- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Multitarget Immunohistochemistry for Confocal and Super-resolution Imaging of Plant Cell Wall Polysaccharides
(*contributed equally to this work) Published: Vol 10, Iss 19, Oct 5, 2020 DOI: 10.21769/BioProtoc.3783 Views: 7678
Reviewed by: Ayelign M. AdalSara Posé Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
The plant cell wall (PCW) is a pecto-cellulosic extracellular matrix that envelopes the plant cell. By integrating extra-and intra-cellular cues, PCW mediates a plethora of essential physiological functions. Notably, it permits controlled and oriented tissue growth by tuning its local mechano-chemical properties. To refine our knowledge of these essential properties of PCW, we need an appropriate tool for the accurate observation of the native (in muro) structure of the cell wall components. The label-free techniques, such as AFM, EM, FTIR, and Raman microscopy, are used; however, they either do not have the chemical or spatial resolution. Immunolabeling with electron microscopy allows observation of the cell wall nanostructure, however, it is mostly limited to single and, less frequently, multiple labeling. Immunohistochemistry (IHC) is a versatile tool to analyze the distribution and localization of multiple biomolecules in the tissue. The subcellular resolution of chemical changes in the cell wall component can be observed with standard diffraction-limited optical microscopy. Furthermore, novel chemical imaging tools such as multicolor 3D dSTORM (Three-dimensional, direct Stochastic Optical Reconstruction Microscopy) nanoscopy makes it possible to resolve the native structure of the cell wall polymers with nanometer precision and in three dimensions.
Here we present a protocol for preparing multi-target immunostaining of the PCW components taking as example Arabidopsis thaliana, Star fruit (Averrhoa carambola), and Maize thin tissue sections. This protocol is compatible with the standard confocal microscope, dSTORM nanoscope, and can also be implemented for other optical nanoscopy such as STED (Stimulated Emission Depletion Microscopy). The protocol can be adapted for any other subcellular compartments, plasma membrane, cytoplasmic, and intracellular organelles.
Background
PCW is an intricate material, which is durable but also undergoes constant structural changes in response to internal and external stimuli such as tissue expansion or pathogen attack. Nevertheless, how these seemingly contradictory features, mechanical strength and structural adaptation, cooperate, remains an unresolved question in plant cell biology. PCW contains cellulose, hemicellulose, different variants of pectin and various proteins, the architecture of which is highly organized (Peaucelle, 2018). The pectin family is composed of several polymers. The most abundant, homogalacturonan, can be demethylated after cell wall insertion. This change in chemistry is a significant step in the process of cell elongation, differentiation, and directional growth (Peaucelle et al., 2012). Several lines of evidence suggest that morphogenesis and cell differentiation are dependent on local changes in cell wall chemistry and cell wall polymer organization (Yang et al., 2016; Anane et al., 2017; Zhao et al., 2019; Haas et al., 2020). Therefore, detailed knowledge of PCW components architecture is essential for understanding plant growth. Historically, the cell wall structure has been studied using biochemical methods that involve disintegrating the tissue and destroying the native organization of its polymers (Höfte and Voxeur, 2017). Other imaging modalities, such as electron microscopy (EM) (Anane et al., 2017) and atomic force microscopy (AFM) (Zhang et al., 2017) are used for in situ cell wall observation, but these techniques often lack chemical contrast, providing only correlative quantification. Multicolor immunohistochemistry (IHC) can reveal multiple targets within a tissue section, and their spatial organization resolved in three dimensions. Despite its widespread use in the cell biology field, multicolor IHC is not yet a standard tool for studies of the cell wall. A broad palette of antibodies and probes against cell wall targets, coupled with multicolor IHC, together with the high resolving power of dSTORM (< 40 nm), permits quantitative in situ chemical analysis of cell wall nanostructure. Other imaging techniques for cell wall analysis on tissue cuts exist, such as Raman (Wightman et al., 2019) and FTIR (Mravec et al., 2017; Cuello et al., 2020). These techniques are based on the characteristic absorption/transmission of different chemical components and can measure some changes in cell wall composition at the cellular level but can lack sensitivity, and their ability to observe changes at the subcellular level is severely limited.
The dSTORM permits localization of biomolecules with the precision of 5-10 nm; however, the final dSTORM resolution, typically around 40 nm, is limited by the size of the antibody complex (~15-30 nm). The immunogold Electron Microscope (iEM) is also used in combination with ICH to study cellular structures at high resolutions. IEM is comparable to dSTORM resolution and is also limited by the antibody complex size. The size of the nanogold particle defines iEM contrast and resolution; > 1 nm gold nanoparticles are available; however, such a small particle limits the contrast, and larger particles are often used. IEM, contrasted with dSTORM, has several additional drawbacks: (1) the primary antibodies probed with protein-A (or G) gold complexes do not penetrate through the resin-embedded sample, and only recognize the surface epitopes, although the serial and ultra-thin cryo-sectioning technique can resolve this problem (50 nm, -120 °C [Slot, 1989]); (2) samples are mostly single-labeled; yet by using different nanoparticle size, two or three epitopes can be tagged, but it requires the technical experiences of ultra-thin cryo-sectioning technique (Slot and Geuze, 2007); (3) IEM has low labeling and detection efficiency (3-5 orders of magnitude less than dSTORM (Majda et al., 2017; Haas et al., 2020), which is also related to the fact, that only the surface epitopes are labeled. Multicolor 3D dSTORM nanoscopy can, therefore, provide unprecedented insights into the nanoarchitecture of the native-structure of the cell wall polysaccharides, beyond the applicability of the aforementioned techniques. DSTORM permits a quantitative three-dimensional (3D) nanoimaging of cells and tissues (Heilemann et al., 2008; Huang et al., 2008; Van De Linde et al., 2011; Sydor et al., 2015; Xu et al., 2018). Applied mainly to cellular systems, it unveiled new structural organizations of proteins, e.g., synaptic nanodomains, trans-synaptic nanocolumns, DNA recombinase nanofilaments, nuclear envelope pore structure, cytoskeleton, mitochondria, adhesion complexes, and chromatin transcriptional landscape (Shroff et al., 2008; Shim et al., 2012; Löschberger et al., 2012; Jakobs and Wurm, 2014; Prakash et al., 2015; Boettiger et al., 2016; Sellés et al., 2017; Dlasková et al., 2018; Haas et al., 2018a and 2018b; Pan et al., 2018; Xia et al., 2019; Chen et al., 2020; Wäldchen et al., 2020). Yet, due to the limitation of single-cell model systems requiring tissue-level imaging, its utilization in plant science is almost absent (Liesche et al., 2013; Komis et al., 2015; Haas et al., 2020). Here we present a detailed protocol for the sample preparation compatible with a standard confocal microscope and a dSTORM nanoscope on thin plant tissue sections.
Materials and Reagents
- 8-well ibidi µ-slides for dSTORM sample preparation and imaging (Ibidi, catalog number: 80827)
- Biopsy cassette 42 x 28 x 6 mm (Leica Biosystems, ID: IP-Biopsy-cassette-III)
- Metal base mold (Leica Biosystems, ID: metal-base-molds)
- Charged slides by poly-L-lysine treatment for confocal imaging sample preparation and imaging such as ProbeOn Plus (Fisher Scientific catalog number: 22-230-900 or PolysineTM Microscope Adhesion Slides (Thermo Scientific catalog number: J2800AMNZ)
- Aluminum foil (you can get it from a local store)
- Coverglass, rectangular, 24 x 60 mm, #1.5 thickness (Knittel Glass, catalog number: 425-2460)
- Eppendorf Safe-lock Microcentrifuge Tubes, 1.5 ml or 2 ml volume (Eppendorf, catalog numbers: 022-36-320-4 [1.5 ml], 022-36-335-2 [2 ml])
- Cardboard Slide Tray (Heathrow Scientific, catalog number: HD9902)
- Paper towels (Divers/Dutcher, catalog number: 475040)
- Empty pipette tips box for homemade humidified chamber for ibidi µ-slides, such as Adamas-Beta 1,000 µl volume tips
- Nitrile gloves, such as FisherbrandTM Comfort Nitrile Gloves (Fisher Scientific, catalog number: 15642367)
- National DiagnosticsTM HistoclearTM Tissue clearing agent (Fisher Scientific, HS-200-1GAL, CAS number: 5989-27-5)
- Embedding medium paraffin (Leica Biosystems, ID: em-400-embedding-medium-paraffin)
- Oxoid Skim Milk Powder (Thermo Fisher Scientific, catalog number: LP0031B)
- CMB3a Crystalline cellulose-binding module, his-tagged and recombinant CBM protein (PlantProbes, catalog number: CMB3a) (Blake et al., 2006; Hernandez-Gomez et al., 2015), store at -20 °C
- Mouse monoclonal antibody against low esterified homogalacturonan (degrees of methyl-esterification (DM) up to 40%) 2F4 (PlantProbes, catalog number: 2F4) (Liners, Thibault and Van Cutsem, 1992), store at 4 °C
- Rat IgM monoclonal antibody against high-esterified homogalacturonan LM20 (PlantProbes, catalog number: LM20) (Verhertbruggen et al., 2009), store at 4 °C
- Rat IgA monoclonal antibody against partially methyl-esterified epitopes of homogalacturonan (PlantProbes, catalog number: JIM7) (Knox et al., 1990), store at 4 °C
- Rat IgG2a monoclonal antibody against xyloglucan binding preferentially to the XLLG motif of xyloglucan LM24 (PlantProbes, catalog number: LM24) (Tanackovic et al., 2016), store at 4 °C
- Anti-His tag polyclonal antibody produced in chicken, Anti-6X-His tag (Abcam, catalog number: ab9107) store at -20 °C
- Anti-His tag polyclonal antibody produced in rabbit (Merck, catalog number: SAB4301134), store at -20 °C
- Rabbit PDM antibody against Mannan was a kind gift by Paul Dupree (Handford et al., 2003; Yang et al., 2016), store at 4 °C. For availability, please contact raymond.wightman@slcu.cam.ac.uk
- Recombinant and His-tagged CBM4 of Cellulomonas fimi (ATCC 484) endoglucanase C (CBD4N1) produced from E. coli. CBM4 was a kind gift by Harry Gilbert (Johnson et al., 1996; Blake et al., 2006), store at 4 °C. For availability, please contact raymond.wightman@slcu.cam.ac.uk
- Goat anti-rat CF568 secondary antibody (Sigma, catalog number: SAB4600086), store at -20 °C
- Goat anti-mouse CF568 conjugated F(ab)’2 secondary antibody fragment (Sigma, catalog number: SAB4600400), store at -20 °C
- Goat anti-mouse F(ab')2 secondary antibody fragment conjugated to Alexa Fluor 647 (Stratech Scientific, catalog number: 115-607-003-JIR), store at 4 °C
- Donkey anti-mouse F(ab')2 secondary antibody fragment conjugated to Alexa Fluor 647 (Abcam, catalog number: ab181292), store at 4 °C
- Donkey anti-Chicken F(ab’)2 secondary antibody fragment conjugated to Alexa Fluor 647 (Jackson immunoresearch, catalog number: 703-606-155), store at 4 °C
- Goat anti-mouse F(ab')2 secondary antibody fragment conjugated to ATTO488 (Hypermole, catalog number: 2402-0.5MG), store at -20 °C
- Goat anti-mouse Alexa 488 conjugated secondary antibody (Sigma, catalog number: SAB4600387), store at 4 °C
- Donkey anti-rat F(ab')2 secondary antibody fragment conjugated to Alexa Fluor 647 (Abcam, catalog number: ab150151), store at 4 °C
- ProLong Gold Antifade Mountant (Thermo Ficher Scientific, catalog number: P36934), store at 4 °C
- Ammonium chloride (NH4Cl) (Sigma-Aldrich, catalog number: 254134), store at RT
- Poly-L-lysinesolution 0.1% (w/v) in H2O (Merck, catalog number: P8920), store at RT
- Ethanol absolute anhydrous (CARLO ERBA reagents, catalog number: 4146082), store at RT
- Acetic Acid ≥ 96% (AnalaR NORMAPUR, catalog number: 20099.324), store at RT
- Formaldehyde (Sigma-Aldrich catalog number: F1635), store at RT
- Arabidopsis inflorescence meristem fixed at 1 cm length stage, and cotyledon of 3 days-old seedling, leaf rachis of star fruit (Supplemental Figure 1AG), immature maize leaf blade at 6th node (Supplemental Figure 1AF)
- Nail polish (you can get it from a local drugstore)
- Pectolyase for enzymatic extraction of pectins (Sigma, catalog number: P3026 or Magazyme,catalog number: E-PLYCJ)
- Calcofluor White Stain for general cell wall staining (Sigma Aldrich, catalog number: 18909-100ML-F)
- 2F4 Buffer (T/Ca/S buffer final concentration) (see Recipes), store at RT
- FAA solution (see Recipes), store at RT
- Formaldehyde diluted in 10x 2F4 buffer (see Recipes)
- Citrate-Phosphate Buffer for Pectolyase incubation (pH 4.8) (see Recipes)
Equipment
- Dispensable microtome knife (Microm Microtech, catalog number: F/MM35p)
- Micro tweezer (Ideal-tek, catalog number: 5-SA)
- Glass Rectangular 250 ml Coplin Staining Jar, with Lid (Wheaton, catalog number: 900620)
- FisherbrandTM Microscope Slide Box for homemade humidified chamber for Polysine microscope slides (Fisher Scientific, catalog number: 22363400)
- Brush for the capture of serial cuts obtained by the microtome (Supplemental Figure 1K, any fine brush from local stationery store, such as Etude, P10531.00, #2)
- HistoCore Arcadia Heated Paraffin Embedding Station (Leica, ID:14039357258)
- Leica EG F Electric Heatable Forceps
- Microtome (Leica, model: RM2265)
For more information, see user’s manual. - Fridge 4 °C
- Incubation chamber, such as Selecta (set at 60 °C), airflow not necessary
- Laboratory freezer (-20 °C), such as Kirsch FROSTER LABO 330 ULTIMATE
- Electronic laboratory heating plate (BIO-OPTICA Milano SpA, catalog number: 40-300-300)
- Laboratory chemical fume hood
- Any Fluorescence Microscope equipped with four laser lines for the detection in UV (405 nm), green (488 nm), red (561 nm) and far red (633 nm) (here we use Zeiss LSM 710, Zeiss Oberkochen Germany)
Brochure available here.
Specification- Stands: Inverted (Axio Observer Z1 with side port port)
- Z drive and XY stage (option): Motorized XY-scanning stage, with Mark & Find function (xyz) and Tile Scan (mosaic scan); the smallest increments 1 μm
- Objectives: x10, x25, x40, x63, x100
- Lasers: Argon laser (458, 488, 514 nm), HeNe laser (633 nm), diode laser 405 nm, and DPSS laser 561 nm
- 405 nm for Calcofluor White
- 488 nm for Alexa 488 and ATTO 488
- 561 nm for CF568 and Alexa 568
- 633 nm for Alexa 647
- Scanning Module
Model: Scanning module with 32 spectral detection channels (QUASAR)
Scanners: Two independent, galvanometric scan mirrors with ultra-short line and frame fly back.
Scanning resolution: 4 x 1 to 6,144 x 6,144 pixels
Scanning speed: 8 frames/sec with 512 x 512 pixels.
Number of fluorescence-spectral detectors: 2
Bright field transmission detector: Installed - Software
Standard Software: ZEN2010
Optional Softwar: Image J or Fiji
Computer specification: HP Z800 Workstation, 64-bit Windows 7 Ultimate 2009, 24 Gb RAM, Intel® Xeon® CPU, X5650, Two processors 2.67 GHz, 2.66 GHz
Wild-field microscope (Nikon N-STORM)
Software
- Grafeo (Custom made software for dSTORM data analysis and visualization, https://github.com/inatamara/Grafeo-dSTORM-analysis- (Haas et al., 2018b)
- Fiji (https://imagej.net/Fiji/Downloads)
Procedure
In this protocol, we use Arabidopsis thaliana cotyledons meristem, star fruit (Averrhoa carambola) rachis (central fiber in the compound leaf), and maize leaf samples.
- Sample Preparation
- Star fruit was grown in soil in the greenhouse for two years, and leaf rachis was harvested at the begging of April 2019. Maize was grown in soil in the field sown in May and harvested at the beginning of July 2019 before its flowering. Arabidopsis was grown on MS (Murashige and Skoog) solid nutrient agar medium without sucrose, in constant light at 21 °C, and seedlings were harvested at three days after germinations (3 DAG) (Peaucelle, Wightman and Höfte, 2015). Arabidopsis meristem was harvested from a plant grown on soil in the growing chamber and harvested when the inflorescence was 1 cm long; flowers with visible sepals were removed, keeping all the closest flower buds (as described in Yang et al., 2016)
- Leaf rachis of star fruit was cut with a dispensable microtome knife into 0.5 cm-length-explant (Supplemental Figures 1AG, 2A). The immature region of the Maize shoot tip was isolated from the plantlet (Supplemental Figure 1AF). Maize immature leaf-blade without midrib at 6th node was cut into 5 mm x 5 mm square (Supplemental Figure 2C).
- A volume range to fix different organs of Arabidopsis is typically between 1:10 to 1:100 (tissue to fixative volume), here we placed 20 seedlings without the excision of cotyledon in 1.5 ml Eppendorf tube filled with a 1.0 ml of FAA solution. The volume ratio for maize leaf explants was 1:3, and the volume ratio of star fruit leaf rachis explants was 1:100. We placed 5 explants of maize leaves and 10 explants of star fruit leaf rachis in 2.0 ml Eppendorf tube filled with 1.5 ml of FAA solution, respectively. The significant protocol steps are shown in the Supplemental Figure 1, and the preparation of tissue explants, and the microtome cutting position in Supplemental Figure 2.
- Fixation and sample embedding
- Wear nitrile gloves.
- Fix plant organs in the FAA solution in the 1.5/2 ml Eppendorf tube for 1 h at room temperature or overnight at 4 °C. Store fixed samples at 4 °C for up to 1 month in 70% EtOH (see Notes). No vacuum treatment is necessary.
- Dehydrate the samples by incubating at room temperature in successive ethanol dilutions for at least 30 min each: 70%, 95%, and twice 100% ethanol. Use approximately 1.5 ml volume in the Eppendorf tube.
- Replace ethanol with 1.5 ml of 50% Histoclear in ethanol in Eppendorf for 1 h at room temperature, followed by 1.5 ml of 100% Histoclear for 1 h each at room temperature (this time depends on the thickness of the sample. Reduce the incubation time for very thin tissue cuts, such as roots or Hypocotyl to 30 min).
- Transfer the sample to a biopsy cassette (IP-Biopsy-Cassette-III, Leica Biosystems). Start to melt the paraffin in an incubation chamber preheated to 60-70 °C, 12 h prior to Procedure B. Completely melted paraffin and new Histoclear should be preheated to 60-70 °C prior to Step B6 to make 50% paraffin in Histoclear. During a typical experiment, we use approximately 600 ml of paraffin and 100 ml Histoclear in Step B6.
Preheat the 250 ml glass jar containing 200 ml of the final of 50% Histoclear and 50% paraffin before Step B6.
Note: We recommend using the EM-400 Embedding Medium Paraffin, which has a low melting point (56-57 °C). This helps the precise positioning of the sample and reduces tissue distortion. - Replace the Histoclear with the paraffin in the incubation chamber by immersing biopsy cassettes in a 250 ml Coplin glass staining jar (Supplemental Figure 1B). Start with a mixture of 50% Histoclear and 50% paraffin in 200 ml for 3 h at 60-70 °C, followed by twice 200 ml of 100 % paraffin for 3 h, and finally 200 ml of 100% paraffin overnight at 60-70 °C.
- Turn on the HistoCore Arcadia H Heated Paraffin Embedding Station (Supplemental Figure 1C) and set the HistoCore Arcadia H in operation mode before Step B8 (Set the temperatures of paraffin tank, dispenser, working surface at ~70 °C and a cold spot at 4 °C. For more information, See the HistoCore Arcadia H user manual).
- Take out biopsy cassettes and hot 100% paraffin in 250 ml Coplin glass staining jar from the incubator. Let biopsy cassettes floating in melted paraffin of heated instrument tray on a hot preparation surface set at ~70 °C of HistoCore Arcadia H before the solidifying the paraffin at room temperature (Supplemental Figure 1D).
- Transfer of the samples from the biopsy cassette to the metal base mold using electric heatable forceps (Supplemental Figure 1E). Samples should be positioned perpendicular to the cutting plane before solidifying the paraffin at room temperature (Supplemental Figures 1F to 1H and 2D).
- Once positioned, rapid solidification of the paraffin could be achieved using a cold spot (small round metallic plate shown in Supplemental Figure 1G), usually at 4 °C.
Note: Rapid cooling of the paraffin will limit the formation of paraffin crystals, and make the preparation transparent. In contrast, slow cooling will lead to crystal formation and the widening of the preparation. The crystallized paraffin is slightly more rigid and limits greatly the ability to localize the sample during microtome cutting. - After Step B10, store the samples at 4 °C overnight. Before cutting samples with the microtome, keep the room temperature bellow 22 °C to prevent the paraffin softening.
- Turn on the microtome. We recommend using the microtome in the manual mode. The use of the motorized mode is possible, but we did not test it. The settings for the cut thickness are: 3-5 µm (for more information: See the user’s manual of Leica, model: RM2265).
- Take out the paraffin block of a tissue specimen from the mold manually, trim the paraffin block, and fix it in the specimen clamps and holder in the microtome (Supplemental Figure 1H). This step will assure a clean and homogenous cut with the formation of a straight ribbon, as shown in the Supplemental Figure 1K.
Trim the paraffin block to get the cutting surface of the specimen facing the knife with a microtome (Supplemental Figure 1I), if you need it. - Capture the sections by using a brush (Supplemental Figure 1K) and put them on a poly-L-lysine treated microscope slide (Supplemental Figure 1L) or 8-well ibidi µ-slides (Supplemental Figure 1M) without inverting sections, i.e., position tissue section side that was facing the knife (the "shiny" side) on the charged slide or ibidi well bottom glass.
- Add a droplet of water between the slide and the serial tissue sections on the slide or a single ibidi well (Supplemental Figures 1N and O, see Note below).
Note: If you use the ibidi multi-well slides for dSTORM sample preparation, add 0.1% Poly-L-Lysine solution instead of water at this stage. - Keep the slides at 42 °C for 3 min.
- Remove the water carefully without touching the cuts. Any drop of remaining water will form a bubble and will lead to the loss of this part of the sample. At this stage, manually spin-dry the slide by swinging it at arm's length 1 or 2 times (Supplemental Figure 1P).
Note: Wearing a face mask and gloves is necessary only during the pandemy. - Leave to dry at least overnight at 37 °C on a heating plate (Supplemental Figure 1Q). The slide can be conserved at room temperature for at least one month. Use the microscope slide boxes for the storage to avoid the dust.
- Deparaffining (Supplemental Figure 1R): Place up to 8 micro slides vertically (or 16 slides back to back) into a 250 ml Coplin glass staining jar and immerse the slide in the three successive treatments with 200 ml of Histoclear each for 30 min, at RT in normal light condition, shaking is not necessary. Then wash the Histoclear-treated microslides with a 200 ml of 100% ethanol using a 250 ml glass staining jar for 20 min.
Note: The jar can be re-used at the next to the following step. Do not wash jar with water; just decant the solution from the jar. - Rehydrate the samples with successive treatments using the same two glass jar used in previous steps, 15 min each, in 100% ethanol, 70% ethanol, 50% ethanol, 25% ethanol, 10% ethanol in 2F4 buffer, and finally 100% 2F4 buffer, at RT in normal light condition, shaking is not necessary. Use approximately 200 ml volume for each step.
Note: The jar can be re-used at the next to the following step. Do not wash the jar with water; just decant the solution from the jar. - Proceed to Step C1 (Immunolabeling) as soon as possible to avoid the drying of sections.
Notes:- In the Step B12, the sample should be prepared in advance and kept at 4 °C overnight. However, samples can also be cut (Steps B12-B18) 1 h after Step B10. In such case, store the samples at -20 °C for 1 h prior to cutting and cut in less than 15 min after taking the sample from -20 °C.
- Use Eppendorf tubes to store tissues after fixation at 70% ethanol and dehydration in 100% ethanol.
- Sample storage breakpoints: After the replacement to 100% Histoclear completely, the sample can be stored for a few days at RT. However, Histoclear is volatile and dissolves the paraffin; thus, it is not recommended for more than a few weeks. After fixation sample could be stored for months a 4 °C. After the embedding sample could be stored for years at a temperature below 25 °C. After cutting with microtome, the sample on microslides could be stored for several weeks at RT.
- Immunolabeling
We present an example of multicolor immunostaining with 2F4 antibody against low methylesterified homogalacturonan binding a dimeric association of homogalacturonans through calcium ions, LM20 for high methylesterified homogalacturonan, JIM7 for partially methylesterified homogalacturonan, CBM3 and CBM4 to recognize mostly crystalline and amorphous cellulose respectively, PDM recognizing mannans, and LM24 recognizing xyloglucans. For all the steps, use 2F4 buffer instead of the PBS, even when not using the 2F4 antibody. It has proven to work correctly with different LM and JIM antibodies, for both CMB3 and CMB4 and microtubule antibodies (for the list of available LM, JIM and others antibodies and plant probes, please look here: www.plantprobes.net) using tissue cut samples of Arabidopsis, rice, star fruit, and maize (Yang et al., 2016). Importantly, if at any stage you wash out this buffer with water or PBS you will lose the 2F4 antibody staining.
Notes:- Prior to immunostaining, optionally, quench the free aldehyde groups using 50 mM NH4Cl in 2F4 buffer for 15 min. Wash 3 times with 2F4 buffer, each time 3-5 min. This step is recommended since any residual aldehyde group from FAA solution used at Sample Fixation Step B2 will react with the amino group of the antibodies leading to unspecific antibody binding.
- We recommend applying all the primary antibodies successively as described in Steps C1-C5, to prevent competition and steric hindrance effects, especially for the closely located epitopes, such as cell wall targets presented here (cellulose, xyloglucan, pectin and mannan, Figure 1). However, performing simultaneous primary antibody incubation works well in most cases.
- To avoid the unspecific antibody binding, we use 5% milk in the 2F4 buffer as a blocking buffer. However, the milk can be contaminated over time; therefore, complete the immunostaining protocol Steps C1-C5 in less than 72 h, and where necessary, perform the overnight antibody incubation at 4 °C. Perform short antibody incubation and washing steps at room temperature (RT).
- Time schedule for staining. For the thin tissue sections (< 5 µm) 2 h at RT primary antibody incubation time is sufficient. For the thicker cuts and for certain antibodies, this time is extended to a minimum 3 h.
- Microwave Treatment. Depending on the manufacturer, selected antibodies may need microwave heating for activation or reaction acceleration (1 to 2 min at 400 watts). This step should be fast to prevent sample boiling–none of the antibodies presented in this protocol display improved efficiency of labeling after microwave heating.
- For enzyme treatments in the case of Aspergillus pectolyase from Sigma, treat sections with 200 μl of pectolyase in the incubation buffer (see Recipes) at the final dilution 0.1% at room temperature for 10 min prior to Step C1 (CBM incubation) or Step C2 (primary antibody incubation). After the enzyme treatments, wash sections with 2F4 buffer 3 times each time 3-5 min. When you use another pectate lyase such as Megazyme E-PLYCJ, check their publica- tion list for the reaction condition.
- How to make the hand made humid chamber (Supplemental Figure 1U). Place 2-3 layers of paper towel on the bottom of an empty micropipette box (10 cm x 14 cm x 9.5 cm) or an empty box for the microscopy slide tray (3 cm x 8.5 cm x 21 cm) and add distilled water (~20 ml), so that paper towels are humid. Cover the box. Use aluminum foil to wrap the humid chamber. If you use micro slides for confocal imaging sample preparation, make sure that the sample on the microslide is not in contact with the wet paper towels. For this, we recommend a microscopy slide tray.
- The cellulose-binding molecule (CBM3 or CBM4) incubation: Dilute 2/100 volume per volume (v/v) CBM3/4 reagent in blocking buffer and add to samples. Incubate for 2 h at RT or overnight at 4 °C in the humid chamber(Supplemental Figure 1U). After removal of CBM3 solution, wash 3 times for 5 min using the blocking buffer. If you perform overnight incubation at 4 °C, the next day, take the humid chamber for samples out of the fridge at least 30 min before the washing steps.
- The first primary antibody incubation: Dilute 20/100 v/v 2F4 antibody in blocking buffer and add to samples. Incubate for 2 h at RT or overnight at 4 °C in the humid chamber. After removal of 2F4 solution, wash 3 times for 5 min using the blocking buffer. If you perform overnight incubation at 4 °C, the next day, take the humid chamber for samples out of the fridge at least 30 min before the washing steps.
- The second primary antibody incubation: Dilute 10/100 v/v LM20 antibody in blocking buffer and add to samples. Incubate for 2 h at RT or overnight at 4 °C in the humid chamber. After removal of LM20 solution, wash 3 times for 5 min using the blocking buffer. If you perform overnight incubation at 4 °C, the next day, take the samples out of the fridge at least 30 min before the washing steps.
- The third primary antibody incubation: Dilute 1/100 v/v rabbit or chicken anti-his tag antibody in blocking buffer and add to samples. Incubate for 2 h at RT or overnight at 4 °C in the humid chamber. After removal of the antibody solution, wash 3 times for 5 min using the blocking buffer. If you perform overnight incubation at 4 °C, the next day, take the humid chamber for samples out of the fridge at least 30 min before the washing steps.
- The secondary antibody incubation: Dilute all of three secondary antibodies 1/100 v/v in the same blocking buffer as a secondary antibody-mixture. Incubate for 2 h at RT or overnight at 4 °C in the humid chamber. Incubate the secondary antibodies in the dark to avoid the photo-bleaching. For this, wrap the humidified chamber in aluminum foil. After removal of the secondary antibody-mixture solution, wash 3 times for 5 min using the blocking buffer. If you perform overnight incubation at 4 °C, the next day, take the humid chamber for samples out of the fridge at least 30 min before the washing steps. The secondary antibody incubation could also be done sequentially (see Troubleshooting subsection), although in the case of Figures 1 to 4, all the secondary antibodies were added simultaneously.
- Steps C1-C5: Perform all the antibody incubation steps in the humidified chamber.
- This protocol can be extended for more than three targets using the antibodies from another species, such as rabbit, guinea pig, horse, and human. However, imaging more than four colors on the confocal microscope requires additional laser lines (> 700 nm) or spectral unmixing procedure.
- Make negative control images without 1st, 2nd, and 3rd primary antibodies like Figure 1A (Only Step C5 with secondary antibody mixture, at least in the case of your first trial.
- For dSTORM imaging, from Step B14, we recommend using the multi-well Ibidi Glass Bottom µ-slides, such as 8-well slides (Ibidi), which can be easily inserted and clipped on many microscope stages. The multi-well slides are convenient for the preparation of several imaging conditions, reduce the quantity of reagent used, and improves the homogeneity of the immunostaining for the semi-quantitative analysis. For 8-well Ibidi slides, we recommend using 70-100 µl of reagent per well at Steps C1-C5 of immuno-labeling.
- Post-Immuno fixation.
This step is important for the dSTORM nanoscopy samples and is not necessary for samples prepared for confocal imaging using mounting media. Since dSTORM samples, contrary to confocal microscopy samples, are not mounted in the mounting media, it reduces the thermal motion of the antibody complex and slows down the antibody dissociation from the epitope.- Incubate the sample for 10 min in 3.7% formaldehyde (see Recipe 4) diluted in an appropriate amount of 10x 2F4 buffer and distilled water at RT. For this, place 200 µl drop of 3.7% formaldehyde solution on the sample located inside the ibidi multi-well slide. Perform this step under the fume hood and wear nitrile gloves.
- Wash 3 times for 3 min with the 300 µl of 2F4 buffer. Perform this step under the fume hood.
- Quench the formaldehyde with 70 µl drop of 50 mM ammonium chloride for 15 min. This step reduces the risk of contact with formaldehyde to your skin while handling the sample during the imaging. Dilute the ammonium chloride in 2F4 buffer (see Recipes). After incubation, wash briefly 3 times with the 300 µl drop of 2F4 buffer. You can also use other aldehyde quenching reagents containing amine groups, such as 50 mM of glycine in 2F4 buffer.
- Sample mounting and storage. Only for confocal imaging.
Note: Do not perform this step for dSTORM sample preparation.
To extend the lifespan of the sample, mount the slide with a coverslip in mounting media containing antifade reagent. We suggest ProLong Gold Antifade Mountant (Thermo Fisher Scientific) (Supplemental Figure 1V). To prevent the formation of bubbles, place a droplet of mounting media on the coverslip, and gently lower it on the slide using a micro tweezer (Supplemental Figure 1W-AA). Seal the coverslip with nail polish to prevent drying (Supplemental Figure 1AB) and put sealed micro slides into Cardboard Slide Tray (Supplemental Figure1AC) and store in the dark at 4°C. - Sample storage. Only for dSTORM imaging.
After completion of Steps C6, add 500 µl of 2F4 buffer to each of 8 wells, seal with parafilm, wrap in the aluminum foil, and keep in a humid chamber at 4 °C as presented in Supplemental Figures 1AD and 1AE.
Note: You can perform this for confocal imaging, but then you cannot mount your sample in the antifade mountant; therefore, it is not recommended.
Choosing the fluorescence dyes.
For confocal imaging, we recommend using bright and photostable dyes, such as Alexa Fluor, ATTO, and CF series. For the dSTORM imaging, only certain dyes work. We recommend ATTO488, CF568, and Alexa647 dyes. For the dSTORM nanoscopy imaging, we recommend using F(ab’)2 secondary antibody fragments or nanobodies, or, when possible, to conjugate the primary antibodies directly with the fluorophore of interest. For densely packed epitopes, we recommend using either single labeled secondary antibodies.
Troubleshooting
High background: Reduce secondary antibody concentration. The good practice is to perform the titration of both primary and secondary antibodies. If, for instance, the recommended antibody dilution is 1:200, a good starting point for the titration test is 1:50, 1:100, 1:200, 1:400, and 1:800 dilution. Before immunostaining, quench free aldehyde groups using 50 mM NH4Cl in the 2F4 buffer for 15 min. Use serum in blocking buffer in which the secondary antibody has been raised, e.g., goat serum for antibodies produced in goat. Incubate the antibodies at 4 °C overnight prior to Steps C1 or C2.
A high degree of colocalization when it is not expected: Use sequential staining for all the secondary antibodies. If possible, use the same host species for all the secondary antibodies (For example, a set of anti-rat antibody, anti-rabbit antibody and anti-mouse antibody from goat and so on). To avoid off-target binding, use highly cross-adsorbed secondary antibodies. We recommend preparing a test sample for which only Step C5 is performed, which is the application of the secondary antibodies without primary antibodies.
Low signal: Increase the primary antibody concentration and/or incubation time. Try to use a fresh batch of the primary and secondary antibodies. Change the secondary antibody (conjugated-dye, host species, or producer for the same type of antibody). Increase the secondary antibody concentration (perform the titration-test at the 2, 4, and 6 fold increased antibody dilution) and/or incubation time (perform tests at 2 h, 4 h, 6 h). Check the pH of the 2F4 buffer. If pH is not 8.0, adjust it.
Confocal imaging tips
1)Counterstaining
For the semi-quantitative analysis of the cell wall component with a confocal microscope, compare the samples from the same experimental preparations. If possible, consider the observation of counterstained sample with the third or fourth channel of the laser scanning confocal microscope, if the counterstaining intensity is expected not to change in different sample conditions. For the cell wall staining, use, for instance, calcofluor white stain. Add 200 µl of calcofluor white stain after the Step C5 at 5 mM concentration in 2F4 buffer and incubate for 5 min at room temperature. Wash briefly twice with 500 µl of 2F4 buffer before applying the antifade mounting media to the sample (Step C7). The counterstaining is useful to normalize the intensity data to size, e.g., area or the cell wall thickness. Prepare all the conditions you want to compare for each experimental preparation.
2)Operation of microscope
Switch on the laser 1h before imaging. During the imaging, make sure not to saturate the signal at the detectors for downstream quantification. Keep the laser power, and detector gains the same across the experiments and keep all other settings constant between comparable experiments. Additionally, in order to avoid the photobleacing during the optimization of the image quality before the final take, consider using a tissue sample or region with less scientific interest, but a similar signal level present on your slide, 1) Find the target point and focus the section in bright field. Differential interference contrast image (DIC) is more convenient to focus the image if DIC is installed to your microscope. 2) Optimize the image quality at the position near the final target point. 3) Stop scanning frequently during the optimization of the image quality. 4) As the initial settings for confocal laser scanning mode, 1 to 2% of laser power is a good starting point to focus on a sample. 5) Faster or maximal scan speed (Arbitrary level of scan speed: > 9 in the case of Zeiss LSM 710) is recommended to focus on the initial conditioning (Live button of ZEN software is convenient in Zeiss LSM 710).
Note: There are fifteen and arbitrary levels of scan speed in LSM710 and the absolute speed at maximum level “15” is 8 frame/s, according to Zeiss local office. When performing quantitative imaging, due to photobleaching, never take the final image of the same area twice. Therefore, move the X-Y-Z stage for the micro slide in a certain direction (from right to left and from front to back) to avoid the observation of the same point. For reliable quantification, 2-3 independent experiments (independent technical replicates) need to be carried out and compared. Perform a pilot experiment to determine the common parameters, such as gain and pixel dwell time, and to abolish/minimize any photobleaching. Sequentially image all the channels, always starting with the longest wavelength and ending with the shortest wavelength.
Data analysis
Figure 1 shows dual- and triple-color IHC of different cell wall epitopes, representing members of three different families of the cell wall polysaccharides: pectin (2F4, LM20, and JIM7), cellulose (CBM4, CBM3) and hemicellulose (PDM and LM24) in the transverse section of star fruit leaf rachis (Supplemental Figure 2, and Table 1).
Comparing Figure 1 panels A and C, the cellulose staining (CBM3 and CBM4, see Table 1) is weak in areas presenting high HG counts and high in tissues with low HG counts. This could suggest that cellulose and pectins occupy exclusive wall compartments. However, the enzymatic extraction of pectin revealed much higher cellulose detection levels with CBM4 (compare Figures 1C and 1E). This indicates that, in the intact cell walls, the majority of cellulose epitopes are masked by pectins. Without pectin extraction, the remaining cellulose staining may correspond to xyloglucans, which are partially detected by CBM probes (see PlantProbes CMB3 reagent description, Hernandez-Gomez et al., 2015). Indeed, Figure 1D shows that CBM3 and LM24 antibody against xyloglucans present a low degree of overlap in the subset of walls.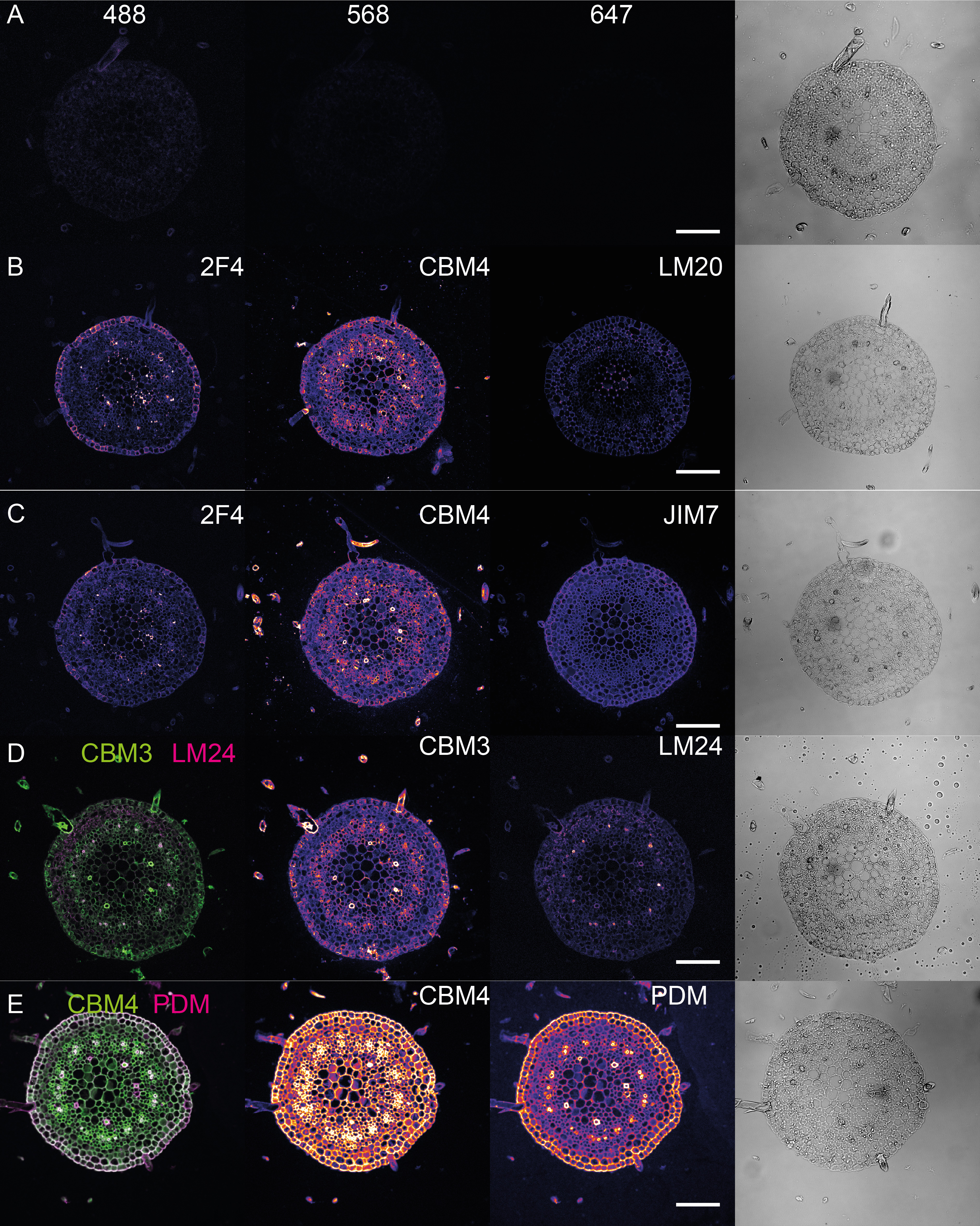
Figure 1. Multicolor confocal imaging of different cell wall epitopes in the star fruit (Averrhoa carambola) leaf rachis. A. Control images with only the secondary antibody staining. B. Triple staining of 2F4, CBM4 and LM20, and (C) 2F4, CBM4, and JIM7 epitopes. D. Double staining of CBM3 and LM24. E. Double staining of CBM4 and PDM after enzymatic pectin extraction. All the images were acquired with the same microscope settings (see Table 2 for suggested settings). The last column represents transmission images of the cuts. Images were visualized in Fiji. Scale bars, 100 µm.
Table 1. The primary antibody, CBM reagents, and the secondary antibody dilutions used in Figures 1-4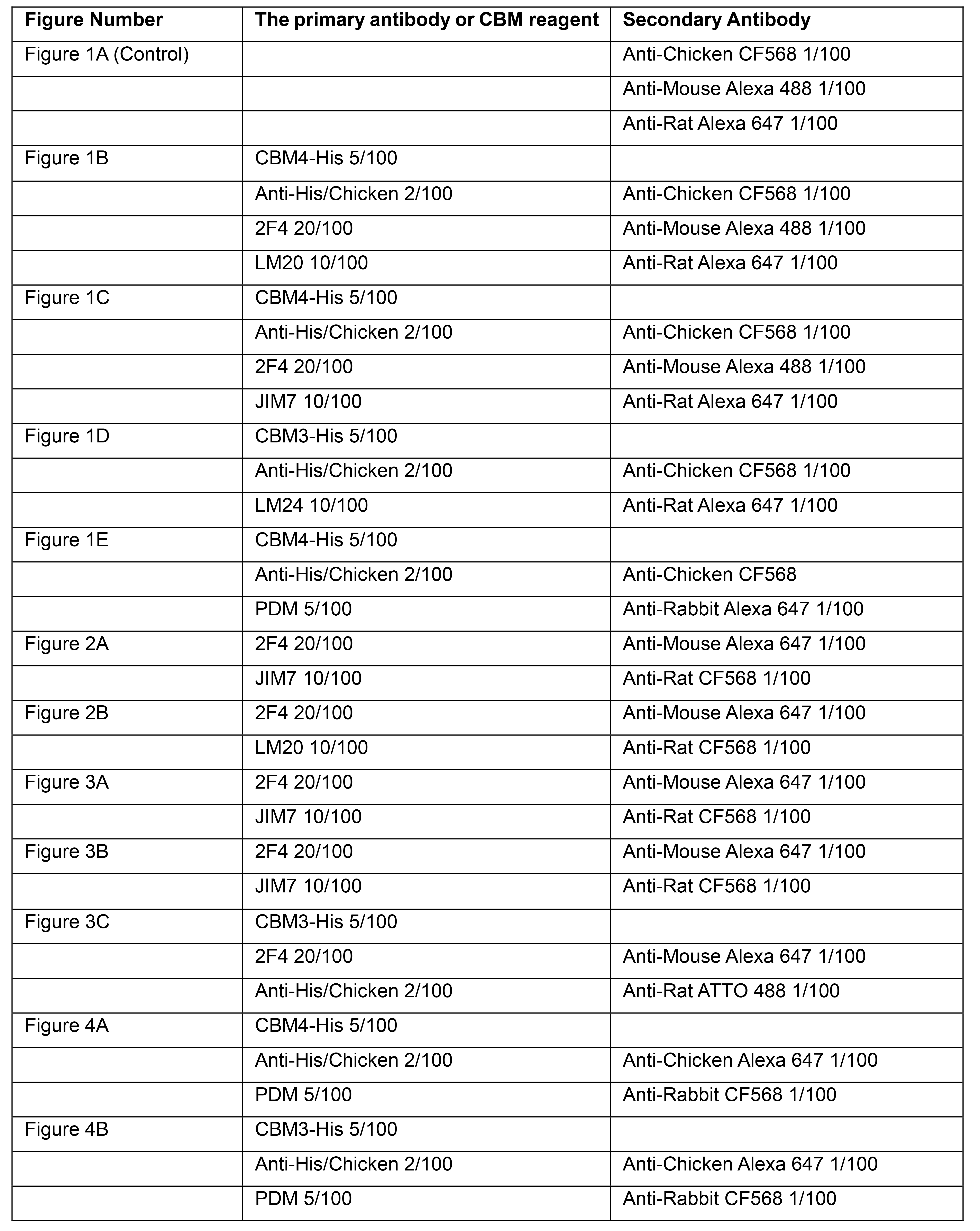
Table 2. The suggested emission filter bandwidth for the triple immunostaining and a counterstaining with calcofluor white using confocal laser scanning microscope optimized to minimize spectral crosstalk. For details on the filter sets for dSTORM please refer to Haas et al. (2020)
Figure 2 shows the 3D dSTORM nanoscopy imaging of the different pectin species in the L2 layer (layer below the epidermis) of the Arabidopsis cotyledon with the longitudinal section described in Supplemental Figure 2E. In Figures 2A and 2B, the typical cell edge staining of low-methylated (2F4), high-methylated (LM20), and partially methylated (JIM7) pectin is observed (see Table 1). The 3D dSTORM images revealed that the staining is weak inside the cell wall (middle lamella) and is mostly limited to the cell wall edges facing the cytoplasm–this highlights one of the biggest limitations of IHC in general: the epitope accessibility and antibody penetrability. Yet, in light of available techniques that possess similar spatial and chemical resolution (e.g., iEM), 3D dSTORM provides overall higher labeling and detection density, and the number of epitopes detected in the middle lamella remains higher for 3D dSTORM than iEM (Cosgrove and Anderson, 2020). One possible way to bypass this limitation is to remove one polymer (for example, structural protein degraded with the recombinant Protease K) from the cell wall to increase antibody penetration. However, this may cause an unspecific wall perturbation. The development of small nanobodies against the cell wall targets will help in the future to address these limitations. The advance in correlative super-resolution light and electron microscopy may help enhance the performance and annihilate the limitations of 3D dSTORM and EM.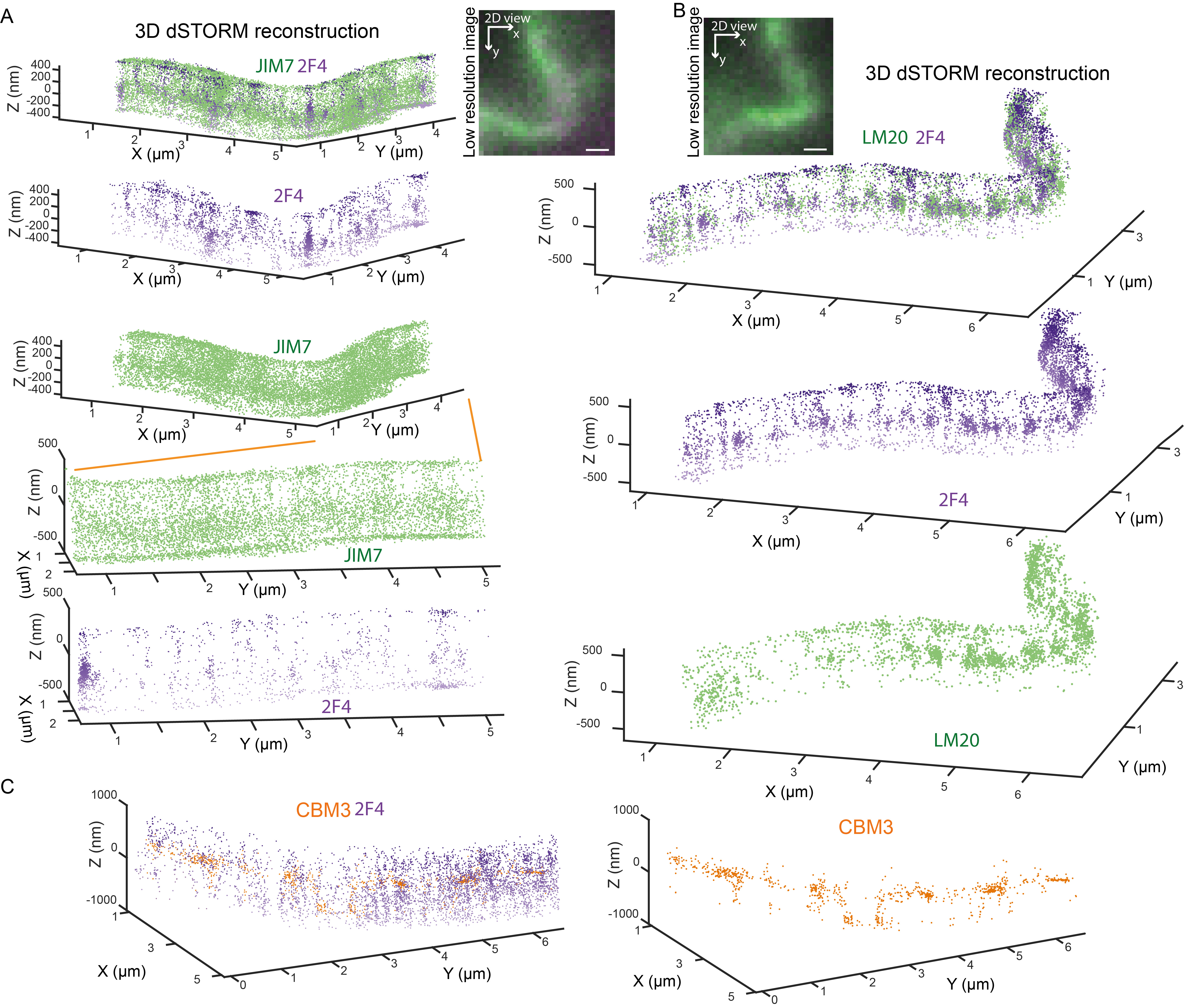
Figure 2. Two-color 3D dSTORM imaging of Homogalacturonan at tri-cellular junctions of L2 (subepidermal cell) layer in Arabidopsis cotyledon. Two-color scatter plots showing the 3D coordinates of localized (A) JIM7 (green) and 2F4 (violet), and (B) LM20 (green) and 2F4 (violet) epitopes. Image insets show a low-resolution oblique illumination image with red square outlining regions shown in the scatter plots. 2D–two dimensional, top view (XY), and 3D–three dimensional, inclined view, where the Z-axis is oriented perpendicular to the conventional image plane. The orange lines show the cell wall region enlarged in the panel below. Image scale bar, 4 µm. Data were visualized using GrafeoV.2.
Figure 3 shows the 3D dSTORM nanoscopy detection of the different pectin species in the epidermal layer of the Arabidopsis cotyledon with a longitudinal section described in Supplemental Figure 2G. In both Figures 3A and 3B, 2F4 and LM20 epitopes form a filamentous pattern of staining in the anticlinal, but not periclinal walls, described previously as the HG nanofilament (Haas et al., 2020). This nanofilament quaternary structure is challenging the canonical view on the pectin in muro architecture as an amorphous gel-like matrix. These unexpected results raise two concerns: (1) that the filamentous pattern represents pectins as a spacer between cellulose microfibrils, or (2) that pectin antibody cannot penetrate between the grooves formed by the cellulose microfibrils (Cosgrove and Anderson, 2020). To address these concerns, we labeled partially methylated HG epitope (JIM7) in the anticlinal walls of the cotyledons, Figure 3A. JIM7 presents a much broader and uniform distribution pattern compared to LM20 and 2F4 HG epitopes. First, the diffuse distribution of JIM7 epitope compared with LM20/2F4 filamentous distribution suggests that the pectin methylation pattern somehow correlates with its localization. Secondly, it implies that HG nanofilaments of LM20 and 2F4 are likely, not due to epitope inaccessibility in the grooves formed by cellulose microfibrils, as JIM7 epitope can be localized there, Figure 3A. Moreover, double staining for cellulose (CBM3) and HG (2F4) in Maize epidermal cells of an immature leaf at 6th node (longitudinal section in Supplemental Figures 2C and F), without prior extraction of pectins, shows that empty spaces between 2F4 nanofilaments are not filled with cellulose and that the overall level of the cellulose staining without pectin extraction is very low, Figure 3C.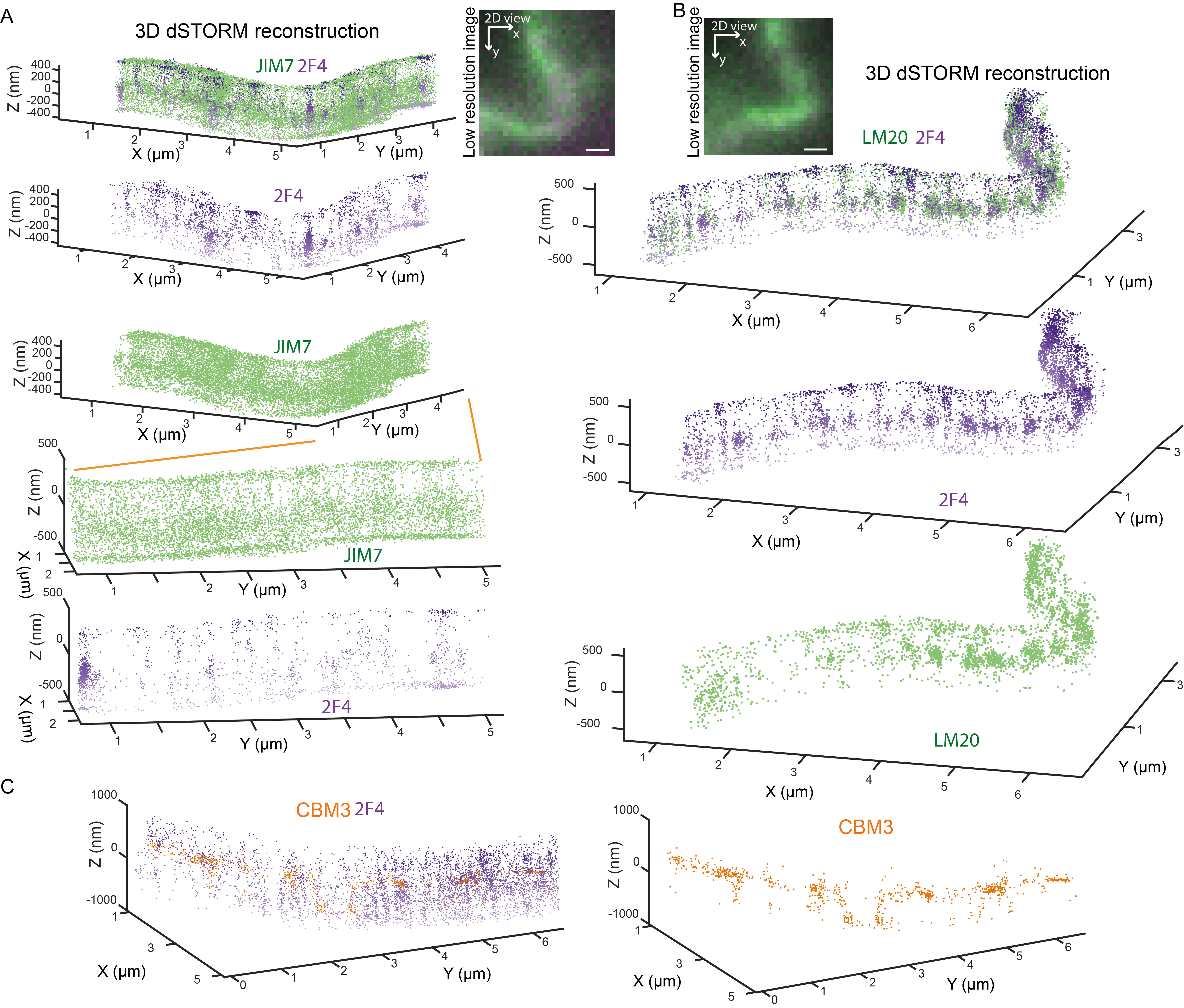
Figure 3. Two-color 3D dSTORM imaging of Homogalacturonan at the lobes of Arabidopsis cotyledons pavement cells and maize leaf pavement cells. Two-color scatter plots showing the 3D coordinates of localized (A) JIM7 (green) and 2F4 (violet), and (B) LM20 (green) and 2F4 (violet) epitopes in the Arabidopsis cotyledons. Image insets show a low-resolution fluorescence image of the regions shown in the 3D dSTORM scatter plots. The orange lines show the cell wall region enlarged in the panels below. (C) Two-color scatter plots showing the 3D coordinates of localized 2F4 (violet), and CBM3 (orange) epitopes in the Maize leaf. Scale bar, 1 µm. Data were visualized using GrafeoV.2.
Figure 4 shows two-color 3D dSTORM imaging of Arabidopsis primordia and meristem directed towards cellulose (CBM3 or CBM4) and (hetero)mannan (PDM) and with prior pectin extraction with a longitudinal section of 1 cm of the inflorescence (Supplemental Figures 2B and 2D, Table 1). Comparing panels A and B show that CBM3, detecting predominantly crystalline cellulose, forms discreet filaments distribution as contrasted with CBM4, tagging amorphous cellulose. The staining for mannans displays different abundance in different cell walls: from extended wall sections mostly exclusive from the cellulose (Figure 4A) to punctate staining decorating extremities of the cellulose nanofibrils (Figure 4B).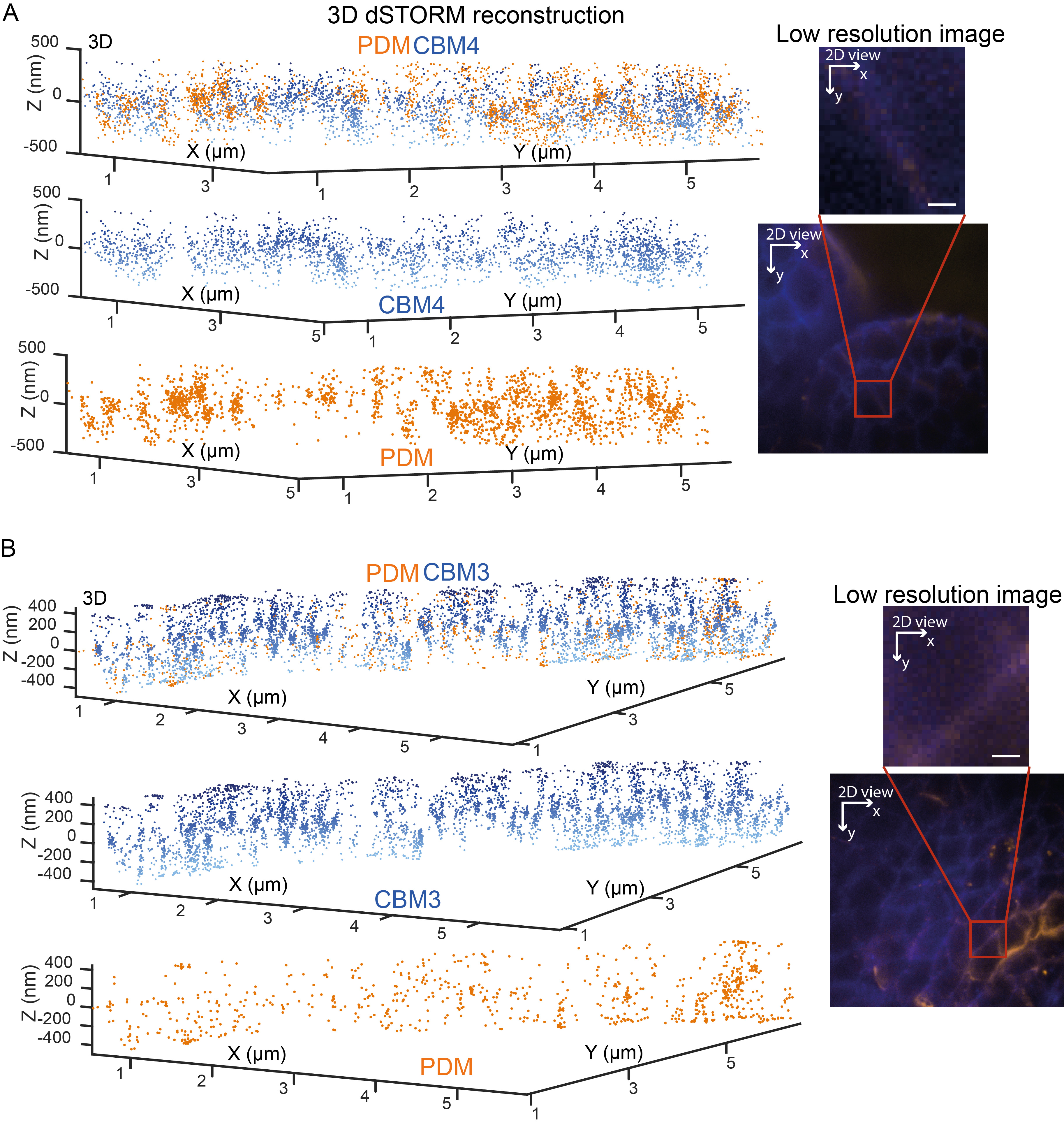
Figure 4. Two-color dSTORM imaging of cellulose (CBM3/4) and (hetero), mannan (PDM) in the Arabidopsis primordia, and meristem. Pectins were enzymatically extracted with pectolyase treatment prior to the immunostaining protocol (Step C1). Two-color scatter plots showing the 3D coordinates of localized (A) CBM4 (blue) and PDM (orange) in a meristem, and (B) CBM3 (blue) and PDM (orange) epitopes in primordia. Insets on the left of the plots show a low-resolution fluorescence image with a red square outlining regions presented in the scatter plots. Scale bar, 1 µm. Data were visualized using GrafeoV.2.
Conclusion
IHC is a powerful tool to study subcellular changes in cell wall chemistry. However, it has several limitations. The number of epitopes that could be explored simultaneously is limited (< 4) due to spectral cross-talk and the number of the antibody species. In addition, only epitopes with well-defined antibodies can be detected. Furthermore, the accessibility of the antibody to the inner cell wall components is limited, except if one of the cell wall components is degraded to help penetrability. Also, the sample preparation for IHC may cause tissue distortion; therefore, spatial scale measurements and topology may not be absolute. Despite this, multicolor IHC coupled with a novel optical nanoscopy offers unprecedented insights into the complex spatial organization of biomolecules. In combination with super-resolution imaging, multicolor IHC is now changing our vision on the cell wall architecture and plant growth. The cell wall mediates plant cell interactions with the environment, and therefore we expect this technique will help us understand plant immunity, fruit ripening, plant-microbe interaction, plant growth, and yield. Constant improvements in fluorescence probes, single-molecule techniques, and data analysis make that 3D dSTORM has huge potential to refine our knowledge on molecular assemblies and their function (Klein et al., 2014; Kim et al., 2019; Gwosch et al., 2020; Zhang et al., 2020). Hence, “Seeing, contrary to popular wisdom, isn't believing. It's where belief stops, because it isn't needed any more”, Terry Prattchet, Pyramids.
Recipes
- 2F4 Buffer (T/Ca/S buffer final concentration)
To dilute all the antibodies, prepare blocking buffer and washing buffer (http://www.plantprobes.net/pp_2F4.pdf).
Tris-HCl 20 mM pH 8.2
CaCl2 0.5 mM
NaCl 150 mM
The final pH should be 8.0 - Formaldehyde Alcohol Acetic Acid FAA solution
50% ethanol
10% acetic acid
3.7% formaldehyde
The percentage indicates the final concentration in FAA solution - 1 M ammonium chloride solution diluted in 2F4 buffer
Mix 53.489 g of ammonium chloride powder in 1 L of 2F4 buffer
Adjust the pH to 8.0 - Formaldehyde diluted in10x 2F4 buffer
Mix 1 volume of 37% formaldehyde solution, 1 volume of 10x 2F4 solution, and 8 volumes of distilled water.
Perform under the fume hood - Citrate-Phosphate Buffer for Pectolyase incubation (pH 4.8, Stoll and Blanchard, 1990)
- 0.2 M Na2HPO4·7H2O
Dissolve 53.65 g in MilliQ water and make 1 L of solution A - 0.1 M citric acid
Dissolve 19.21 g in MilliQ water and make 1 L of solution - Mix 24.8 ml of solutionA and 25.2 ml of solution B and dilute to a total of 100 ml for pH 4.8
If you need, check pH value
- 0.2 M Na2HPO4·7H2O
Acknowledgments
This work presents an extended protocol, which was published in Haas et al., 2020. The dSTORM was performed at the MRC Laboratory of Molecular Biology, Cambridge, and we thank Nick Barry and Jonathan Howe for their support. We Herman Höfte for help with the fundraising and discussion of the results.
Funding: AP has received the support of the French National Research Agency (ANR) GoodVibration ANR-17-CE13-0007 and of the EU in the framework of the Marie-Curie FP7 COFUND People Program, through the award of an AgreenSkills+ fellowship (under grant agreement n° 201310). The IJPB benefits from the support of Saclay Plant Sciences-SPS (ANR-17-EUR-0007). The Microscopy Facility at the Sainsbury Laboratory is supported by the Gatsby Charitable Foundation. This work has benefited from the support of IJPB's Plant Observatory technological platforms.
Competing interests
The authors declare no competing interests.
References
- Anane, R. F., Sun, H., Zhao, L., Wang, L., Lin, C. and Mao, Z. (2017). Improved curdlan production with discarded bottom parts of Asparagus spear. Microb Cell Fact 16(1): 59.
- Blake, A. W., McCartney, L., Flint, J. E., Bolam, D. N., Boraston, A. B., Gilbert, H. J. and Knox, J. P. (2006). Understanding the biological rationale for the diversity of cellulose-directed carbohydrate-binding modules in prokaryotic enzymes. J Biol Chem 281(39): 29321-29329.
- Boettiger, A. N., Bintu, B., Moffitt, J. R., Wang, S., Beliveau, B. J., Fudenberg, G., Imakaev, M., Mirny, L. A., Wu, C. T. and Zhuang, X. (2016). Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 529(7586): 418-422.
- Chen, B., Gong, W., Yang, Z., Pan, W., Verwilst, P., Shin, J., Yan, W., Liu, L., Qu, J. and Kim, J. S. (2020). STORM imaging of mitochondrial dynamics using a vicinal-dithiol-proteins-targeted probe. Biomaterials 243: 119938.
- Cosgrove, D. J. and Anderson, C. T. (2020). Plant Cell Growth: Do Pectins Drive Lobe Formation in Arabidopsis Pavement Cells? Curr Biol 30(11): R660-R662.
- Cuello, C., Marchand, P., Laurans, F., Grand-Perret, C., Laine-Prade, V., Pilate, G. and Dejardin, A. (2020). ATR-FTIR microspectroscopy brings a novel insight into the study of cell wall chemistry at the cellular level. Front Plant Sci 11: 105.
- Dlasková, A., Engstova, H., Spacek, T., Kahancova, A., Pavluch, V., Smolkova, K., Spackova, J., Bartos, M., Hlavata, L. P. and Jezek, P. (2018). 3D super-resolution microscopy reflects mitochondrial cristae alternations and mtDNA nucleoid size and distribution. Biochim Biophys Acta Bioenerg 1859(9): 829-844.
- Gwosch, K. C., Pape, J. K., Balzarotti, F., Hoess, P., Ellenberg, J., Ries, J. and Hell, S. W. (2020). MINFLUX nanoscopy delivers 3D multicolor nanometer resolution in cells. Nat Methods 17(2): 217-224.
- Haas, K. T., Compans, B., Letellier, M., Bartol, T. M., Grillo-Bosch, D., Sejnowski, T. J., Sainlos, M., Choquet, D., Thoumine, O. and Hosy, E. (2018a). Pre-post synaptic alignment through neuroligin-1 tunes synaptic transmission efficiency. Elife 7: e31755.
- Haas, K. T., Lee, M., Esposito, A. and Venkitaraman, A. R. (2018b). Single-molecule localization microscopy reveals molecular transactions during RAD51 filament assembly at cellular DNA damage sites. Nucleic Acids Res 46(5): 2398-2416.
- Haas, K. T., Wightman, R., Meyerowitz, E. M. and Peaucelle, A. (2020). Pectin homogalacturonan nanofilament expansion drives morphogenesis in plant epidermal cells. Science 367(6481): 1003-1007.
- Handford, M. G., Baldwin, T. C., Goubet, F., Prime, T. A., Miles, J., Yu, X. and Dupree, P. (2003). Localisation and characterisation of cell wall mannan polysaccharides in Arabidopsis thaliana. Planta 218(1): 27-36.
- Heilemann, M., van de Linde, S., Schuttpelz, M., Kasper, R., Seefeldt, B., Mukherjee, A., Tinnefeld, P. and Sauer, M. (2008). Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew Chem Int Ed Engl 47(33): 6172-6176.
- Hernandez-Gomez, M. C., Rydahl, M. G., Rogowski, A., Morland, C., Cartmell, A., Crouch, L., Labourel, A., Fontes, C. M., Willats, W. G., Gilbert, H. J. and Knox, J. P. (2015). Recognition of xyloglucan by the crystalline cellulose-binding site of a family 3a carbohydrate-binding module. FEBS Lett 589(18): 2297-2303.
- Höfte, H. and Voxeur, A. (2017). Plant cell walls. Curr Biol 27(17): R865-R870.
- Huang, B., Wang, W., Bates, M. and Zhuang, X. (2008). Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 319(5864): 810-813.
- Jakobs, S. and Wurm, C. A. (2014). Super-resolution microscopy of mitochondria. Curr Opin Chem Biol 20: 9-15.
- Johnson, P. E., Tomme, P., Joshi, M. D. and McIntosh, L. P. (1996). Interaction of soluble cellooligosaccharides with the N-terminal cellulose-binding domain of Cellulomonas fimi CenC 2. NMR and ultraviolet absorption spectroscopy. Biochemistry 35(44): 13895-13906.
- Kim, J., Wojcik, M., Wang, Y., Moon, S., Zin, E. A., Marnani, N., Newman, Z. L., Flannery, J. G., Xu, K. and Zhang, X. (2019). Oblique-plane single-molecule localization microscopy for tissues and small intact animals. Nat Methods 16(9): 853-857.
- Klein, T., Proppert, S. and Sauer, M. (2014). Eight years of single-molecule localization microscopy. Histochem Cell Biol 141(6): 561-575.
- Knox, J. P., Linstead, P. J., King, J., Cooper, C. and Roberts, K. (1990). Pectin esterification is spatially regulated both within cell walls and between developing tissues of root apices. Planta 181(4): 512-521.
- Komis, G., Samajova, O., Ovecka, M. and Samaj, J. (2015). Super-resolution Microscopy in Plant Cell Imaging. Trends Plant Sci 20(12): 834-843.
- Liesche, J., Ziomkiewicz, I. and Schulz, A. (2013). Super-resolution imaging with Pontamine Fast Scarlet 4BS enables direct visualization of cellulose orientation and cell connection architecture in onion epidermis cells. BMC Plant Biol 13: 226.
- van de Linde, S., Loschberger, A., Klein, T., Heidbreder, M., Wolter, S., Heilemann, M. and Sauer, M. (2011). Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat Protoc 6(7): 991-1009.
- Liners, F., Thibault, J. F. and Van Cutsem, P. (1992). Influence of the degree of polymerization of oligogalacturonates and of esterification pattern of pectin on their recognition by monoclonal antibodies. Plant Physiol 99(3): 1099-1104.
- Löschberger, A., van de Linde, S., Dabauvalle, M. C., Rieger, B., Heilemann, M., Krohne, G. and Sauer, M. (2012). Super-resolution imaging visualizes the eightfold symmetry of gp210 proteins around the nuclear pore complex and resolves the central channel with nanometer resolution. J Cell Sci 125(Pt 3): 570-575.
- Majda, M., Grones, P., Sintorn, I. M., Vain, T., Milani, P., Krupinski, P., Zagórska-Marek, B., Viotti, C., Jönsson, H., Mellerowicz, E. J., Hamant, O. and Robert, S. (2017). Mechanochemical polarization of contiguous cell walls shapes plant pavement cells. Dev Cell 43(3): 290-304 e294.
- Mravec, J., Guo, X., Hansen, A. R., Schückel, J., Kračun, S. K., Mikkelsen, M. D., Mouille, G., Johansen, I. E., Ulvskov, P., Domozych, D. S. and Willats, W. G. T. (2017). Pea border cell maturation and release involve complex cell wall structural dynamics. Plant Physiol 174(2): 1051-1066.
- Pan, L., Yan, R., Li, W. and Xu, K. (2018). Super-Resolution Microscopy Reveals the Native Ultrastructure of the Erythrocyte Cytoskeleton. Cell Rep 22(5): 1151-1158.
- Peaucelle, A. (2018). Cell wall expansion: Case study of a biomechanical process. In: Sahi, V. P. and Baluška, F. (Eds.). Plant Cell Monographs. Springer. Cham: Springer International Publishing, pp. 139-154.
- Peaucelle, A., Braybrook, S. and Hofte, H. (2012). Cell wall mechanics and growth control in plants: the role of pectins revisited. Front Plant Sci 3: 121.
- Peaucelle, A., Wightman, R. and Hofte, H. (2015). The Control of Growth Symmetry Breaking in the Arabidopsis Hypocotyl. Curr Biol 25(13): 1746-1752.
- Prakash, K., Fournier, D., Redl, S., Best, G., Borsos, M., Tiwari, V. K., Tachibana-Konwalski, K., Ketting, R. F., Parekh, S. H., Cremer, C. and Birk, U. J. (2015). Superresolution imaging reveals structurally distinct periodic patterns of chromatin along pachytene chromosomes. Proc Natl Acad Sci U S A 112(47): 14635-14640.
- Sellés, J., Penrad-Mobayed, M., Guillaume, C., Fuger, A., Auvray, L., Faklaris, O. and Montel, F. (2017). Nuclear pore complex plasticity during developmental process as revealed by super-resolution microscopy. Sci Rep 7(1): 14732.
- Shim, S. H., Xia, C., Zhong, G., Babcock, H. P., Vaughan, J. C., Huang, B., Wang, X., Xu, C., Bi, G. Q. and Zhuang, X. (2012). Super-resolution fluorescence imaging of organelles in live cells with photoswitchable membrane probes. Proc Natl Acad Sci U S A 109(35): 13978-13983.
- Shroff, H., Galbraith, C. G., Galbraith, J. A., White, H., Gillette, J., Olenych, S., Davidson, M. W. and Betzig, E. (2007). Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes. Proc Natl Acad Sci U S A 104(51): 20308-20313.
- Slot, J. W., Posthuma, G., Chang, L. Y., Crapo, J. D. and Geuze, H. J. (1989). Quantitative aspects of immunogold labeling in embedded and in nonembedded sections. Am J Anat 185(2-3): 271-281.
- Slot, J. W. and Geuze, H. J. (2007). Cryosectioning and immunolabeling. Nat Protoc 2(10): 2480-2491.
- Stoll, V. S. and Blanchard, J. S. (1990). Buffers: principles and practice. Methods Enzymol 182: 24-38.
- Sydor, A. M., Czymmek, K. J., Puchner, E. M. and Mennella, V. (2015). Super-Resolution Microscopy: From Single Molecules to Supramolecular Assemblies. Trends Cell Biol 25(12): 730-748.
- Tanackovic, V., Rydahl, M. G., Pedersen, H. L., Motawia, M. S., Shaik, S. S., Mikkelsen, M. D., Krunic, S. L., Fangel, J. U., Willats, W. G. and Blennow, A. (2016). High throughput screening of starch structures using carbohydrate microarrays. Sci Rep 6: 30551.
- Verhertbruggen, Y., Marcus, S. E., Haeger, A., Ordaz-Ortiz, J. J. and Knox, J. P. (2009). An extended set of monoclonal antibodies to pectic homogalacturonan. Carbohydr Res 344(14): 1858-1862.
- Wäldchen, F., Schlegel, J., Gotz, R., Luciano, M., Schnermann, M., Doose, S. and Sauer, M. (2020). Whole-cell imaging of plasma membrane receptors by 3D lattice light-sheet dSTORM. Nat Commun 11(1): 887.
- Wightman, R., Busse-Wicher, M. and Dupree, P. (2019). Correlative FLIM-confocal-Raman mapping applied to plant lignin composition and autofluorescence. Micron 126: 102733.
- Xia, S., Lim, Y. B., Zhang, Z., Wang, Y., Zhang, S., Lim, C. T., Yim, E. K. F. and Kanchanawong, P. (2019). Nanoscale Architecture of the Cortical Actin Cytoskeleton in Embryonic Stem Cells. Cell Rep 28(5): 1251-1267 e1257.
- Xu, J., Ma, H. and Liu, Y. (2017). Stochastic Optical Reconstruction Microscopy (STORM). Curr Protoc Cytom 81: 12 46 11-12 46 27.
- Yang, W., Schuster, C., Beahan, C. T., Charoensawan, V., Peaucelle, A., Bacic, A., Doblin, M. S., Wightman, R. and Meyerowitz, E. M. (2016). Regulation of meristem morphogenesis by cell wall synthases in Arabidopsis. Curr Biol 26(11): 1404-1415.
- Zhang, T., Vavylonis, D., Durachko, D. M. and Cosgrove, D. J. (2017). Nanoscale movements of cellulose microfibrils in primary cell walls. Nat Plants 3: 17056.
- Zhang, Y., Schroeder, L. K., Lessard, M. D., Kidd, P., Chung, J., Song, Y., Benedetti, L., Li, Y., Ries, J., Grimm, J. B., Lavis, L. D., De Camilli, P., Rothman, J. E., Baddeley, D. and Bewersdorf, J. (2020). Nanoscale subcellular architecture revealed by multicolor three-dimensional salvaged fluorescence imaging. Nat Methods 17(2): 225-231.
- Zhao, F., Chen, W., Sechet, J., Martin, M., Bovio, S., Lionnet, C., Long, Y., Battu, V., Mouille, G., Moneger, F. and Traas, J. (2019). Xyloglucans and microtubules synergistically maintain meristem geometry and phyllotaxis. Plant Physiol 181(3): 1191-1206.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Haas, K. T., Rivière, M., Wightman, R. and Peaucelle, A. (2020). Multitarget Immunohistochemistry for Confocal and Super-resolution Imaging of Plant Cell Wall Polysaccharides. Bio-protocol 10(19): e3783. DOI: 10.21769/BioProtoc.3783.
- Haas, K. T., Wightman, R., Meyerowitz, E. M. and Peaucelle, A. (2020). Pectin homogalacturonan nanofilament expansion drives morphogenesis in plant epidermal cells. Science 367(6481): 1003-1007.
Category
Plant Science > Plant cell biology > Cell imaging
Developmental Biology > Morphogenesis > Cell structure
Cell Biology > Cell imaging > Fixed-tissue imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.