- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Analysis of Gram-negative Bacteria Peptidoglycan by Ultra-performance Liquid Chromatography
Published: Vol 10, Iss 19, Oct 5, 2020 DOI: 10.21769/BioProtoc.3780 Views: 5702
Reviewed by: Alexandros AlexandratosErkin KuruAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Bacteria are surrounded by a protective peptidoglycan cell wall. Provided that this structure and the enzymes involved are the preferred target for our most successful antibiotics, determining its structural and chemical complexity is of the highest interest. Traditionally, high-performance liquid chromatography (HPLC) analyses have been performed, but these methods are very time consuming in terms of sample preparation and chromatographic separation. Here we describe an optimized method for preparation of Gram-negative bacteria peptidoglycan and its subsequent analysis by ultra-performance liquid chromatography (UPLC). The use of UPLC in peptidoglycan analyses provides a dramatic reduction of the sample volume and hands-on time required and, furthermore, permits in-line mass spectrometry (MS) of the UPLC resolved muropeptides, thus facilitating their identification. This method improves our capability to perform high throughput analysis to better understand the cell-wall biology.
Keywords: UPLCBackground
Bacteria are surrounded by a peptidoglycan (PG) cell wall that, in addition to a structural role, conveys cell shape and protects bacteria from external damage, acting as a barrier against biological, chemical and physical stresses. The murein sacculus or PG is the major component of the cell wall. Gram-negative bacteria present a monolayer in the periplasmic space (Gan et al., 2008), while it constitutes a thick mesh work with several pilled and crosslinked layers in Gram-positive bacteria, (Pasquina-Lemonche et al., 2020). The cell wall is a three-dimensional meshwork of crosslinked glycan strands that encloses the cell body (Glauner et al., 1988; Typas et al., 2011; Egan et al., 2020). The canonical monomeric subunit consists of the disaccharide pentapeptide GlcNAc-(β1-4)-MurNAc-L-Ala-D-Glu-(γ)-(di-amino acid)-D-Ala-D-Ala, where meso-diaminopimelic acid and L-lysine are the most prevalent di-amino acids (Glauner et al., 1988; Vollmer et al., 2008). These monomers are converted into linear polymers by means of MurNAc-(β1-4)-GlcNAc glycosidic bonds and can be crosslinked by peptide chains.
The cell wall is subject to various enzymatic modifications that affect the chemical properties of its subunits, their relative abundance, and the structure of the cell wall (Vollmer et al., 2008). A detailed knowledge of the muropeptide composition under particular conditions is key to understanding to what extent PG variations influence bacterial adaptation to environmental challenges, resistance to antibacterial agents, immune-modulatory activity and toxin release and signaling (Alvarez et al., 2014; Cava and de Pedro, 2014; Yadav et al., 2018). Our method allows us to study the effect of genetic perturbations on cell-wall composition in a high-throughput manner. Furthermore, combining high-throughput PG analysis with genetic perturbations, we can explore the functions of different cell-wall-modifying enzymes. In fact, we have recently studied how class-A Penicillin-Binding Proteins (aPBPs) contribute to cell-wall integrity in Escherichia coli (Vigouroux et al., 2020). By combining PG analysis, morphology analysis and single-molecule tracking, we have demonstrated that the major aPBP PBP1b contributes to cell-wall integrity by repairing cell-wall defects, while the Rod complex governs rod-like cell shape.
Here we describe in detail the procedure for Gram-negative bacteria sacculi isolation, muramidase digestion and muropeptide separation by liquid chromatography. We also provide some general instructions on compositional and structural analysis. The isolation of PG relies on the insolubility of the sacculi in SDS, which allows a relatively easy method to obtain high amounts of PG, and on the availability of specific enzymes (muramidases or lysozymes) that split the MurNAc-(β1-4)-GlcNAc glycosidic bonds which hold the structure together and disassemble it into its individual subunits. The next step requires the use of sensitive and reliable methods that permit the resolution, identification and quantification of the different PG subunits. Since the 80’s, HPLC has been traditionally used for this purpose (Glauner et al., 1988). While this technology was a revolution at the time and revealed an unexpected complexity in PG structure and composition, it has been used essentially unchanged for more than 30 years. However, there are three critical limitations: i) the requirement for inorganic buffers, incompatible with mass spectrometry in-line analysis that would facilitate identification of subunits; ii) the very low sample through-put, which requires a few days for sample preparation and several hours of HPLC run time per sample; and iii) the requirement for large sample volumes (injection volumes of 100-500 µl), due to the relatively low sensitivity of the chromatographic systems. The introduction of UPLC replacing its predecessor HPLC permits to dramatically reduce sample size (100x), while increasing the speed (20x), without compromising the quality of the data. The new systems allow the use of new and improved materials for reverse phase chromatography, including stationary phases with a very small particle size (in the range of 2 µm) that withstand very high pressures, increasing resolution, speed and sensitivity, which are essential requisites for high throughput analysis.
We have developed a new transformative approach that circumvents the limitations of the already available methods (e.g., Desmarais et al., 2013; Kühner et al., 2014). Our protocol i) can be easily adapted for the more frequently available UPLC machines by anyone with a basic knowledge of UPLC techniques; ii) has cut down sample preparation and run times dramatically, allowing for the processing and analysis of large numbers of PG samples per day; iii) is MS-compatible, samples can be collected and subjected to MS analysis without prior desalting steps or can even be analyzed in MS-systems coupled to the UPLC for a faster muropeptide identification, thus revealing a much higher PG chemical complexity than previously anticipated (i.e., identification of minor muropeptides); iv) and requires much less of sample, which is particularly important for in vivo samples (low sample size amount).
Materials and Reagents
- Disposable pipette tips (VWR, catalog numbers: 613-1083 , 613-1079, 613-1077)
- 1.5 ml microtubes (Eppendorf, catalog number: 00 30120086 )
- 2 ml microtubes (Eppendorf, catalog number: 00 30120094 )
- 15 ml conical test tubes (Sarstedt, catalog number: 62.554.502 )
- 50 ml conical test tubes (Sarstedt, catalog number: 62.547.205 )
- Thickwall polycarbonate ultracentrifuge 3.5 ml tubes, 13 x 51 mm (Beckman, catalog number: 349622 )
- pH-indicator strips: pH range 5.0-10.0 and pH range 0.0-6.0 (Merck, catalog numbers: 109531 , 109533)
- 96-well V-shaped-bottom microplates (Corning, Falcon, catalog number: 353263 )
- Pierceable adhesive seal for microplates (Waters, catalog number: 186006336 )
- Analytical column: Kinetex C18 UPLC Column 1.7 µm particle size, 100 Å pore size, 150 x 2.1 mm (Phenomenex, catalog number: 00F-4475-AN )
- Precolumn filter or guard column: SecurityGuard ULTRA Cartridges C18 for 2.1 mm ID columns (Phenomenex, catalog number: AJ0-8782 )
- Precolumn holder: SecurityGuard ULTRA Holder, for UHPLC Columns 2.1 (Phenomenex, catalog number: AJ0-9000 )
- MilliQ water
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S7653-250G )
- Potassium chloride (KCl) ((Sigma-Aldrich, catalog number: P9333-500G )
- Sodium phosphate monobasic dihydrate (NaH2PO4•2H2O) (Sigma-Aldrich, catalog number: 71505-250G )
- Sodium phosphate dibasic dihydrate (Na2HPO4•2H2O) (Sigma-Aldrich, catalog number: 71643-250G )
- Potassium phosphate monobasic (KH2PO4) (Sigma-Aldrich, catalog number: P5655-100G )
- Trizma base (Tris(hydroxymethyl)aminomethane) (Sigma-Aldrich, catalog number: T6791-100G )
- Ortho-phosphoric acid 85% (H3PO4) (Merck, catalog number: 1005731000 )
- Hydrochloric acid fuming 37% (HCl) (Merck, catalog number: 1003171000 )
- Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: L6026-50G )
- Boric acid (H3BO3) (Sigma-Aldrich, catalog number: B6768-500G )
- Sodium hydroxide pellets (NaOH) (Merck, catalog number: 1064821000 )
- Sodium borohydride (NaBH4) (Merck, catalog number: 1063710100 )
- Formic acid (CH2O2) (Merck, catalog number: 5330020050 )
- Acetonitrile (CH3CN) (Merck, catalog number: 1000291000 )
- Proteinase K 20 mg/ml (Thermo Scientific, catalog number: EO0491 ), store enzyme at -20 °C
- Mutanolysin from Streptomyces globisporus ATCC 21553 (Sigma-Aldrich, catalog number: SAE0092-10KU). Resuspend in 2.5 ml digestion buffer (stock concentration ~1 mg/ml) and store enzyme at -20 °C
- Phosphate buffer saline (PBS) (see Recipes)
- Lysis buffer [SDS 5% (w/v)] (see Recipes)
- Tris HCl 1 M pH 8 buffer (see Recipes)
- Tris HCl 100 mM pH 8 buffer (see Recipes)
- SDS 10% (w/v) solution (see Recipes)
- Digestion buffer (see Recipes)
- NaOH 2 M solution (see Recipes)
- Borate buffer (see Recipes)
- NaBH4 solution (see Recipes)
- Ortho-phosphoric acid 25% (v/v) solution (see Recipes)
- Organic buffer A (see Recipes)
- Organic buffer B (see Recipes)
Equipment
- Pipettes (Gilson, catalog numbers: F144563 , F144565 , F144566 )
- Glassware: bottles, measurement cylinders, beakers
- pH-meter (VWR, catalog number: 662-1422 )
- Hot plate stirrer (VWR, catalog number: 97042-598 )
- 12 x 6 mm magnetic stirring bars (VWR, catalog number: 442-4501 )
- Centrifuge for microtubes (Eppendorf, Centrifuge 5418, catalog number: 5418000017 )
- Centrifuge for 15/50 ml conical tubes (Eppendorf, Centrifuge 5920 R, catalog number: 5948000914 )
- Beckman Optima MAX-TL Ultracentrifuge (Beckman, catalog number: A95761 )
- Ultracentrifuge TLA-100.3 rotor (Beckman, catalog number: 349490 )
- Vacuum pump
- Laminar flow cabinet
- Acquity UPLC system (Waters) or similar (e.g., Agilent)
Software
- Waters Empower 3, build 3471 (Waters, https://www.waters.com/waters/en_US/Empower-3-Chromatography-Data-Software/nav.htm?locale=en_US&cid=10190669)
- Microsoft Excel Version 2004 (Microsoft, Office 365, https://www.microsoft.com/en-us/microsoft-365/excel?rtc=1)
- Prism 8 Version 8.0.2 (GraphPad, https://www.graphpad.com/scientific-software/prism/)
Procedure
- Cell lysate preparation
- Grow cultures to the desired optical density in the appropriate culture medium for the bacteria. Record the culture optical density for normalization. For optimal sample preparation, aim for 1010 bacteria or higher (e.g., 10 ml at 1 unit OD600, 50 ml at 0.2 units OD600). Samples can be scaled up but require more reagents and are more time-consuming.
For statistical analysis, prepare and process samples in triplicates. - Transfer the cultures to 50 ml conical tubes and harvest the cells at 3,000 x g for 15 min. Remove the supernatant and resuspend the pellet in 1.5 ml of its own media. Alternatively, pellets can be resuspended in PBS or Tris HCl buffers. Final volume is not critical, but lower volumes are preferred to shorten the number of washes required later (Figure 1A).
Note: Resuspension media containing potassium (e.g., M63 minimal medium) can interfere with sacculi preparation as potassium salts react with SDS producing insoluble KDS. - Transfer the samples to 15 ml conical tubes. The tubes must be suitable for boiling. Place a small stirring bar (magnet) per tube (Figure 1A).
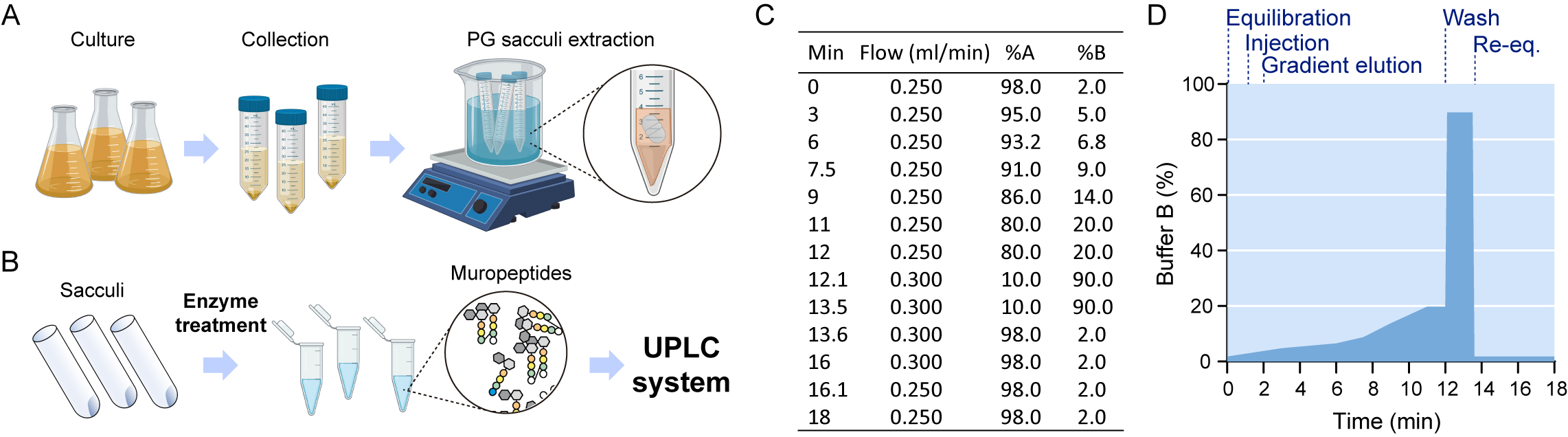
Figure 1. Pipeline for peptidoglycan sample preparation. A. Lysate preparation: bacterial cultures are collected and SDS is added to the resuspended pellets, which are boiled and stirred to facilitate solubilization of all cell components except for the peptidoglycan sacculus. B. Sacculi are washed repeatedly with water and submitted to enzymatic treatment. Released muropeptides in the supernatant are injected in a chromatographic system. C. Mobile phase gradient applied during UPLC separation. D. Steps of the gradient separation of muropeptides by UPLC. - Add 1.5 ml lysis buffer and place the tubes on a beaker with boiling water on a magnetic hot stirrer plate. Boil the samples for 30 min-2 h, then switch off the hot plate and let the lysate stir overnight. Cells will immediately lyse upon boiling in SDS, but longer stirring times are recommended for shearing the DNA (Figure 1A).
- Boiled lysates can be stored at room temperature for long time. Make sure the lid is tightly closed to avoid desiccation.
- Grow cultures to the desired optical density in the appropriate culture medium for the bacteria. Record the culture optical density for normalization. For optimal sample preparation, aim for 1010 bacteria or higher (e.g., 10 ml at 1 unit OD600, 50 ml at 0.2 units OD600). Samples can be scaled up but require more reagents and are more time-consuming.
- Isolation of Gram-negative bacteria peptidoglycan
- To completely remove SDS, perform several washes with water by centrifugation. Spin down the samples 10 min and 150,000 x g using 3 ml polycarbonate ultracentrifuge tubes. Use 20 °C or higher since SDS precipitates at low temperature. Fill the tubes with 3 ml lysate. Make sure the rotor is properly balanced. After centrifugation, all soluble compounds will remain in the supernatant. The resulting pellet is usually transparent and hard to distinguish. It is recommended to mark one side of the tube and use this as reference to locate the pellet (Figure 1B).
Note: If during the washes a white precipitate (likely insoluble KDS) is observed, use Tris HCl 100 mM pH 8.0 buffer instead of water. - Carefully discard the supernatant with a vacuum pump without removing the pellet. Resuspend the pellet in 900 µl MilliQ water and check for the presence of SDS by assessing the production of bubbles. If needed, add 2 ml MilliQ water, mix and centrifuge again. Repeat this wash step until SDS is completely removed (no bubbles are formed after the pellet is resuspended in water, typically 2-4 washes).
- To remove Braun’s lipoprotein or other PG-associated proteins, treat the washed sacculi with a protease. This step is optional, but highly recommended. Transfer the resuspended pellet (~900 µl) to 2 ml microtubes. Add 100 µl Tris HCl 1 M pH 8 buffer and 1 µl proteinase K 20 mg/ml. Incubate samples at 37 °C during 1 h. Stop the reaction by adding 110 µl SDS 10% (w/v) and boil for 5 min in water.
Note: Proteinase K is a broad-spectrum serine protease used for removal of PG bound proteins such as Braun’s lipoprotein. The enzyme retains activity in SDS 1% (w/v). Some bacteria do not require Proteinase K treatment, but it is recommended for removal of other PG-associated proteins.Other proteases as chymotrypsin (EC 3.4.21.1) or trypsin (EC 3.4.21.4) can also be used. - Let the sample cool down before transferring to the ultracentrifuge tubes again. Wash the Proteinase K digested sacculi by adding 2 ml MilliQ water. Mix and ultracentrifuge as described before. Carefully discard the supernatant and, after total removal of SDS, resuspend the pellet in 50-100 µl digestion buffer. The final volume is critical, since soluble muropeptides will remain in the soluble fraction after the muramidase digestion. Washed sacculi should be stored at 4 °C to avoid degradation or contamination. Storing the samples at -20 °C is not recommended since sacculi can break due to freeze/thaw processes.
- To completely remove SDS, perform several washes with water by centrifugation. Spin down the samples 10 min and 150,000 x g using 3 ml polycarbonate ultracentrifuge tubes. Use 20 °C or higher since SDS precipitates at low temperature. Fill the tubes with 3 ml lysate. Make sure the rotor is properly balanced. After centrifugation, all soluble compounds will remain in the supernatant. The resulting pellet is usually transparent and hard to distinguish. It is recommended to mark one side of the tube and use this as reference to locate the pellet (Figure 1B).
- Muramidase digestion
- Transfer resuspended sacculi (50-100 µl) to 1.5 ml microtubes.
- Add 2 µl mutanolysin 1 mg/ml and let the reaction work 2-16 h at 37 °C. The reaction is almost complete after 2 h, but to ensure total digestion, let it proceed for longer times.
- To inactivate the enzyme, boil the samples for 5 min. Do not add any detergent to the sample.
- Centrifuge 15 min at room temperature and 20,000 x g in a benchtop centrifuge and transfer the supernatant to new 1.5 ml microtubes. This supernatant contains the soluble muropeptides released by the mutanolysin (Figure 1B). These samples are very stable and can be stored at 4 or -20 °C.
- Sample reduction
Note: Sample reduction is optional but highly recommended. Digestion with mutanolysin produces a terminal reducing NAM. Sample reduction avoids the different anomeric configurations and the consequent appearance of two to four peaks per muropeptide in the PG profile, as the NAM is reduced to muramitol. Perform this procedure in a laminar flow cabinet. If no reduction is performed, adjust the sample pH to 2.0-4.0 with diluted orthophosphoric acid instead (0.25%, v/v).- Add borate buffer to the sample to adjust pH to 8.5-9.0. For a 100 µl reaction, 15-20 µl borate buffer are typically used. Check pH using indicator strips: take 0.5 µl sample and drop on the indicator strip. Check the color/pH on the reference table. Do not leave samples at high pH for too long, since it leads to alkaline β-elimination (Tipper, 1968).
- Add 20 µl freshly prepared NaBH4 2 M and let the sample reduce at room temperature for 30 min. This reaction produces H2 and bubbles are accumulated, do not close the tube lids to prevent gas accumulation.
- Adjust sample pH to 2.0-4.0 with orthophosphoric acid 25% (v/v). Addition of the acid will cause a violent reaction and sudden bubble formation. First add 4 µl and check the pH with the appropriate indicator strip as indicated before. If needed, carefully add acid µl by µl to ensure the sample reaches the desired pH.
- Spin the tubes for 1 min at maximum speed to ensure maximum sample recovery. Transfer the samples to a 96-well V-shaped-bottom microplate. Seal the plate using a pierceable adhesive seal. Samples can be stored long-term at -20 °C. Insoluble precipitates might form and if observed, filter the samples prior to injection in the UPLC.
- UPLC separation
Note: settings will depend on the software, model and manufacturer of the UPLC. For muropeptide profiling, reverse-phase (RP) columns, typically C18-bonded silicas that are able to retain and separate medium-polar and non-polar metabolites provide a good separation pattern. For RPLC, maximum retention of analytes is ensured by loading samples onto the column using solvents of low eluotropic strength (i.e., composed mainly or entirely of water). Elution of retained metabolites is accomplished using a gradient of increasing acetonitrile content.- Set the column temperature to 45 °C.
- Prepare mobile phases and refill bottles A and B with organic buffer A [formic acid 0.1% (v/v)] and organic buffer B [formic acid 0.1% (v/v) in acetonitrile] respectively. Purge pumps and tubes according to the UPLC system instructions.
- Equilibrate the column with organic buffer A, flow 0.25 ml/min until pressure is stabilized.
- Using the system auto-sampler, inject 10 µl sample.
- Perform the LC run using the gradient described in Figures 1C and 1D and measure absorbance at 204 nm.
- Run a blank injection to monitor and subtract the baseline.
- Troubleshooting
- Carefully control parameters such as flow rate and column temperature following the manufacturers indications to ensure repeatability of the separations. Typically, ~2,000 samples can be injected in a chromatographic column before separation efficiency is lost, peaks degrade and retention times change. If the chromatogram shows poor peak shapes, this is indicative of column degradation or sample overloading. To solve this problem, dilute the sample or improve sample preparation. If the problem persists, consider cleaning or replacing the column.
- The injection volume can be modified depending on the sample concentration:
- For concentrated samples (e.g., PG from large starting cultures), inject less volume, ensuring the system is working within its detection limits.
- If there are no or few peaks or the signal is too low, increase the injection, concentrate the sample using a Speedvac concentrator or prepare new sample by resuspending the washed sacculi in a lower volume prior to mutanolysin digestion.
- Changes in the retention time can be due to improper sample pH or presence of detergent (SDS) in the sample. When sample pH is greater than 5, it results in shifted chromatograms. Adjust pH with orthophosphoric acid 25% (v/v) and rerun the sample. If there is detergent in the sample, either prepare new sample increasing the amount of washing steps or wash the column after each run to get rid of the retained detergent.
- Improper sample reduction results in profiles with altered retention times. Repeat the sample preparation adding an excess of NaBH4.
- Contamination with other components or short re-equilibration time between runs also contribute to the appearance of ghost peaks.
- When sample concentration is low, the baseline drift and noise become more evident. Run a blank injection with water and subtract the baseline during the data processing.
Data analysis
Prepare and run samples in triplicates for statistical analysis.
For optimal comparison conditions, use the same volumes, washes and treatments for all samples.
- Peptidoglycan profile
- Extract the raw data: retention time (min) and absorbance at 204 nm (arbitrary units).
- For baseline correction, subtract the data of a blank injection of water (Figure 2A).
- Define the chromatographic processing regions removing not useful data, typically the injection front and the wash at the end of the run (datapoints ≤ 2-3 min, datapoints ≥ 12-13 min) (Figure 2A).
- Represent the chromatogram by plotting absorbance at 204 nm (arbitrary units) against retention time (min) (Figure 2A).
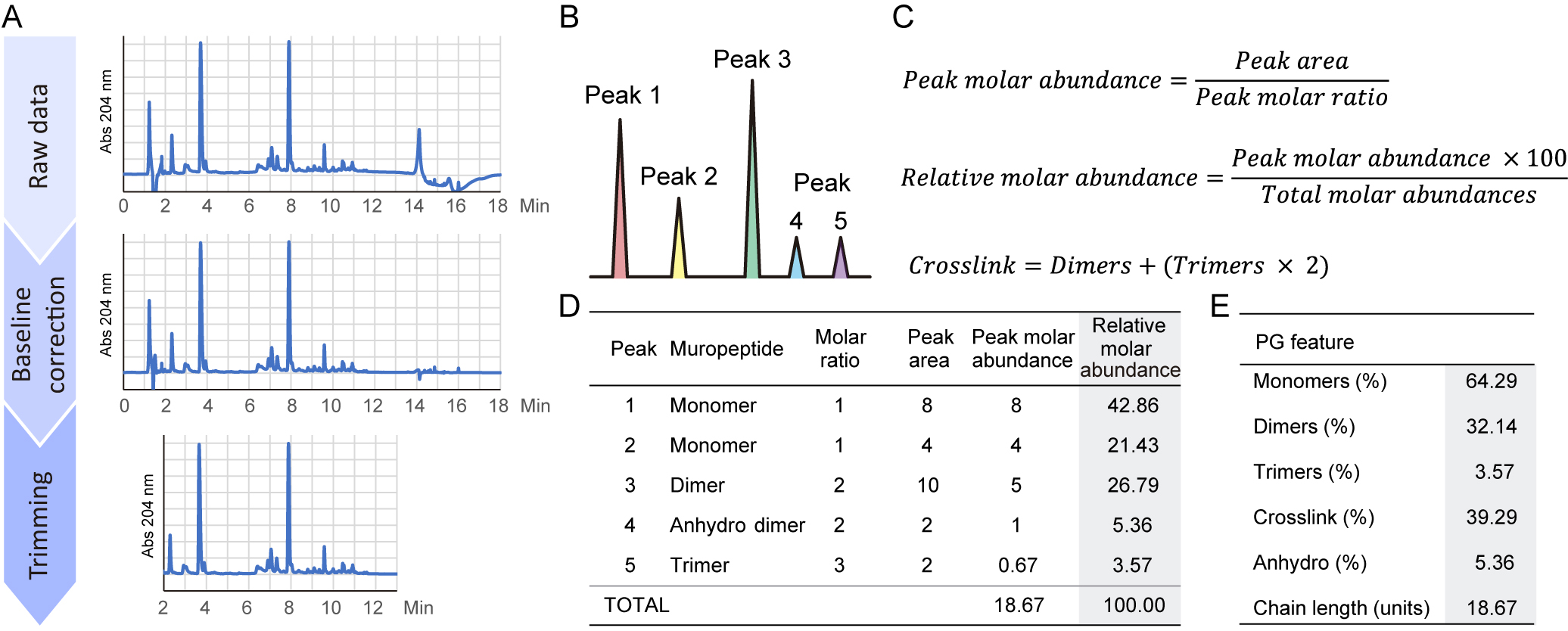
Figure 2. Schematics for analysis of peptidoglycan data. A. Transformation of the raw data for optimal absorbance versus time representation. Subtract a blank injection for baseline correction and trim the datapoints to be shown. B. Example of a simple peptidoglycan chromatogram. C. Formulas employed for the quantifications of muropeptide relative molar abundances and crosslink. D. Table showing the muropeptide abundances in the example profile. E. Peptidoglycan features calculated from the table of muropeptide abundances. - Identify the muropeptides by comparison to a known reference sample run in the same chromatographic system. Unknown or novel peaks can only be identified by MS analysis.
- Relative total PG amount
- For normalization, calculate the relative cell amounts by dividing the recorded optical densities at the time of harvesting by the average optical density of the control samples.
- Calculate the total area of the chromatogram by summing all the intensity values in the time range of interest (min 2-3 to min 12-13).
- Normalize the total area by dividing the total area of each sample by its relative cell amount.
- Divide the normalized total area by the average value of the control samples, and then multiply the result by 100 to calculate the relative percent.
- Perform an unpaired t-test analysis to determine the statistical significance of the results.
- Muropeptide quantifications
- For quantifications, calculate the area of each peak by integration using the appropriate software (e.g., UPLC manufacturers software, MATLAB) (Figure 2B).
- To calculate the peak molar abundance, divide each area by the muropeptide molar ratio: for simplification, 1 if it is a monomer, 2 if it is a dimer, 3 if it is a trimer, … (Figure 2C).
- Determine the relative molar abundances for each peak: divide the molar abundance of every peak by the total of the chromatogram (sum of all individual molar abundances) and multiply the result by 100 to calculate the relative percentage (Figure 2C).
- Perform an unpaired t-test analysis to determine the statistical significance of the results.
- Represent the results as a muropeptide table that typically contains retention time and relative molar abundance for all detected muropeptides (Figure 2D).
- Calculation of peptidoglycan main features
- Monomers (%): sum the relative molar abundances of all monomeric muropeptides.
- Dimers (%): sum the relative molar abundances of all dimeric muropeptides.
- Trimers (%): sum the relative molar abundances of all trimeric muropeptides.
- Crosslink (%): calculate the percentage of crosslink as Dimers + (Trimers × 2). There is a crosslink in a dimer, 2 in a trimer (Figure 2C).
- Anhydro (%): sum the relative molar abundances of all anhydro muropeptides (muropeptides with (1-6anhydro)MurNAc).
- Chain length (glycan units): calculate the average glycan chain length by dividing 100 by the percentage of anhydro muropeptides.
- Perform an unpaired t-test analysis to determine the statistical significance of the results.
- Represent the results as a PG feature table (Figure 2D).
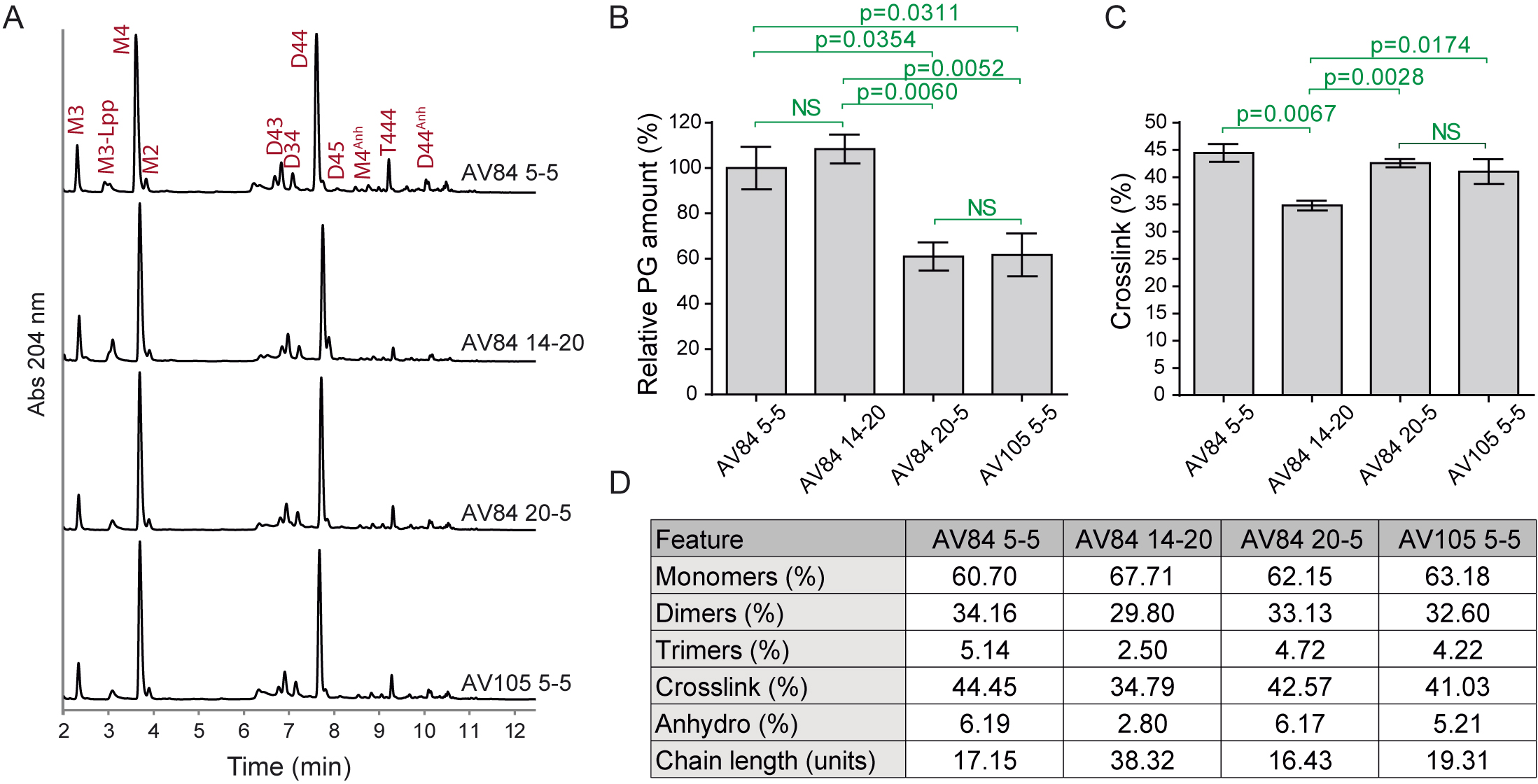
Figure 3. Peptidoglycan analysis of Escherichia coli samples with different repression levels of PBP1b and/or PBP1a. A. Representative UPLC profiles. Identified muropeptides are indicated. B. Relative PG amount (compared to AV84 5-5). C. Percentage of crosslink. Calculated as: dimers + (trimers × 2). D. Main PG features showing relative molar abundance of monomers, dimers and trimers, overall crosslink values, relative molar amount of anhydro muropeptides and estimated chain length. Statistical analysis: unpaired t-test. NS: not significant. Data represents mean ± SEM, 3 replicas. Data modified from Vigouroux et al., 2020.
Notes
- This protocol is not recommended for preparation of PG from most Gram-positive bacteria, since the existence of associated cell-wall polymers such as wall teichoic acids or lipoteichoic acids requires a more elaborated procedure for their removal (Mann et al., 2016).
- This protocol also works with lysed samples (e.g., treatment with an antibiotic such as D-cycloserine), as long as an ultracentrifugation step is implemented before sample collection for sacculi preparation:
- Spin down the samples at 4 °C and 200,000 x g for 30 min and discard the supernatant.
- Resuspend the pellet corresponding to the insoluble fraction in 1.5 ml supernatant or alternative media.
- Proceed with Step A3.
Recipes
Prepare all solutions with fresh MilliQ water. Use ultrapure water from a distillation or deionization unit with a resistance of 8 MΩ/cm at 25 °C. For the UPLC buffers, use only analytical grade reagents. Since pH is critical in most solutions used, make sure to gently stir the solutions and adjust the pH at a temperature as similar as possible to the working conditions. Solutions are stored at room temperature.
- Phosphate buffer saline (PBS)
- Dissolve 8 g NaCl, 0.2 g KCl, 1.77 g Na2HPO4·2H2O and 0.24 g KH2PO4 in 800 ml MilliQ water
- Stir until completely dissolved
- Adjust to pH 7.4 using HCl
- Adjust final volume to 1 L with MilliQ water
- Sterilize by autoclaving
- Lysis buffer [SDS 5% (w/v)]
Dissolve 5 g SDS in 100 ml MilliQ water
Note: Use protective gear since SDS powder may cause irritation of the respiratory tract. - Tris HCl 1 M pH 8.0 buffer
- Dissolve 12.11 g Tris base in 80 ml MilliQ water
- Stir until completely dissolved
- Adjust to pH 8.0 using HCl
- Adjust final volume to 100 ml with MilliQ water
- Tris HCl 100 mM pH 8.0 buffer
- Dilute 10 ml of Tris HCl 1 M pH 8 buffer in 80 ml MilliQ water
- Adjust final volume to 100 ml with MilliQ water
- SDS 10% (w/v) solution
Dissolve 10 g SDS in 100 ml MilliQ water
Note: Use protective gear since SDS powder may cause irritation of the respiratory tract. - Digestion buffer (50 mM phosphate buffer pH 4.9)
- Dissolve 887.6 mg NaH2PO4•2H2O and 9.5 mg Na2HPO4•2H2O in 80 ml MilliQ water
- Stir until completely dissolved
- Adjust pH to 4.9 with orthophosphoric acid 25% (v/v) solution to finely adjust pH
- Adjust volume to 100 ml with MilliQ water
- NaOH 2 M solution
Dissolve 8 g NaOH in 100 ml MilliQ water and stir until completely dissolved
Notes:- This reaction is highly exothermic, safety measures must be taken.
- The NaOH solution is relatively unstable and its concentration can change over time, so for better accuracy, it is better prepared fresh.
- Borate buffer (borate buffer 0.5 M pH 9.0)
- Dissolve 3.1 g boric acid in 80 ml MilliQ water and stir until completely dissolved
- Adjust pH to 9.0 with NaOH 2 M solution
- Adjust final volume to 100 ml with MilliQ water
- NaBH4 solution (NaBH4 2 M)
Dissolve 76 mg NaBH4 in 1 ml MilliQ water
Note: Do not close any tube lids, since this reducing agent reacts with the solvents producing H2 in a violent reaction. This solution must be prepared fresh, do not store. - Orthophosphoric acid 25% (v/v) solution
- Dilute 29.4 ml orthophosphoric acid 85% in 50 ml MilliQ water
- Stir until completely diluted
- Adjust final volume to 100 ml with MilliQ water
- Organic buffer A [formic acid 0.1% (v/v)]
Dilute 1 ml HPLC grade formic acid in 1 l MilliQ water and mix - Organic buffer B [formic acid 0.1% (v/v) in acetonitrile]
Dilute 1 ml HPLC grade formic acid in 1 l acetonitrile and mix
Acknowledgments
This work reports in detail the sample preparation and analysis of peptidoglycan samples from Gram-negative bacteria. This work was supported by the Knut and Alice Wallenberg Foundation, the Swedish Research Council, the Kempe Foundation and the Laboratory for Molecular Infection Medicine Sweden. A project grant from Stiftelsen Clas Groschinskys Minnesfond is also acknowledged. Work at the Institut Pasteur was supported by the European Research Council (ERC) under the Europe Union’s Horizon 2020 research and innovation program [Grant Agreement No. (679980)], the French Government’s Investissement d’Avenir program Laboratoire d’Excellence ‘Integrative Biology of Emerging Infectious Diseases’ (ANR-10-LABX-62-IBEID), the Mairie de Paris ‘Emergence(s)’ program, and the Volkswagen Foundation. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication. This protocol was derived from the E. coli PG analysis method published in our work (Vigouroux et al., 2020).
Competing interests
The authors declare no conflict of interest or competing interest.
References
- Alvarez, L., Espaillat, A., Hermoso, J. A., de Pedro, M. A. and Cava, F. (2014). Peptidoglycan remodeling by the coordinated action of multispecific enzymes. Microb Drug Resist 20(3): 190-198.
- Cava, F. and de Pedro, M. A. (2014). Peptidoglycan plasticity in bacteria: emerging variability of the murein sacculus and their associated biological functions. Curr Opin Microbiol 18: 46-53.
- Desmarais, S. M., De Pedro, M. A., Cava, F. and Huang, K. C. (2013). Peptidoglycan at its peaks: how chromatographic analyses can reveal bacterial cell wall structure and assembly. Mol Microbiol 89(1): 1-13.
- Gan, L., Chen, S. and Jensen, G. J. (2008). Molecular organization of Gram-negative peptidoglycan. Proc Natl Acad Sci U S A 105(48): 18953-18957.
- Glauner, B., Höltje, J. V. and Schwarz, U. (1988). The composition of the murein of Escherichia coli. J Biol Chem 263(21): 10088-10095.
- Egan, A. J. F., Errington, J. and Vollmer, W. (2020). Regulation of peptidoglycan synthesis and remodelling. Nat Rev Microbiol.
- Kühner, D., Stahl, M., Demircioglu, D. D. and Bertsche, U. (2014). From cells to muropeptide structures in 24 h: peptidoglycan mapping by UPLC-MS. Sci Rep 4: 7494.
- Pasquina-Lemonche, L., Burns, J., Turner, R. D., Kumar, S., Tank, R., Mullin, N., Wilson, J. S., Chakrabarti, B., Bullough, P. A., Foster, S. J. and Hobbs, J. K. (2020). The architecture of the Gram-positive bacterial cell wall. Nature 582(7811): 294-297.
- Mann, B., Loh, L. N., Gao, G. and Tuomanen, E. (2016). Preparation of Purified Gram-positive Bacterial Cell Wall and Detection in Placenta and Fetal Tissues. Bio-protocol 6(23): e2037.
- Tipper, D. J. (1968). Alkali-catalyzed elimination of D-lactic acid from muramic acid and its derivatives and the determination of muramic acid. Biochemistry 7(4): 1441-1449.
- Typas, A., Banzhaf, M., Gross, C. A. and Vollmer, W. (2011). From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat Rev Microbiol 10(2): 123-136.
- Vigouroux, A., Cordier, B., Aristov, A., Alvarez, L., Ozbaykal, G., Chaze, T., Oldewurtel, E. R., Matondo, M., Cava, F., Bikard, D. and van Teeffelen, S. (2020). Class-A penicillin binding proteins do not contribute to cell shape but repair cell-wall defects. Elife 9: e51998.
- Vollmer, W., Blanot, D. and de Pedro, M. A. (2008). Peptidoglycan structure and architecture. FEMS Microbiol Rev 32(2): 149-167.
- Yadav, A. K., Espaillat, A. and Cava, F. (2018). Bacterial Strategies to Preserve Cell Wall Integrity Against Environmental Threats. Front Microbiol 9: 2064.
Article Information
Copyright
![]() Alvarez et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Alvarez et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Alvarez, L., Cordier, B., van Teeffelen, S. and Cava, F. (2020). Analysis of Gram-negative Bacteria Peptidoglycan by Ultra-performance Liquid Chromatography. Bio-protocol 10(19): e3780. DOI: 10.21769/BioProtoc.3780.
- Vigouroux, A., Cordier, B., Aristov, A., Alvarez, L., Ozbaykal, G., Chaze, T., Oldewurtel, E. R., Matondo, M., Cava, F., Bikard, D. and van Teeffelen, S. (2020). Class-A penicillin binding proteins do not contribute to cell shape but repair cell-wall defects. Elife 9: e51998.
Category
Microbiology > Microbial biochemistry > Other compound
Microbiology > Microbial cell biology > Cell-based analysis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.