with highlighted the two peak intensities (red) arrows on the right. C. Analysis of spindle pole movements. The kymograph of the γ-tubulin::GFP channel (left) and the fluorescence intensity profiles are shown (right). D. Analysis of the autosome-to-autosome distance over time for 31 data sets aligned at anaphase onset (t0). Circles represent the mean, shaded area the standard deviation. All mean values are connected by a solid line. E. Analysis of the pole-to-pole distance over time for 31 data sets. F. Analysis of the pole-to-autosome distance over time for 31 data sets.
Data analysis
For each spindle, the pole-to-pole, the autosome-to-autosome and the pole-to-autosome distance was calculated for each time point from the kymograph data. In addition, the time of anaphase onset was determined as time zero (t0) by the first frame showing a clear separation of the autosomes. Accordingly, spindle dynamics in all data sets were then aligned to t0. By calculating the average distance (± standard deviation) for each time point the dynamic properties of both meiotic spindles were calculated, i.e., the initial and final spindle length, the initial rate of elongation and the duration of spindle elongation or segregation.
Recipes
- NGM-agar plates (Stiernagle, 2006)
0.25% (w/v) peptone
51 mM NaCl
25 mM KPO4 buffer
0.005 g/L cholesterol
1 mM CaCl2
1 mM MgCl2
2% (w/v) agar
The solution was autoclaved for 15 min at 120 °C, afterwards plates were poured - KPO4 buffer (1 M)
0.8 M KH2PO4 and 0.8 M K2HPO4 in double-distilled water
Adjust the pH to 6.0
Filter sterilize the solution by using a vacuum filtration system (0.22 µm filter mesh size) - Bleaching solution (Stiernagle, 2006)
500 mM NaOH
2.6% (v/v) NaOCl in double-distilled water
Protect solution from light and renew solution every 3-6 month as NaOCl gets less reactive over time - M9-buffer (Brenner, 1974)
22 mM KH2PO4
42.3 mM Na2HPO4
85.6 mM NaCl
1 mM MgSO4
The solution was filtered afterwards (filter pore size 0.2 μm)
Acknowledgments
The authors would like to thank the members of the Core Facility Cellular Imaging of the Faculty of Medicine “Carl Gustav Carus” (TU Dresden, Germany), especially Silke Tulok and Dr. Anja Nobst, and the light microscopy facility at the MPI-CBG (Dresden, Germany) for technical assistance. We also would like to thank Drs. Leocadia Paliulis and Robert Haase for the critical reading of this manuscript. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). Research in the Müller-Reichert laboratory is supported by the Deutsche Forschungsgemeinschaft (MU 1423/10–1 to TMR).
This protocol was part of and derived from Fabig et al. (2020).
Competing interests
FL and CG are employed by arivis AG. This company develops commercial image processing software (Vision4D). This software was utilized to process fluorescence microscopy data used in this protocol.
References
- Ault, J. G. (1986). Stable versus unstable orientations of sex chromosomes in two grasshopper species. Chromosoma 93(4): 298-304.
- Bonaccorsi, S. and Gatti, M. (2017). Drosophila Male Meiosis. Methods Mol Biol 1471: 277-288.
- Boring, A. (1909). A small chromosome in Ascaris megalocephala. Arch f Zellf 4: 120-131.
- Boveri, T. (1909). Über Geschlechtschromosomen bei Nematoden. Arch f Zellf 4: 132-141.
- Brenner, S. (1974). The genetics of Caenorhabditis elegans. Genetics 77(1): 71-94.
- Burger, W., and Burge, M. J. (2013). Principles of Digital Image Processing-Advanced Methods. In: Burger, W., and Burge, M. J. (Eds). Undergraduate Topics in Computer Science Springer-Verlag.
- Fabig, G., Kiewisz, R., Lindow, N., Powers, J. A., Cota, V., Quintanilla, L. J., Brugués, J., Prohaska, S., Chu, D. S. and Müller-Reichert, T. (2020). Male meiotic spindle features that efficiently segregate paired and lagging chromosomes. ELife 9: e50988.
- Fang-Yen, C., Wasserman, S., Sengupte, P., and Samuel, A. D. T. (2009). Agarose immobilization of C. elegans. Worm Breeder’s Gazette.
- Fegaras, E. and Forer, A. (2018). Chromosomes selectively detach at one pole and quickly move towards the opposite pole when kinetochore microtubules are depolymerized in Mesostoma ehrenbergii spermatocytes. Protoplasma 255(4): 1205-1224.
- Felt, K. D., Lagerman, M. B., Ravida, N. A., Qian, L., Powers, S. R. and Paliulis, L. V. (2017). Segregation of the amphitelically attached univalent X chromosome in the spittlebug Philaenus spumarius. Protoplasma 254(6): 2263-2271.
- Forer, A., Ferraro-Gideon, J. and Berns, M. (2013). Distance segregation of sex chromosomes in crane-fly spermatocytes studied using laser microbeam irradiations. Protoplasma 250(5): 1045-1055.
- Forer, A., Sheykhani, R. and Berns, M. W. (2018). Anaphase Chromosomes in Crane-Fly Spermatocytes Treated With Taxol (Paclitaxel) Accelerate When Their Kinetochore Microtubules Are Cut: Evidence for Spindle Matrix Involvement With Spindle Forces. Front Cell Dev Biol 6: 77.
- Gleason, E. J., Hartley, P. D., Henderson, M., Hill-Harfe, K. L., Price, P. W., Weimer, R. M., Kroft, T. L., Zhu, G. D., Cordovado, S. and L'Hernault, S. W. (2012). Developmental genetics of secretory vesicle acidification during Caenorhabditis elegans spermatogenesis. Genetics 191(2): 477-491.
- Golding, A. E. and Paliulis, L. V. (2011). Karyotype, sex determination, and meiotic chromosome behavior in two pholcid (Araneomorphae, Pholcidae) spiders: implications for karyotype evolution. PLoS One 6(9): e24748.
- Goldstein, P. (1977). Spermatogenesis and spermiogenesis in Ascaris lumbricoides Var. suum. J Morphol 154(3): 317-337.
- Howe, M., McDonald, K. L., Albertson, D. G. and Meyer, B. J. (2001). HIM-10 is required for kinetochore structure and function on Caenorhabditis elegans holocentric chromosomes. J Cell Biol 153(6): 1227-1238.
- Hu, J., Cheng, S., Wang, H., Li, X., Liu, S., Wu, M., Liu, Y. and Wang, X. (2019). Distinct roles of two myosins in C. elegans spermatid differentiation. PLoS Biol 17(4): e3000211.
- Janicke, M. A. and LaFountain, J. R., Jr. (1982). Chromosome segregation in crane-fly spermatocytes: cold treatment and cold recovery induce anaphase lag. Chromosoma 85(5): 619-631.
- Kim, E., Sun, L., Gabel, C. V. and Fang-Yen, C. (2013). Long-term imaging of Caenorhabditis elegans using nanoparticle-mediated immobilization. PLoS One 8(1): e53419.
- King, J. M. and Nicklas, R. B. (2000). Tension on chromosomes increases the number of kinetochore microtubules but only within limits. J Cell Sci 113 Pt 21: 3815-3823.
- LaFountain, J. R., Jr., Cohan, C. S. and Oldenbourg, R. (2012). Pac-man motility of kinetochores unleashed by laser microsurgery. Mol Biol Cell 23(16): 3133-3142.
- LaFountain, J. R., Jr., Cole, R. W. and Rieder, C. L. (2002). Partner telomeres during anaphase in crane-fly spermatocytes are connected by an elastic tether that exerts a backward force and resists poleward motion. J Cell Sci 115(Pt 7): 1541-1549.
- Nicklas, R. B. and Koch, C. A. (1969). Chromosome micromanipulation. 3. Spindle fiber tension and the reorientation of mal-oriented chromosomes. J Cell Biol 43(1): 40-50.
- Nicklas, R. B. and Staehly, C. A. (1967). Chromosome micromanipulation. I. The mechanics of chromosome attachment to the spindle. Chromosoma 21(1): 1-16.
- Peters, N., Perez, D. E., Song, M. H., Liu, Y., Muller-Reichert, T., Caron, C., Kemphues, K. J. and O'Connell, K. F. (2010). Control of mitotic and meiotic centriole duplication by the Plk4-related kinase ZYG-1. J Cell Sci 123(Pt 5): 795-805.
- Nicklas, R. B., Waters, J. C., Salmon, E. D. and Ward, S. C. (2001). Checkpoint signals in grasshopper meiosis are sensitive to microtubule attachment, but tension is still essential. J Cell Sci 114(23): 4173-4183.
- Rivera Gomez, K. and Schvarzstein, M. (2018). Immobilization of nematodes for live imaging using an agarose pad produced with a Vinyl Record. MicroPubl Biol 2018.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P. and Cardona, A. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7): 676-682.
- Schvarzstein, M., Pattabiraman, D., Bembenek, J. N. and Villeneuve, A. M. (2013). Meiotic HORMA domain proteins prevent untimely centriole disengagement during Caenorhabditis elegans spermatocyte meiosis. Proc Natl Acad Sci U S A 110(10): E898-907.
- Shakes, D. C., Wu, J. C., Sadler, P. L., Laprade, K., Moore, L. L., Noritake, A. and Chu, D. S. (2009). Spermatogenesis-specific features of the meiotic program in Caenorhabditis elegans. PLoS Genet 5(8): e1000611.
- Sheykhani, R., Berns, M. and Forer, A. (2017). Elastic tethers between separating anaphase chromosomes in crane-fly spermatocytes coordinate chromosome movements to the two poles. Cytoskeleton (Hoboken) 74(2): 91-103.
- Stiernagle, T. (2006). Maintenance of C. elegans. WormBook: 1-11.
- Sulston, J. and Hodgkin, J. (1988). Methods. In: Wood W. B. (Ed.). The Nematode Caenorhabditis elegans. (pp. 587-606). Cold Spring Harbor Laboratory Press.
- Vielle, A., Callemeyn-Torre, N., Gimond, C., Poullet, N., Gray, J. C., Cutter, A. D. and Braendle, C. (2016). Convergent evolution of sperm gigantism and the developmental origins of sperm size variability in Caenorhabditis nematodes. Evolution 70(11): 2485-2503.
- Watt, H. A. (1999). 3D Computer Graphics, Addison Wesley.
Article Information
Copyright
![]() Fabig et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Fabig et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Fabig, G., Löffler, F., Götze, C. and Müller-Reichert, T. (2020). Live-cell Imaging and Quantitative Analysis of Meiotic Divisions in Caenorhabditis elegans Males. Bio-protocol 10(20): e3785. DOI: 10.21769/BioProtoc.3785.
- Fabig, G., Kiewisz, R., Lindow, N., Powers, J. A., Cota, V., Quintanilla, L. J., Brugués, J., Prohaska, S., Chu, D. S. and Müller-Reichert, T. (2020). Male meiotic spindle features that efficiently segregate paired and lagging chromosomes. ELife 9: e50988.
Category
Cell Biology > Cell imaging > Live-cell imaging
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.