- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Workflow for Ultra-rapid Analysis of Histone Post-translational Modifications with Direct-injection Mass Spectrometry

Published: Vol 10, Iss 18, Sep 20, 2020 DOI: 10.21769/BioProtoc.3756 Views: 7043
Reviewed by: Rohit JainZijian ZhangAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Chromatin modifications, like histone post translational modifications (PTMs), are critical for tuning gene expression and many other aspects of cell phenotype. Liquid chromatography coupled to mass spectrometry (LC-MS) has become the most suitable method to analyze histones and histone PTMs in a large-scale manner. Selected histone PTMs have known functions, and their aberrant regulation is linked to a wide variety of diseases, including cancer. However, histone analysis is scarcely used in diagnostics, partially due to the limited throughput and not ideal reproducibility of LC-MS based analysis. We describe a workflow that allows for high-throughput sample preparation is less than a day using 96-well plates. Following preparation, samples are sprayed into MS without LC, using an automated direct injection (DI-MS) method. Each analysis provides accurate quantification for 29 peptide sequences with 45 PTMs (methylations, acetylations and phosphorylations) for a total of 151 histone marks plus 16 unmodified histone peptides for relative quantification of histone variants. This workflow allows for < 1 min MS runs and higher reproducibility and robustness due to the absence of carryover or LC-based batch effects. Finally, we describe an engineered peptide sequence used to accurately monitor the efficiency of sample preparation, which can be detected during the DI-MS run.
Keywords: HistoneBackground
Histones are basic proteins with a globular head and a N-terminal tail rich in arginine and lysine residues. A pair each of canonical histone H2A, H2B, H3 and H4 (called core histones) form an octomer around which 147 bp of DNA winds to form a nucleosome. The tails from these core histones are extensively modified by covalent post translational modifications (PTMs) including methylations, phosphorylations, and acyl groups including acetylations, sugar derivatives and by hundreds of other groups mostly at the lysine residues (Figure 1) (Zhao and Garcia, 2015). These histone PTMs modulate chromatin compaction by altering the charge at the DNA-histone interface and by acting as docking sites for chromatin-associated proteins.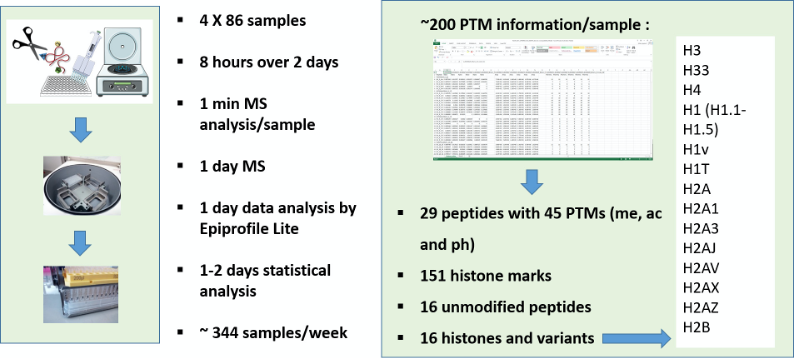
Figure 1. Merits of DI-MS workflow. Multiplexing sample preparation from extraction to derivatization/digestion to desating in a highthroughput manner. Sample preparation still requires at least one day to get histones ready for analysis. However, the workflow allows to multiplex potentially thousands of samples in a single batch, and they can then be analyzed at the rate of one per minute. Virtually, about 344 samples can be analyzed in a regular week (5-days work), when four 96-well plates are processed simultaneously. This capacity is higher than any existing diagnostics facility analyzing histone modifications. Each sample yields data with 220 relative quantifications from 29 peptides, belonging to 16 histone types, both canonical and variant, bearing single or multiple PTMs such as methylations, acetylations and phosphorylations in a total of 45 loci is obtained within a week.
Every eukaryotic cell has a tightly tuned chromatin state, whether it is pluripotent or differentiated, healthy or diseased. Actively transcribed chromatin domains are commonly referred as euchromatin, while silenced sequestered chromatin domains are defined as heterochromatin. While acetylation is generally assigned to accessible chromatin, heterochromatin is associated to more precise histone marks. For instance, methylation of lysine 9 on histone H3 (H3K9me) is the target of the heterochromatin protein 1 (HP1) and it is well known to create constitutive silenced chromatin domains (Nakayama et al., 2001). Other lysine methylations have other biological roles; H3K27 trimethylation is another heterochromatic marker, while methylation of H3K4, H3K36, or H3K79 are associated with active chromatin (Maison and Almouzni, 2004; Peterson and Laniel, 2004; Bannister et al., 2005). Increasingly, we are recognizing cross-talk between different modifications as an extra level of regulation of chromatin function (Kouzarides, 2007). In this context, the ability to differentially quantify e.g., the relative abundance on chromatin of H3K9me3 vs H3K9me3K14ac allows to define the co-existence frequency of histone PTMs.
Conventional antibody-based approaches are very limited for histone PTM analysis due to the following reasons: (i) only very few antibodies recognize multiple PTMs; (ii) only known modifications can be studied with this approach; (iii) antibody-based techniques rarely can be adopted in a large-scale manner, necessary to investigate combinatorial codes; (iv) the specificity of antibodies for histone PTMs, even if commercial, is sometimes not sufficient. Egelhofer and coworkers reported that “more than 25% of commercial antibodies failed specificity tests by dot blot or western blot; among specific antibodies, more than 20% failed in chromatin immunoprecipitation experiments” (Egelhofer et al., 2011). Mass spectrometry (MS) is currently the most suitable analytical tool to study novel and/or combinatorial PTMs, especially due to the high sensitivity, high mass accuracy and the possibility to perform large-scale analyses.
The most widely adopted proteomics workflow for histone analysis is the so-called “bottom-up” approach, where histones are digested into 4-20 amino acid long peptides. We and others have optimized a protocol that allows for the use of trypsin as digestion enzyme (cleaves after K/R) despite the large frequency of arginine (R) and lysine (K) residues on histone proteins, i.e., about 30% of all amino acids. This is then followed up with derivatizing unmodified and monomethylated lysine residues with propionylation (Sidoli et al., 2015) or heavy-labeled acetylation (Bonaldi et al., 2004) to prevent lysine cleavage by trypsin. This yields proper size peptides for best ionization in liquid chromatography coupled online with mass spectrometry (LC-MS). Nevertheless, there are some inherent problems in using LC such as batch effects resulting from column conditions, long gradients to fractionate peptides online, thereby limited sample throughput and unpredictable ionization efficiency for different peptides. While some batch effect arising from performing the sample preparation at different times (even by the same person) and the calibration status of the mass spectrometer still persists, the huge variability that HPLC contributed to MS-aided PTM analysis has been greatly reduced using this DI-MS (Sidoli et al., 2015). Good lab practices such as preparing and analyzing all samples pertaining to a study in a high throughput manner will reduce batch effects. We also recommend ensuring that samples are relatively free of ionizable contaminants as these “background” interferences lead to sample-specific signal suppression in mass spectrometry. Sample clean-up is very crucial as injecting contaminants reduce the radio frequency-guided ion transmission by the S-lens, resulting in poor data acquisition and expensive instrument maintenance costs. In all, DI-MS is straightforward to implement in a mass spectrometry lab or core facility that has the technical expertise for handling LC-MS instruments.
Once high throughput quantitative histone PTM analysis is established as we describe in this protocol, there are exciting opportunities for researchers to examine the missing epigenetic aspects of biological processes and diseases, in the comfort of their own laboratories. Moreover, about 200 pieces of information can be generated for over 300 samples/week making it especially amenable for discovery of epigenetic biomarkers for diseases.
Though DI-MS can be optimized for any analyte including metabolites (Kirwan et al., 2014) and nucleic acids (Quinn et al., 2013), we leveraged a high-throughput infusion workflow for histone PTM analysis because of our familiarity with both the advantages and challenges associated with it as well as the importance for basic science and disease phenotypes. Some properties of histones lend itself for clean extraction, ideal proteolysis and data-independent acquisition (DIA) in the mass spectrometer. The abundance of histones in the nuclear compartment in association with DNA (chromatin) and their basic nature due to arginine and lysine residues have been exploited for extraction, while the specificity of trypsin to the multiple arginine and lysine sites that are unmodified post-translationally provide right-sized (4-20 aminoacids long) peptides for bottom-up strategy. Extracting histones from cell lysates after RIPA or other common cell lysis buffers is not ideal for histones quantification; therefore, we combined cell lysis with nuclear fractionation to extract the chromatin-bound histones. This chromatin-enriched fraction represents 11% of the total cellular proteins and consists predominantly of histone proteins (H1, H2A, H2B, H3, and H4) and other minor components (Shiio et al., 2003). Though chromatin isolation can also be performed by high salt extraction (Rodriguez-Collazo et al., 2009), it is not ideal for mass spectrometry if salt clean-up is not effective. High salts notoriously interfere with derivatization steps, form salt adducts and be detrimental to electrospray ionization. Therefore, we and others use a mild detergent lysis step followed by centrifugation to isolate nuclei, reducing sample handling and thereby loss (Das et al., 2009; Bhanu et al., 2019). While extraction of histones from eukaryotic cells is straightforward, organisms with the cell wall like yeast need harsher pretreatment with homogenization, alkali, sonication, hypotonic swelling and boiling (Kizer et al., 2006). After chromatin isolation, we then extract histones in acids like H2SO4 or HCl, in which they are highly soluble due to their high isoelectric point. H2SO4 is the most used acid; however, selected histones and histone PTMs are more soluble and stable in other acids. For example, extraction of H1 is more favorable in HCl, while labile PTMs like phosphorylations, differential extraction of isoforms as well as novel PTMs may benefit from high salt extraction in neutral pH.
The protocol we present here is focused for human myoblasts and myotubes, but it is easily translatable for most cell types. We describe a propionylation procedure to derivatize lysine side chains prior trypsin digestion. We also recommend the addition of a synthetic peptide for quality control (QC) of the sample preparation, and its products can be monitored for assessing over- and under-propionylation, and trypsin digestion (Sidoli et al., 2019).
Following derivatization and digestion, the peptides are desalted off-line in an easy-to-assemble, in-house stage tip. Because of the high hydrophilicity of the peptides generated with our protocol (Sidoli et al., 2016), we use a Porous Graphitic Carbon (PGC) resin on top of a C18 plug for desalting, thereby increasing the dynamic range of detection and quantification. Desalting can be performed using 96-well plates to multiplex the sample preparation. After eluting peptides from the desalting column, the samples are ready to spray. We recommend to utilize automated direct injection (DI-MS) methods like the TriVersa NanoMate (Advion), so samples can be injected in a robotized manner. We describe in the protocol the details to prepare a dedicated acquisition method to analyze the described histone peptides, including differential quantification of isobaric forms. In addition, we describe our software EpiProfile Lite (Sidoli et al., 2019) for automated signal extraction and relative quantification of histone PTMs. Finally, we give helpful indications for correlating PTM analysis to their epigenetic significance, so that follow-up studies can be designed by the investigators.
To sum up, we outline a standard improved one-pot workflow for ultra-rapid analysis of histone modifications with direct-injection mass spectrometry. In this protocol, we simplified the most laborious steps in sample preparation and allow for efficient sample preparation with a QC peptide. We demonstrate high-throughput analysis of 344 samples by multiplexed desalting and the DI-MS acquisition that allows for < 1 min runs into the mass spectrometer. Rapid data analysis was also possible due to about 2 MB RAW files generated by DI-MS, as against 1,300 MB file sizes from nLC/MS2 analyses. This workflow allows comprehensive systems level analysis yielding quantitative information on 220 histone peptides and their PTMs for each of the 344 samples, all within 2-3 days (Figure 1), making it particularly suitable for biomarker discovery and validation of epigenetic diseases.
Materials and Reagents
- Glass pasteur pipettes (Neta Scientific, catalog number: 7095B-9 )
- Microcentrifuge tube adapter (GL Sciences, catalog number: 501021514 )
- 3M EmporeTM Solid Phase Extraction Disks C18 (CDS Analytical, catalog number: 98-0604-0218-1 )
- Hypercarb® 30-40 µm Carbon 150-300 Å (Thermo Scientific, catalog number: 60106-402 )
- Fused silica capillary 75 µm ID x 363 µm OD (Molex (Polymicro), catalog number: TSP075375 )
- 1.7 ml microcentrifuge tubes, Posi-click (Denville, catalog number: C2170 )
- 15 and 50 ml conical centrifuge tubes (Falcon, catalog numbers: 352196 , 352070 )
- TipOne tips for P1000, 200, 20 and 10 pipettes (USA Scientific)
- MS sample vial, LaPhaPack, Snap, 12 x 32 mm (LEAP PAL Parts, catalog number: LAP.11190933 )
- MS sample vial Snap Cap, PE, Natural, Pre-Slit (LEAP PAL Parts, catalog number: LPP.011-03-8303 )
- Deep Well, 96-Well Microplate, 2.0 ml (Thomas Scientific, catalog number: 89237526 )
- 96-well plate, V-Bottom 600 µl (Axygen, catalog number: P-DW-500-C-S )
- 96-well flat bottom plate (Fisher Scientific, catalog number: 12565501 )
- Sealing Mat for 96 Well PCR Microplates (Axygen, catalog number: 14-222-024 )
- Reprosil-Pur 120 C18-AQ 3 µm, 3 g (ESI Source Solutions, r13.aq.0003)
- 10x Dulbecco’s PBS without Ca2+/Mg2+ (Mediatech, catalog number: MT21031CM )
- Cell dissociation buffer (Thermo Fisher, catalog number: 13151014 )
- NP-40 Alternative (Sigma-Millipore, catalog number: 492016 )
- Dithiothreitol (DTT) (Sigma-Millipore, catalog number: 111474 )
- AEBSF (Sigma-Millipore, catalog number: 101500 )
- Microcystin (Sigma-Millipore, catalog number: 475818 )
- Sodium butyrate (Sigma-Millipore, catalog number: 817500 )
- Sulfuric acid (H2SO4) (Fisher Scientific, catalog number: A484-212 )
- Trichloroacetic acid (Fisher Scientific, catalog number: A322-100 )
- Acetone (Fisher Scientific, catalog number: A18-500 ), store at 20 °C
- Hydrochloric acid (HCl) (Fisher Scientific, catalog number: A144S-500 )
- Ammonium bicarbonate (NH4HCO3) (Fisher Scientific, catalog number: A643-500 )
- Bovine Serum Albumin (BSA) (Sigma-Millipore, catalog number: A7906-100G )
- Coomassie Brilliant Blue G-250 (Fisher Scientific, catalog number: BP100-25 )
- Acetonitrile, Optima LC/MS grade (Fisher Scientific, catalog number: A955-1 )
- Propionic anhydride (Sigma-Millipore, catalog number: 8006080100 )
- Ammonium hydroxide (Fisher Scientific, catalog number: A669S-500 )
- Modified Trypsin, sequencing grade (Fisher Scientific, catalog number: PRV5113 )
- Glacial acetic acid (Fisher Scientific, catalog number: 18-602-907 )
- Trifluoroacetic acid, Optima LC/MS grade (Fisher Scientific, catalog number: A116-05AMP )
- Formic Acid, Optima LC/MS grade (Fisher Scientific, catalog number: A117-50 )
- Water, Optima LC/MS grade (Fisher Scientific, catalog number: W6-4 )
- Methanol, Optima LC/MS grade (Fisher Scientific, catalog number: A456-4 )
- Trypsin-digested BSA MS standard (CAM-modified) (New England Biolabs, catalog number: P8108S )
- Sodium chloride (NaCl) (Fisher Scientific, catalog number: BP358-1 )
- Potassium chloride (KCl) (Fisher Scientific, catalog number: AC424090250 )
- Magnesium chloride (MgCl2) (Fisher Scientific, catalog number: AC223211000 )
- Calcium chloride (CaCl2) (Fisher Scientific, catalog number: C79-500 )
- Nuclei isolation buffer (NIB-250) (see Recipes)
- NP-40 Alternative (see Recipes)
- Protease inhibitors (see Recipes)
- Phosphatase inhibitor (see Recipes)
- HDAC inhibitor (see Recipes)
- 0.2 M (0.4 N) sulfuric acid (see Recipes)
- 100% Trichloroacetic acid (TCA) (w/v) in ddH2O (see Recipes)
- Acetone + 0.1% hydrochloric acid (see Recipes)
- 100 mM Ammonium bicarbonate (see Recipes)
Caution: Avoid direct contact with reagents and research material; always use gloves and other appropriate personal protective equipment (PPE). Use automatic pipetting to prevent accidental ingestion.
Liquid nitrogen: Training required before use. Use face shield, cryogenic gloves, cryogenic apron and closed toed shoes while dispensing.
H2SO4, TCA, HCl and acetic acid are corrosive acids. Always open stock bottles of concentrated acid in a chemical fume hood to avoid inhalation of fumes; open dilute acid in a well-ventilated workspace.
Ammonium hydroxide is a strong base and should also be used in fume hood to avoid inhaling fumes.
Acetone, acetonitrile and methanol are flammable. Use precautions for fire hazard. Exercise standard exposure precautions. Store all acetone-containing reagents in -20 °C. It is also a strong organic solvent and will leach polystyrene pipettes. Use glass pipettes for dispensing.
Propionic anhydride is an irritant and is corrosive. Degrades in contact with atmospheric air and so it is stored under argon. Higher exposures are likely when topping with argon gas. Use face shield and goggles in addition to gloves and lab coat when topping. Note: This is DEA-controlled substance that needs approvals and not be available for prompt delivery.
Compressed gas (nitrogen, argon): Training required for proper securing and safe handling. Argon spray cans can also be used, instead of 14 L tanks.
Equipment
- -80 °C freezer
- Hypersep cartridge (Thermo Scientific, catalog number: 60109-404 )
- Ceramic scoring wafer (Restek, catalog number: 20116 )
- Pipetman, P1000, 200, 20 and 10 (Rainin)
- Cold centrifuge (700 x g-18,000 x g) (NuAire, model: Nuwind , catalog number: NU-C200V )
- Argon (Airgas, catalog number: AR HP 300 )
- Nitrogen (Airgas, catalog number: NI UHP300 )
- Dounce tissue grinder & pestle (Kimble Kontes, catalog number: 885300 )
- Mass spectrometer (Thermo Scientific, model: Orbitrap Fusion )
- Nanodrop (Thermo Scientific, model: ND3300 )
- pH indicator strips, Instachek (pH 0-13), Micro Essential Lab, model: Hydrion , catalog number: JR-113 )
- Pressure Injection Cell (Next Advance, model: PC77 )
- SpeedVac+ vacuum pump and plate rotor (Savant, model: SC210A )
- Tube rotator (Thermo Scientific, catalog number: 88881001 )
- Vortex Mixer (Thermo Scientific, catalog number: 88880017 )
- Triversa Nanomate (Advion, model: TR263 )
Software
- EpiProfile Lite (Sidoli et al., 2019). Available in GitHub at https://github.com/zfyuan/EpiProfileLite
Procedure
- Cell culture and harvest
A 10 cm2 plate containing about 10 million cells yields approximately 100 μg histones from human myoblast cell line, LHCN-M2 as well as from most cell lines. This is more than sufficient for DI-MS analysis, considering that few μgs of material at 1 μg/μl is injected. However, depending on the cell state (e.g., proliferation vs differentiation) and treatment, variable amounts of histones may be obtained. For example, we found that the yield of histones is more for a plate of myotubes compared to myoblasts, as myotubes are multi-nucleate cells, carrying a larger amount of chromatin. Lower histone yields may result from DNA gelling during nuclei isolation step and this can be avoided by following precautions as below (i) detach cells using cell dissociation buffer rather than trypsin-EDTA (ii) use fresh pellet instead of ones snap-frozen in liquid nitrogen.- Collect dissociated cultured cells (> 5 x 106 cells/ml) with 10 ml phosphate buffered saline-free of Ca2+ and Mg2+ (PBS) in a 15 ml tube.
- Pellet cells by spinning for 5 min at 300 x g. Discard supernatant.
- Wash traces of dissociation buffer/culture media with 10 ml PBS for 5 min at 300 x g. Discard supernatant.
- If tissue or biopsies are used, dice them using a clean razor blade to < 1 mm piece and collect in 5 ml pf PBS; repeat Step A3.
- Estimate the packed cell pellet volume and mark with permanent ink at the bottom of the tube.
- Flash-freeze cells in liquid nitrogen at this stage and store in -80 °C until histone extraction.
- Isolation of cellular nuclei (Time needed: up to 1 h)
Batch-to batch variations from sample preparation are very minimal when a single person processes 344 samples simultaneously. Most users experience lesser yields due to loss of histones from the chromatin during nuclear isolation. This loss can be greatly reduced by carefully optimizing the lysis step, as we describe below.- Prepare buffers as follows:
- NIB wash buffer: Take 50 volumes of NIB for every volume of packed cell pellet (100 μl cell pellet: 5 ml NIB). Add 5 μl of 1 M DTT, 10 μg each of 2.5 μM microcystin and 5 M sodium butyrate, as well as 12.5 μl of 200 mM AEBSF. Henceforth, NIB means NIB + phosphatase and HDAC inhibitors. This buffer is for washes.
- NIB lysis buffer: Take about 1/4th of NIB wash buffer prepared as above into a new tube and add 0.1% (v/v) of NP-40 Alternative. This buffer is for lysis.
- NIB wash buffer: Take 50 volumes of NIB for every volume of packed cell pellet (100 μl cell pellet: 5 ml NIB). Add 5 μl of 1 M DTT, 10 μg each of 2.5 μM microcystin and 5 M sodium butyrate, as well as 12.5 μl of 200 mM AEBSF. Henceforth, NIB means NIB + phosphatase and HDAC inhibitors. This buffer is for washes.
- Thaw frozen cells on ice.
Note: Group samples based on cell pellet sizes and transfer to same rows or columns, so that similar volumes can be dispensed using multichannel pipettes. - Gently resuspend cells in 10 volumes of NIB wash buffer on ice. Transfer to 96-well plate.
Note: Use plate with well capacity of 200 μl for pellet size up to 20 μl. For larger pellets such as 60 μl-200 μl, use 600 μl-2 ml deep-well plates respectively. - Wash cells by centrifugation at 700 x g for 5 min at 4 °C.
- Tilt the plate to an angle and remove supernatant from the sides carefully using a multichannel pipette (do not use vacuum aspirator as the pellet is loose at this stage and will be lost).
- Mark the packed cell volumes in wells that are easy for visual inspection such as column A and row H using a permanent marker.
- Add 10 volumes of NIB lysis buffer from a reagent reservoir and mix gently by pipetting the cells up and down to break clumps.
- Incubate on ice for 5 min.
Note: Use 96-well ice block placed on ice for 200 μl wells to ensure uniform temperature conduction. - Centrifuge cells at 700 x g for 5 min at 4 °C.
- Check pellet size. This should be less than half of the initial packed cell volume (shown with marker at the bottom of the tube). If not, resuspend the contents of selected wells gently and follow one of the two options:
- increase incubation time in increments of 5 mins or
- increase concentration of NP-40 Alternative in increments of 0.05% followed by centrifugation
- Repeat Steps B8-B10 until optimal pellet size is obtained.
Caution: Over lysis will result in loss of histones from the nuclei. - increase incubation time in increments of 5 mins or
- Remove NIB lysis buffer after centrifugation once pellet size reduces, as in Step B5.
- Wash traces of NP-40 Alternative in at least 8 volumes of NIB relative to initial pellet size.
Note: Use fresh/dedicated (if reusing) polypropylene reagent reservoir for each reagent. - Spin at 700 x g/5 min/4 °C and remove supernatant as in Step B5.
- Repeat Steps B12-B13, two or more times until no bubbles from the detergent are seen in the buffer.
- Save pellet (chromatin with histones) after pipetting off supernatant. Do not use vacuum aspirator.
- If not used immediately, lid the plate with sealing mat and snap-freeze in liquid nitrogen to store wrapped in cling wrap (indefinitely) at -80 °C.
- Prepare buffers as follows:
- Acid extraction of histones
- To the nuclei, add 5 volumes (based on the original washed cell pellet size) of cold 0.4 N H2SO4 and resuspend the pellet by gentle trituration.
- Incubate the sample with gentle shaking for 2 h at 4 °C.
Note: Five volumes of acid is added to one volume of initial cell pellet. This ratio is good for up to 200 μl pellet size. Incubation for 2 h is critical as longer time in acid will result in extraction of non-histone chromatin proteins. - Mix once in between (after an hour of start of incubation) using a multichannel pipette.
- Centrifuge at 3,400 x g/5 min/4 °C and gently transfer supernatant without disturbing pellet (pellet contains non-histone proteins, DNA, RNA etc.) to a new 96-well plate. This acid-extract now contains histones.
- To the supernatant, add cold 100% TCA such that the final concentration is 30%. The sample will appear cloudy indicating the presence of proteins, in this case histones. Mix quickly and gently by pipetting several times.
- Secure the mat to the plate and leave the protein to precipitate on ice undisturbed, from few hours to up to overnight.
- Centrifuge at 3,400 x g/5 min/4 °C. Histones would be seen as a whitish coating on the wall, with sediment at the bottom of the tube.
- Remove the mat carefully and place a thick wad of tough-task tissue paper, to completely cover all the wells that have samples.
- Invert the plate, by quick flick of the wrist so that TCA supernatant is blotted to the tissue paper.
- Repeat Step C9 to remove any leftover TCA.
- Using a glass Pasteur pipette, aliquot cold acetone + 0.1% HCl (premixed and stored in -20 °C) to a fresh reagent reservoir.
- Using a multichannel pipette, add the acidified acetone up to the level of the whitish coating on the wall (histones) to wash off impurities.
- Repeat Steps C7-C10.
- Add cold 100% acetone (stored in -20 °C) up to the level of histones and scrape the walls to collect histones to the bottom of the well.
Note: Acetone degrades plastic. Use glass Pasteur pipette to dispense to reservoir. Use new reservoir for each time. For transferring to samples, use plastic pipette tips but be as quick as you can and use fresh pipette tips for each dispensation. - Spin at the highest speed possible for 5 min at 4 °C.
- Remove supernatant by gently turning the plate upside down on tissue paper.
- Repeat the acetone rinse so that no trace of detergent or acid remains (Steps C14-C16).
- Airdry pellet to complete dryness on benchtop, from 30 min to up to overnight.
- Resuspend the dried pellet with about 30 μl of MS-grade water, pipetting vigorously. Histones are very easily soluble in water and the insoluble material is mostly non-histone proteins.
- Quickly spin at high speed to sediment the insoluble non-histone protein.
- Carefully transfer the purified histones in the clear supernatant to a fresh 96-well plate.
- Check pH of rows or columns using < 1 μl sample picked up by just touching with tip, to avoid loss.
- Adjust to about pH 8.0 with 100 mM ammonium bicarbonate.
- Estimate protein concentration by Bradford assay, using a plate reader at 595 nm.
Note: BCA assay is not suitable for histone estimation as histones have very few cysteines and barely any tryptophan and tyrosine. - Dry histones on the plate using speedvac.
- Resuspend histones in MS-grade water such that concentration is 1 μg/μl.
- Transfer 20 μg to a fresh 96-well plate (200 μl capacity) for further processing.
- Seal with mat and store at -80 °C until further use. Alternatively, the histones can be dried completely in a speedvac concentrator and stored on bench indefinitely.
- Protein estimation by Bradford assay
Bradford assay measures the protein-Coomassie dye complex formation in acidic conditions at 595 nm. In fact, basic aminoacids such as arginine, lysine and histidine of the protein form this complex. SDS and detergents strongly interferes with the formation of this complex and thereby compromise protein quantification. To avoid this, always perform the multiple NIB washes and if needed, acetone washes. As TCA precipitation also eliminates NP-40 Alternative to an extent, do not skip this step.- Prepare up to 8 dilutions of BSA standards for a range of 0.1-10 mg/ml with MilliQ water.
- Equilibrate Bradford reagent and samples to room temperature.
- Make dilutions of the stock reagents in MilliQ water.
- Make dilutions of the BSA standard, as instructed by the manufacturer.
- Aliquot diluted Bradford to a 96-well flat bottom plate (Fisher Scientific).
- Add standard dilutions and samples each to the diluted Bradford reagent.
- Mix without making bubbles.
- Follow the manufacturer’s instruction for assay.
- Follow instructions for operation of spectrophotometer and read absorbance at 595 nm.
- Estimate protein concentration based on the BSA standard curve.
- Chemical derivatization of histones
Tagging a chemical group such as propionyl to ε-amino groups of unmodified/monomethylated lysine served two purposes in the conventional “bottom-up” histone analysis coupling chromatography with mass spectrometry: a) mimicking ArgC digestion despite using trypsin as proteolytic enzyme and b) increase hydrophobicity of the small histone peptides improving their retention by reversed-phase chromatography. In DI-MS, there is no need to increase hydrophobicity of histone peptides, and therefore the derivatization of peptide N-termini after trypsin digestion is not used in this protocol.
To assist quality control (QC) of the sample preparation, we engineered a synthetic peptide with no homology to any known endogenous histone peptide for quality control. This peptide with sequence, GVKFRGSTGGKAPRGKAPATSGMVGPHR mimics histones in having arginine and lysine in the same frequency and acts like a histone peptide during propionylation and trypsin digestion. Products and byproducts of derivatization and digestion of this peptide can be detected and quantified during the DI-MS acquisition to assess the quality of the sample preparation as listed in table (Table 1):
Table 1. Synthetic peptide with sequence engineered for quality control. The peptide sequence, GVKFRGSTGGKAPRGKAPATSGMVGPHR can be propionylated and digested by trypsin, generating fragments of different lengths with characteristic m/z for various problems in the sample processing listed. Column named “Relative ratios” is an example of an ideal profile where the sum from the first three peptides indicating “Complete derivatization and digestion” should be over 0.95 (or 95%); this run passes process QC.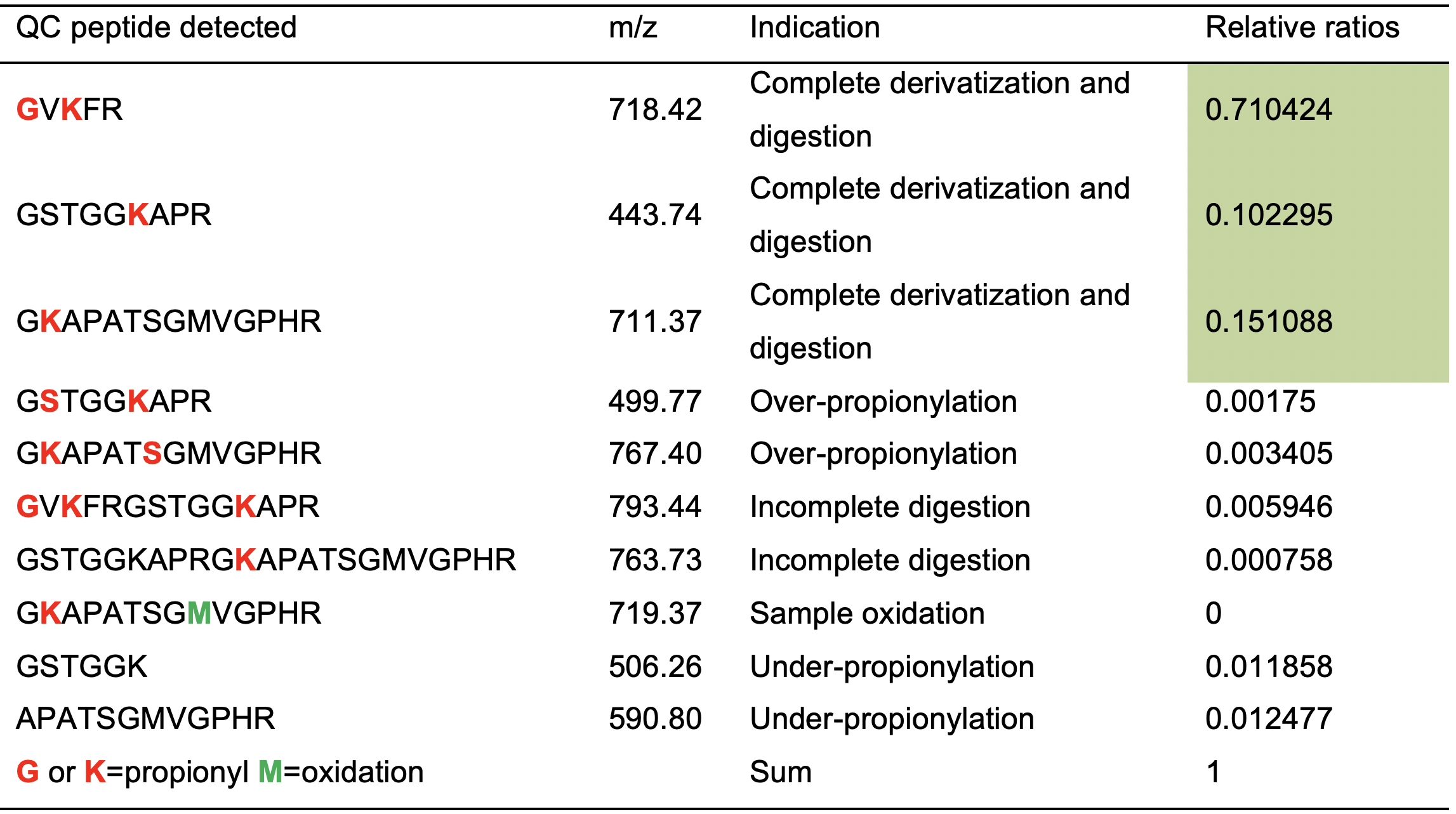
In a fume hood, set up the reagents (ammonium hydroxide, acetonitrile and propionic anhydride), multichannel pipettes, P200 and P10 tip boxes (as many as needed), dedicated/new reagent reservoirs and pH paper roll (Figure 2A).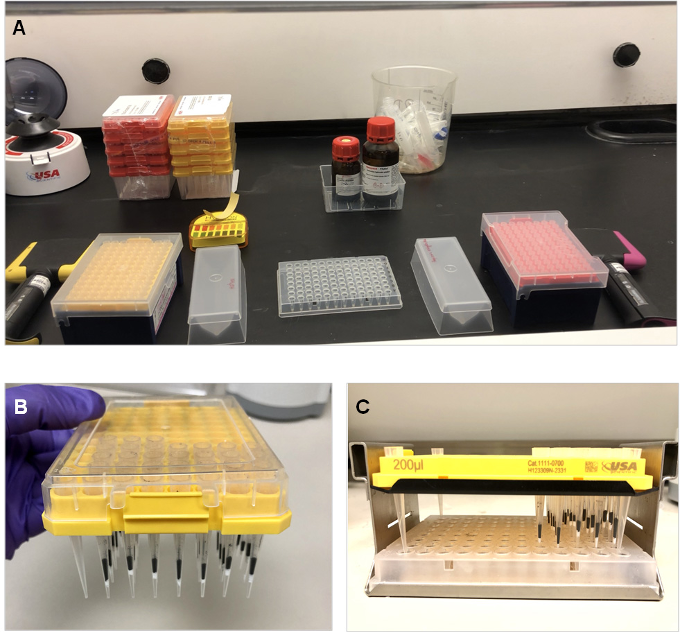
Figure 2. Histone derivatization and desalting. A. Set up of supplies in chemical hood for rapid performance of derivatization. B. Prepare desalting stage-tips with C18 (white plug) and PGC (black particles packed as a slurry in acetonitrile using a multichannel pipette) housed in a pipette tip wafer. Note that the first row (row A) is not usually used as we normally reserve these to check BSA standard and desalting buffers during MS analysis. This stage-tip wafer can be prepared ahead of time and stored on the bench with a lid in an empty tip box. C. Positioning the stage-tip wafer over wells in a 96-well plate. Note that the four corners are tethered by tips, if all the 84 wells are not used. Desalt wafer is positioned in the wells of the microplate and is placed in a speedvac plate holder for elution.- Use about 10 μg of purified histones resuspended in MS-grade water, in a plate. Ensure pH 8.0 by adjusting with 100 mM ammonium hydroxide.
- Thaw one vial of QC peptide stored as 1 μg/μl aliquots in -20 °C.
Note: Use completely after dilution. Do not refreeze-thaw. - Make 1:10 dilution of 1 μg/μl QC peptide in MS-grade water and add 0.1 μg (1 μl) to 10 μg of histones/well in the plate. Mix well.
- Take MS-grade acetonitrile in a dedicated reagent reservoir and using a multichannel pipette, add 5 μl to sample well and mix contents.
- Move to the chemical hood where reagents, reservoirs, pipettes, tip box, pH paper strip etc. are arranged on either side of the plate so there is minimum crossing of hands while dispensing (Figure 2A).
Caution: From this step onward, all steps have to be performed in a chemical hood since propionic acid is an irritant and ammonium hydroxide fumes are an inhalation hazard. When dispensing either of these reagents, avoid pipettes and tips so as not to contaminate stocks; instead decant reagents to reservoir. Direct infusion MS is very sensitive to contaminants such as detergents and polymers as there is no LC in to filter them off; these contaminants, if built-up in stocks will be carried over to samples and will suppress sample-specific peptide signals. - Perform derivatization Steps E7-E10 as demonstrated (Video 1).
 Video 1. Rapid derivatization by a single user
Video 1. Rapid derivatization by a single user - Add 5 μl propionic anhydride to the first row/column using the multi-channel pipette in one hand and mix by pipetting.
Caution: Work swiftly so that propionic anhydride is fresh and active when added to samples. In the presence of atmospheric moisture, this reagent immediately forms acetic acid after propionylating the free amines in the unmodified and monomethylated lysines of the histones. - To neutralize the acidity, quickly follow up with 12-17 μl of ammonium hydroxide using multichannel pipette in the other hand (might need to be adjusted depending on the potency of the ammonium hydroxide and the sample type) (Video 1).
Note: Alternatively, two persons can work in tandem to accomplish these steps rapidly. - Repeat Steps E5 and E8 for each row/column.
- Check pH of a couple of random rows/columns to verify that the pH is around 8.0. If not, add ammonium hydroxide in few μl increments and mix well by pipetting.
Note: Check pH using < 1 μl sample picked up by just touching the sample with tip, to avoid sample loss. - Incubate in the fume hood for 15 min for the reaction to complete.
- Repeat Steps E4-E9 to ensure propionyl amidation of all free amines on unmodified lysine.
Caution: Store propionic anhydride under argon (refer to notes under Buffer and reagent preparation and storage: Compressed gas) so that it doesn’t produce acetic acid with atmospheric air and lose activity. - Dry the sample using a speedvac centrifuge adapted for plates. This might easily take > 1 h.
- Add trypsin to sample at a 1:20 ratio (0.5 μg trypsin to 10 μg histones) in 20-30 μl of 100 mM ammonium bicarbonate.
- Adjust to pH 8.0 using ammonium hydroxide.
- Incubate on bench from 6 h to overnight.
- Store at -80 °C or follow through subsequent steps.
- Desalting propionylated peptides using C18/PGC stage-tip
There is accumulation of salts with each step of propionylation and in the histone extract as well. Salts ionize easily compared to peptides and appear as adducts, with higher signal intensities compared to samples, leading to sample signal suppression. Prior to MS analysis, the samples should be desalted and buffers removed by passing through C18/PGC. This matrix also retains the hydrophobic peptides, enabling elution in a weakly acidified organic solvent, thereby allowing pure peptides to be obtained for MS analysis.- Prepare stage tip at room temperature on the bench as demonstrated (Video 2). Video 2. Fashioning in-house desalting columns
- Cut about 0.5 cm from the end of a P1000 tip, using a razor blade, such that the bore is about 2 mm in diameter.
- Using this cut tip, punch a small disk of C18.
- Using a piece of fused silica, plug a P200 pipette tip with the C18 punch.
- Arrange these plugged tips in an empty 8 x 12 format P200 pipette tip refill wafer.
- Remove porous graphite carbon (PGC) particles out of the Hypersep cartridge (Thermo Scientific), by displacing the plug using forceps, to a dedicated reagent reservoir.
- Make a slurry of PGC in 100% acetonitrile and add to the plugged P200 tip such that about 0.5 cm PGC material settles above the C18 plug, after acetonitrile drains. This is called the stage tip (Figure 2B).
Note: The slurry tends to sediment and so pipette hard to mix before adding to C18 tips. Acetonitrile prewets the C18/PGC and activates them by exposing polar groups, so that the peptides bind to C18/PGC. - Spin the stage-tip assembly at low speed in a speedvac without vacuum to remove 100% acetonitrile.
Notes:- All spins in speedvac to remove buffers (Steps F8-F21) should be carried out in the speedvac without vacuum.
- The stage-tips or desalting columns can be prepared ahead of time and stored in an empty tip box. We have not tested reusing these stage tip, as there might be carryover of samples.
- All spins in speedvac to remove buffers (Steps F8-F21) should be carried out in the speedvac without vacuum.
- Wash the residual acetonitrile from the stage-tip with about 70 μl of 0.1% trifluoroacetic acid (TFA, wash solution), taking care to add it in the middle of tip so that it seeps through the PGC evenly.
- Spin gently at lowest speed for 30 s to let the wash solution drip down.
- Repeat Steps F8-F9.
- Add 100 μl of 0.1% TFA to derivatized peptide. This will acidify the sample to about pH 4.0.
- Now gently add the acidified, derivatized peptide sample over the washed stage tip in the center.
- Spin at low speed for 30 s. to let liquid flow-through.
- Wash the bound peptides on the C18/PGC material with 100 μl of 0.1% TFA.
- Spin at low speed for 30 s.
- Repeat Steps F15-F16.
- Place the stage-tip wafer assembly over a fresh 96-well plate for elution. Make sure the tips are aligned to the wells correctly.
- Take care to tether the four corners of the wafer and plate with tips (Figure 2C).
- Add 30 μl of 60% acetonitrile/0.1% formic acid gently into the center of the stage-tip.
- Spin gently at low speed for 30 s
- Dry eluted samples using a speedvac concentrator with vacuum turned on.
Note: Do not turn on heat in the speedvac at any time as peptides may aggregate. - Resuspend in 13 μl water and perform BCA peptide assay at 480 nm to ensure total peptide concentration of at least 10 μg/sample.
- Dry the plate in speedvac.
- Cover the microplate with mat to store it in -80 °C.
- Just before injecting in mass spectrometer using nanomate, resuspend samples in a volume of 60% acetonitrile/0.1% formic acid that gives the final concentration of sample as 1 μg/μl for optimal spraying.
- Prepare stage tip at room temperature on the bench as demonstrated (Video 2).
- Nanoelectrospray mass spectrometric analysis
Samples are analyzed in the NanoMate Triversa instrument using a microfluidics electrospray ionization (ESI) chip and Chipsoft software. A conductive pipette tip (emitter size 5 μm ID) picks sample from the 96 well-plate and the sample-filled tip aligns with a nozzle inlet on the back of the disposable ESI chip, creating a tight seal and a unique path into the mass spectrometer. As pipette tip and nozzle are used once per sample, there is no sample carryover. This part of the robotic operation is programmed into Nanomate Triversa using Chipsoft. As the sample resuspended in formic acid gets sprayed through a charged tip, the peptides get protonated and enters a hot (170 °C) ion transfer tube, evaporating on its exit to the gas phase where it is analyzed by the mass spectrometer. We describe an acquisition method for the Orbitrap Fusion Tribrid (Thermo Scientific), but other mass spectrometers can also be utilized. We recommend to acquire the signals using a hybrid mass spectrometer with a selective analyzer (like the quadrupole or the ion trap) and a high resolution analyzer (like the time-of-flight or the orbitrap). The selective analyzer is needed to perform the acquisition of the peptide signals using a scan named targeted selected ion monitoring (tSIM) to enhance the sensitivity of the scan that compensates for the lack of liquid chromatography. In tSIM, quantification is performed on the MS1 data. High resolution is required to discriminate nearly isobaric modifications such as acetylation (42.010 Da) vs trimethylation (42.047 Da). MS/MS fragmentation should be performed for quantifying the peptides that have isobaric forms. All the scans should be performed by sending the ions into the high resolution mass analyzer.
Below, we describe the method designed for the Orbitrap Fusion Tribrid (Thermo Scientific) with Instrument Control Software 3.0. These global settings can be used as a start point to generate methods. We recommend optimizing parameters according to the instruments used (Supplementary Table S1 and Supplementary Table S2). Supplementary Table S3 lists m/z signals to acquire in targeted mode for the quantification of (un)modified histone peptides.
While some batch effect arising from performing the sample preparation at different times (even by the same person) and the calibration status of the mass spectrometer still persists, the huge variability that HPLC contributed to MS-aided PTM analysis has been greatly reduced using this DI-MS (Sidoli et al., 2019). We recommend preparing and analyzing all samples pertaining to a study in a high throughput manner. We also recommend ensuring that samples are relatively free of ionizable contaminants as these “background” interferences lead to sample-specific signal suppression in mass spectrometry. Furthermore, injecting contaminants reduce the radio frequency-guided ion transmission by the S-lens, resulting in poor data acquisition and expensive instrument maintenance costs.
Check if the Xcalibur raw file size is about 2MB (megabyte) for the parameters we use. This file size indicates relatively dense spectrum, with 2 sets of MS1 and one MS2.- Visualize the total ion chromatogram and base peak in “chromatogram view” of RAW file in Thermo Xcalibur Qual Browser (Figure 3).
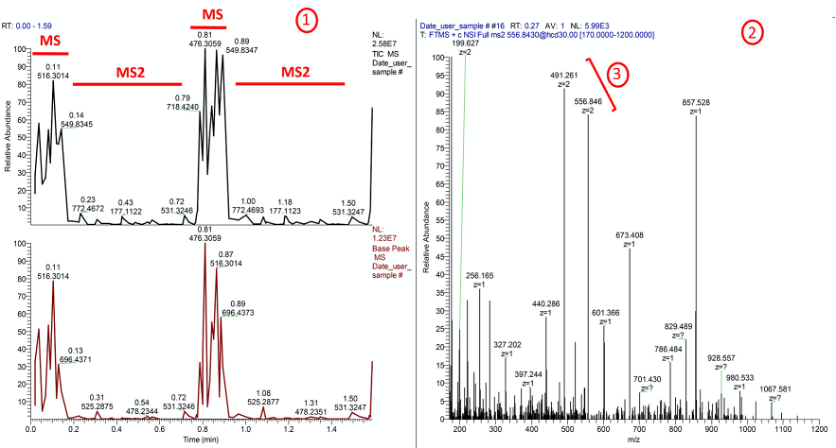
Figure 3. Checking quality of DI-MS run by inspecting mass spectra in Xcalibur Qual Browser 3.1. A good MS analysis showing summed intensities of all ions, both from background noise as well as sample components in the scanned mass range (top left), base peak of the mass spectrum above (bottom left) and a representative MS2 spectrum (right). In orange, #1 indicates a good run QC where a pair each of MS and MS2 have been acquired in less than 2 min. Though the method is for 2 min, as soon as two MS-MS2 events have been recorded, the nanomate stops acquisition and the leftover sample is returned to the well. In order to get this good quality spectrum, a minimum of 10 μl of peptides at 1 mg/ml concentration is needed. #2 indicates densely ionized sample without polymer adducts, #3 shows the +2 charged abundant ions. - Scan the fragmentation ion abundance and presence of +2 ions in the “Spectrum view” on the right.
- Visualize the total ion chromatogram and base peak in “chromatogram view” of RAW file in Thermo Xcalibur Qual Browser (Figure 3).
- Data Analysis for Histone PTMs
The high-throughput quantitative histone PTM analysis we describe in this protocol will allow researchers to examine the missing epigenetic aspects of biological processes and diseases, in the comfort of their own laboratories. About 220 relative quantifications from 29 peptides bearing PTMs such as methylations, acetylations and phosphorylations in a total of 45 loci. Roughly, 38 peptides had monomethylations, 23 peptides had dimethylations, 21 had trimethylations, 91 had acetylations and 28 phosphorylations. These marks occurred in all combinations, either singly or as multiple marks at different loci of the same peptide. These 29 peptides belong to 16 histone types as shown in Figure 1. Thus, a rich mine of PTM information can be generated for over 300 samples/week making it especially amenable for discovery of epigenetic biomarkers for diseases. Spectra can be directly uploaded into EpiProfile Lite or analyzed manually by extracting the signal intensity of a given peptide from the spectrum where this was acquired. EpiProfile Lite extracts the signals of the peptides presented in Supplementary Table S2, including the differential quantification of isobaric forms.
EpiProfile Lite provides a result table (called “histone ratios”) with approximately 150-200 peptide isoforms depending on the species. Because the list can be overwhelming for manual inspection, it is very helpful to process the data using the proper statistics, in order to detect the most significant and reliable changes. When using a limited number of replicates (< 5), we recommend the use of t-test to estimate the significant differences between conditions. Non-parametric statistics is generally more appreciated, as it can be applied also to replicates that do not have a Gaussian distribution; however, it is not sufficiently powerful to deal with such small number of data points. In case an overall trend of acetylation increase/decrease is observed, we recommend to correlate the observation with the levels of acetyl-CoA before jumping to conclusions like “we observe an overall higher activity of enzymes catalyzing histone acetylation”.- Open the “histone ratios” table containing the raw intensities of the identified and quantified histone peptides.
- Normalize each modified histone peptide by the sum intensity of all peptides sharing the same sequence (Figure 4). For instance, the peptide containing H3K9me2 has the sequence KSTGGKAPR in the H3 9_17 peptide in human (and most other eukaryotes). The relative abundance of the peptide modified as H3K9me2 is calculated as: Intensity of K9me2STGGKAPR/Sum of intensities of (KSTGGKAPR + K9me1STGGKAPR + K9me2STGGKAPR + K9me3STGGKAPR + K9acSTGGKAPR + KSTGGK14acAPR + K9me1STGGK14ac APR + K9me2STGGK14ac APR + K9me3STGGK14ac APR + K9acSTGGK14acAPR). The sum of relative abundance of all of these PTM forms will be 1.
Note: We strongly recommend to perform the extracted ion chromatography of histone peptides using EpiProfile Lite, as this calculation is already performed, and the software automatically deals with isobaric peptides.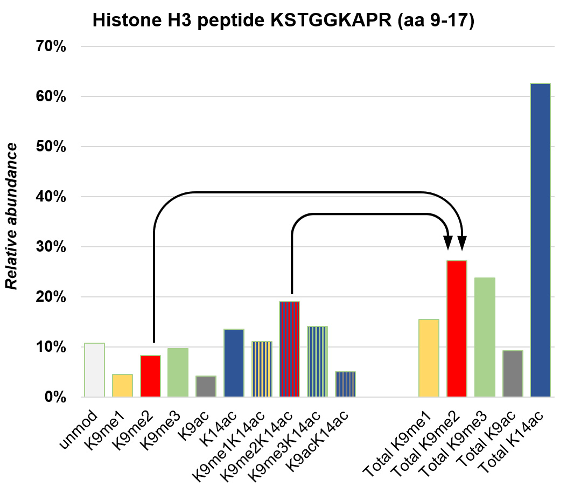
Figure 4. Difference between relative ratios and single PTM abundance, illustrated for H3 9-17 peptide and H3K9me2 respectively. The single PTM abundance of H3K9me2 is calculated as: sum of relative ratios of (H3K9me2 + H3K9me2K14ac). - Perform the t-test when comparing two conditions, or ANOVA when comparing more than two conditions. As the range of relative ratios is in several logs, we use log values for both fold change and P-values.
- To be stringent, we recommend significance based on both fold change and P-value, displaying data as a volcano plot, using on the x-axis, the log2 fold change between two conditions (such as treated vs untreated) and on the y-axis, the -log2 of the t-test P-value. Conventionally, the significance threshold is set at a P-value < 0.05, which when transformed as -log2 becomes > 4.32. For fold change significance, any threshold can be set, such as log2 of 1 being two-fold change (Figure 5A).
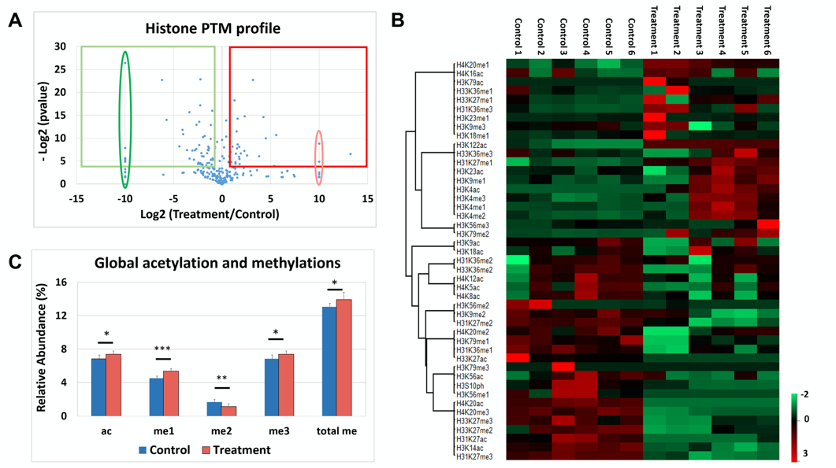
Figure 5. Visualization of histone PTM analysis data. A. Volcano plot provides group information by plotting log fold change between two conditions [x-axis, log2(Treatment/Control)] as a function of their significance [y-axis, -log2(0.05)]. The elongated ovals represent imputed Zscore values for expression only in treatment (red) or in control (green) conditions. Threshold for significance is 4.32 [= log2(0.05)] and that for fold change is 1 (= log2 of 2). The red square represents the histone marks upregulated in the treatment and the green square encompasses histone PTMs upregulated in the control. B. Post-analysis representation of “histone ratios_single PTMs” table from EpiProfile Lite. Heat map was constructed in Perseus 1.5.3.2 version using Zscore normalized values; this heat map provides sample expression levels, along with co-trending marks shown by hierarchial clustering, with samples in the columns and histone marks in rows. Hierarchial clustering was based on histone marks with samples left unclustered. The samples here display expression ranging from -2 (downregulation, green) to 3 (upregulation, red). A definite pattern of regulation (expression from red to green and vice versa) between control and treated samples is seen in two blocks of cotrending marks. C. Abundance of global acetylations (ac) and methylations (valencies me1, me2 and me3 and total me) can be represented as graph. Horizontal bar over conditions denotes significant changes by t-test: *P = 0.04, **P = 0.01 and ***P = 0.0004. - Copy the “histone ratios” table to a fresh sheet. Add up the relative abundance of all acetylated peptides, and compare them with the relative changes of acetyl-CoA. If the correlation is linear and positive, a possible biological interpretation of the data is that the acetyl-CoA levels change affecting the abundance of histone acetylation. An example of this analysis is illustrated previously (Simithy et al., 2017).
- Once identified, the PTMs that are significantly regulated between the analyzed conditions, a potential next step can be determining which histone writer is potentially responsible for this regulation. This enzyme then is a potential target for complementary treatment using either inhibitors or other post-transcriptional regulation (e.g., knock-down).
- Correlate histone marks to writers to understand the biological relevance of changes observed in your data set. Supplementary Table S4 summarizes some of the known epigenetic players that can be investigated in mechanistic studies.
- The normalized intensities are also called relative ratios as they indicate the ratios of each modified form within the peptide. This is also called the relative ratio table for combinatorial marks, to distinguish it from single PTMs, a table that summarizes the occurrence of a PTM within a peptide, regardless of being single or in combination with another mark. For example, the single PTM abundance of H3K9acK14ac is calculated as: sum of intensities of (H3K9ac + H3K14ac + H3K9acK14ac + H3K9me1K14ac + H3K9me2K14ac + H3K9me3K14ac). Another example of single PTM abundance for H3K9me2 is illustrated in Figure 4.
- In EpiProfile Lite, single PTM ratios for H3 and H4 are computed and available as a “single PTM” table. This can be normalized as z-score and heat map made using Perseus (Figure 5B) (Bhanu et al., 2019).
- Global acetylations and methylations of different conditions can also be compared (Figure 5C).
- Refer to Supplementary Table S4 for relevance of histone PTMs to epigenetic mechanisms.
- Follow-up the MS-aided PTM analysis with assays for writers and erasers, functional studies, genome-wide enrichment studies using ChIP-Seq or gene-specific enrichments using ChIP-qPCR (Bhanu et al., 2019).
The DI-MS method we describe for a 96-well format can easily be adapted to a 384-or 1356-well formats with Triversa Nanomate ESI chips for even higher throughput needed in life science applications. Besides the histone PTM analysis described here, DI-MS can be performed for any analyte by optimizing protocols tailored to the physicochemical properties of the sample. While the method is robust compared to LC-MS, there is still room to increase sensitivity. To achieve high signal-to-noise ratio, improvements for cleaner analyte extraction and desalting procedures are desirable. Supercharging agents such as m-nitro benzyl alcohol (m-NBA), dimethyl sulfoxide (DMSO) and tetramethylene sulfone (sulfolane) enhance signal abundance and are worth investigating. One of the problems we encounter with direct injection is spray stability even with the use of ESI chip. In general, electrospray ionization is susceptible to charge competition from interferences, while other modes such as atmospheric pressure chemical ionization (APCI) are not. Histones may lend favorably to APCI as they are thermally stable and are positively charged. With hardware changes by Advion, APCI may also be explored for direct infusion to improve MS sensitivity. - Open the “histone ratios” table containing the raw intensities of the identified and quantified histone peptides.
Recipes
- Nuclei isolation buffer (NIB-250)
15 mM Tris-HCl (pH 7.5)
15 mM NaCl
60 mM KCl
5 mM MgCl2
1 mM CaCl2
250 mM sucrose
Make a stock and store in -20 °C as aliquots - NP-40 Alternative
10% (v/v) in ddH2O
Store at room temperature - Protease inhibitors (add fresh to buffers prior to use)
1 M dithiothreitol (DTT) in ddH2O (1,000x)
200 mM AEBSF in ddH2O (400x)
Stocks stored as aliquots in -20 °C - Phosphatase inhibitor (add fresh to buffers prior to use)
2.5 μM microcystin in 100% ethanol (500x)
Stocks stored as aliquots in -20 °C - HDAC inhibitor (add fresh to buffers prior to use)
5 M sodium butyrate, adjust with NaOH to pH 7.0 (500x)
Stocks stored as aliquots in -20 °C - 0.2 M (0.4 N) sulfuric acid (H2SO4) (v/v)
In a fume hood, add 5.54 ml of concentrated H2SO4 to 1 L of ddH2O
Store at 4 °C - 100% Trichloroacetic acid (TCA) (w/v) in ddH2O
Add 227 ml of ddH2O to 500 g of TCA. This clear solution is approximately 1.45 g/ml
Store at 4 °C - Acetone + 0.1% hydrochloric acid (HCl)
0.1% (v/v) HCl in acetone
Store in amber bottle at -20 °C - 100 mM Ammonium bicarbonate (NH4HCO3) (w/v) pH 8.0
Dissolve 79 mg NH4HCO3 salt in 8 ml ddH2O
Adjust pH to 8.0 using formic acid
Bring up the volume to 10 ml
Make this buffer in small volume and store tightly capped in fridge
Check pH before use as pH shifts over time
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants NIH AI118891, CA196539, St. Jude Collaborative A11576 and GM110104. We also acknowledge the original research paper by Sidoli et al. (2019) from which this protocol is derived.
Competing interests
The authors declare no competing interests.
References
- Bannister, A. J., Schneider, R., Myers, F. A., Thorne, A. W., Crane-Robinson, C. and Kouzarides, T. (2005). Spatial distribution of di-and tri-methyl lysine 36 of histone H3 at active genes. J Biol Chem 280(18): 17732-17736.
- Bhanu, N. V., Sidoli, S., Yuan, Z. F., Molden, R. C. and Garcia, B. A. (2019). Regulation of proline-directed kinases and the trans-histone code H3K9me3/H4K20me3 during human myogenesis. J Biol Chem 294(20): 8296-8308.
- Bonaldi, T., Imhof, A. and Regula, J. T. (2004). A combination of different mass spectroscopic techniques for the analysis of dynamic changes of histone modifications. Proteomics 4(5): 1382-1396.
- Das, C., Lucia, M. S., Hansen, K. C. and Tyler, J. K. (2009). CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 459(7243): 113.
- Egelhofer, T. A., Minoda, A., Klugman, S., Lee, K., Kolasinska-Zwierz, P., Alekseyenko, A. A., Cheung, M. S., Day, D. S., Gadel, S., Gorchakov, A. A., Gu, T., Kharchenko, P. V., Kuan, S., Latorre, I., Linder-Basso, D., Luu, Y., Ngo, Q., Perry, M., Rechtsteiner, A., Riddle, N. C., Schwartz, Y. B., Shanower, G. A., Vielle, A., Ahringer, J., Elgin, S. C., Kuroda, M. I., Pirrotta, V., Ren, B., Strome, S., Park, P. J., Karpen, G. H., Hawkins, R. D. and Lieb, J. D. (2011). An assessment of histone-modification antibody quality. Nat Struct Mol Biol 18(1): 91-93.
- Kirwan, J. A., Weber, R. J., Broadhurst, D. I. and Viant, M. R. (2014). Direct infusion mass spectrometry metabolomics dataset: a benchmark for data processing and quality control. Sci Data 1: 140012.
- Kizer, K. O., Xiao, T. and Strahl, B. D. (2006). Accelerated nuclei preparation and methods for analysis of histone modifications in yeast. Methods 40(4): 296-302.
- Kouzarides, T. (2007). Chromatin modifications and their function. Cell 128(4): 693-705.
- Maison, C. and Almouzni, G. (2004). HP1 and the dynamics of heterochromatin maintenance. Nat Rev Mol Cell Biol 5(4): 296-304.
- Nakayama, J. I., Rice, J. C., Strahl, B. D., Allis, C. D. and Grewal, S. I. (2001). Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292(5514): 110-113.
- Peterson, C. L. and Laniel, M. A. (2004). Histones and histone modifications. Curr Biol 14(14): R546-551.
- Quinn, R., Basanta-Sanchez, M., Rose, R. E. and Fabris, D. (2013). Direct infusion analysis of nucleotide mixtures of very similar or identical elemental composition. J Mass Spectrom 48(6): 703-712.
- Rodriguez-Collazo, P., Leuba, S. H. and Zlatanova, J. (2009). Robust methods for purification of histones from cultured mammalian cells with the preservation of their native modifications. Nucleic Acids Res 37(11): e81-e81.
- Shiio, Y., Eisenman, R. N., Eugene, C. Y., Donohoe, S., Goodlett, D. R. and Aebersold, R. (2003). Quantitative proteomic analysis of chromatin-associated factors. J Am Soc Mass Spectrom 14(7): 696-703.
- Sidoli, S., Bhanu, N. V., Karch, K. R., Wang, X. and Garcia, B. A. (2016). Complete workflow for analysis of histone post-translational modifications using bottom-up mass spectrometry: from histone extraction to data analysis. J Vis Exp (111): e54112.
- Sidoli, S., Kori, Y., Lopes, M., Yuan, Z.-F., Kim, H. J., Kulej, K., Janssen, K. A., Agosto, L. M., da Cunha, J. P. C. and Andrews, A. J. (2019). One minute analysis of 200 histone posttranslational modifications by direct injection mass spectrometry. Genome Res 29(6): 978-987.
- Sidoli, S., Yuan, Z. F., Lin, S., Karch, K., Wang, X., Bhanu, N., Arnaudo, A. M., Britton, L. M., Cao, X. J. and Gonzales-Cope, M. (2015). Drawbacks in the use of unconventional hydrophobic anhydrides for histone derivatization in bottom-up proteomics PTM analysis. Proteomics 15(9): 1459-1469.
- Simithy, J., Sidoli, S., Yuan, Z. F., Coradin, M., Bhanu, N. V., Marchione, D. M., Klein, B. J., Bazilevsky, G. A., McCullough, C. E., Magin, R. S., Kutateladze, T. G., Snyder, N. W., Marmorstein, R. and Garcia, B. A. (2017). Characterization of histone acylations links chromatin modifications with metabolism. Nat Commun 8(1): 1141.
- Zhao, Y. and Garcia, B. A. (2015). Comprehensive catalog of currently documented histone modifications. Cold Spring Harb Perspect Biol 7(9): a025064.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Bhanu, N. V., Sidoli, S. and Garcia, B. A. (2020). A Workflow for Ultra-rapid Analysis of Histone Post-translational Modifications with Direct-injection Mass Spectrometry. Bio-protocol 10(18): e3756. DOI: 10.21769/BioProtoc.3756.
- Bhanu, N. V., Sidoli, S., Yuan, Z. F., Molden, R. C. and Garcia, B. A. (2019). Regulation of proline-directed kinases and the trans-histone code H3K9me3/H4K20me3 during human myogenesis. J Biol Chem 294(20): 8296-8308.
Category
Biochemistry > Protein > Posttranslational modification
Molecular Biology > Protein > Detection
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.