- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Probe-Seq: Method for RNA Sequencing of Specific Cell Types from Animal Tissue

Published: Vol 10, Iss 18, Sep 20, 2020 DOI: 10.21769/BioProtoc.3749 Views: 7576
Reviewed by: Gal HaimovichKarthik KrishnamurthyYi Zhang
Original research article
The authors used this protocol in:

Dec 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

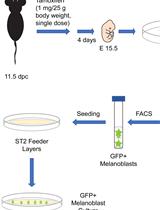
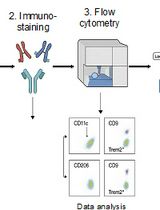
Abstract
Most organs and tissues are composed of many types of cells. To characterize cellular state, various transcription profiling approaches are currently available, including whole-tissue bulk RNA sequencing, single cell RNA sequencing (scRNA-Seq), and cell type-specific RNA sequencing. What is missing in this repertoire is a simple, versatile method for bulk transcriptional profiling of cell types for which cell type-specific genetic markers or antibodies are not readily available. We therefore developed Probe-Seq, which uses hybridization of gene-specific probes to RNA markers for isolation of specific types of cells, to enable downstream FACS isolation and bulk RNA sequencing. We show that this method can enable isolation and profiling of specific cell types from mouse retina, frozen human retina, Drosophila midgut, and developing chick retina, suggesting that it is likely useful for most organisms.
Keywords: RNA-sequencingBackground
Transcriptional profiling using RNA-Seq and microarrays has become ubiquitous in biological research in the past two decades. Profiling is now one of the main tools used to understand cells and cellular states across most organisms. It is applied to studies of normal development, abnormal development and disease, and has greatly expanded our understanding of evolutionary relationships. In particular, scRNA-Seq has led to the identification of novel cell types at an unprecedented rate (Picelli et al., 2013; Jaitin et al., 2014; Klein et al., 2015; Macosko et al., 2015). To allow a deeper appreciation of such newly described cell types, a method that allows their isolation, without need for a transgenic marker or specific antigen, would be of great benefit. Although scRNA-Seq can be used to gain deeper coverage of all cell types, this approach entails a great deal of sequencing of abundant cell types to gain even modest coverage of less abundant cell types.
To complement single cell modalities, we developed Probe-Seq, which enables transcriptional profiling of potentially any cell type from any organism, leveraging the newly-available RNA markers defined by scRNA-Seq efforts. Probe-Seq uses gene-specific probe sets designed for SABER Fluorescent in situ Hybridization (FISH) (Kishi et al., 2019) to fluorescently label specific RNA markers in dissociated, fixed cells or nuclei for downstream FACS isolation and RNA sequencing. While FISH has been previously used for FACS and RNA sequencing of cell cultured in vitro (Klemm et al., 2014), Probe-Seq works with cells taken directly from tissue, as we show for the mouse retina, human frozen retina, Drosophila midgut, and developing chick retina. To date, it has worked with all organisms and tissues tested, suggesting that it may be useful for any organism (Amamoto et al., 2019). This method is also cost and time effective (Figure 1). Libraries ready for sequencing can be made by Probe-Seq in less than three days with less than 6 hours of hands-on time. Moreoever, the whole procedure does not require any specialized equipment, and most of the reagents can be found in a general molecular biology lab.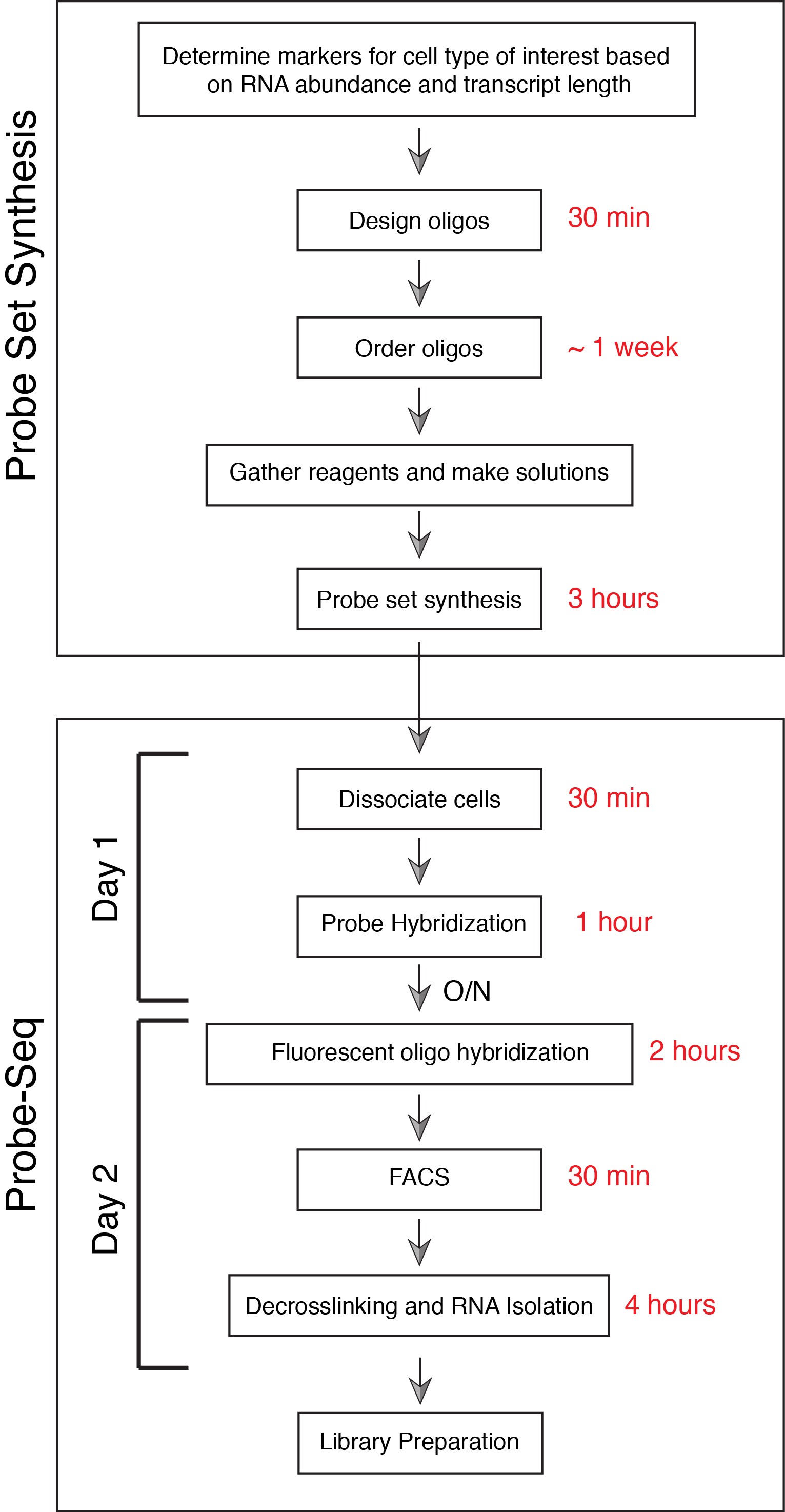
Figure 1. Flow Chart for Probe-Seq protocol
Materials and Reagents
Reagents for Probe Synthesis
- 10x PBS (Thermo Fisher Scientific, catalog number: AM9624 ). Store at RT
- 100 mM MgSO4 (NEB, catalog number: B1003S ). Store at -20 °C
- dNTP (A, C, T 6 mM each) (NEB, catalog number: N0446S ). Store at -20 °C. Do not mix dGTP
- Clean.G (1 μM), store at -20 °C
Clean.G is ordered through IDT with the following sequence (standard desalting, diluted with UltraPure water): CCCCGAAAGTGGCCTCGGGCCTTTTGGCCCGAGGCCACTTTCG - Bst DNA Polymerase (McLab, catalog number:BPL-300). Store at -20 °C
- IDTE pH 7.5 (IDT, catalog number: 11-05-01-05 ). Store at RT
- Sterile reagent reservoir (i.e., MilliporeSigma, catalog number: CLS4870 ). Store at RT
- MinElute PCR Purification Kit (Qiagen, catalog number: 28004 ). Store columns at 4 °C, everything else at RT. Other purification kits have been tested, but this kit has provided the most reliable results
Reagents for Tissue Dissociation (optional)
- HBSS with calcium/magnesium (Thermo Fisher Scientific, catalog number: 14025092 ). Store at RT
- FBS (Thermo Fisher Scientific, catalog number: 10437028 ). Store aliquots at -20 °C. Thawed aliquots can be stored at 4 °C for up to 2 weeks
- PBS pH 7.4 without calcium/magnesium (Thermo Fisher Scientific, catalog number: 10010023 ). Store at RT
- DMEM (Thermo Fisher Scientific, catalog number: 11995065 ). Store at 4 °C
- BSA (MilliporeSigma, catalog number: A9418 ). Store at 4 °C
- 1 M HEPES pH 7.0 (Thermo Fisher Scientific, catalog number: 15630080 ). Store at RT
- 50 mM L-Cysteine (MilliporeSigma, catalog number: 168149 )
L-Cysteine is stored at RT. The solution is prepared fresh before experiment. Dissolve 15 mg of L-Cysteine in 2.5 ml of UltraPure water. - 0.5 M EDTA pH 8.0 (Thermo Fisher Scientific, catalog number: AM9260G ). Store at RT
- Papain, Suspension (Worthington, catalog number: 00 3124 ). Store at 4 °C
- 5-ml Polypropylene Tube (Thermo Fisher Scientific, catalog number: 14-959-11A ). Store at RT
- HBSS/FBS (see Recipes)
- DMEM/BSA (see Recipes)
- Papain Mix (see Recipes)
Reagents for Probe Hybridization
- UltraPure Water (Thermo Fisher Scientific, catalog number: 10977015 ). Store at RT
- 16% Paraformaldehyde Ampule (Thermo Fisher Scientific, catalog number: 28908 ). Store at RT
Prepare 4% PFA by diluting in PBS before each experiment. Mix whole ampule (10 ml) with 30 ml of PBS. Dilute fresh 4% PFA before experiment and discard after use. - Hoechst 33342 (10 mg/ml) (Thermo Fisher Scientific, catalog number: H3570 ). Store at 4 °C
- 10% Triton X-100 (MilliporeSigma, catalog number: T8787 ). Store at RT
100% Triton X-100 is diluted in UltraPure water, and dissolved overnight on a shaker at RT. - 20x SSC (Thermo Fisher Scientific, catalog number: 15557044 ). Store at RT
- Deionized formamide (MilliporeSigma, catalog number: S4117 ). Aliquot and store at -20 °C. Do not thaw and refreeze
- Dextran sulfate sodium salt (MilliporeSigma, catalog number: D8906 ). Store powder at 4 °C
To make a 50% solution, dissolve 5 g of powdered dextran sulfate in 8 ml of UltraPure water overnight, then bring total volume to 10 ml. Store 50% dextran sulfate at -20 °C - RNasin Plus (Promega, catalog number: N2615 ). Store at -20 °C
- 5 ml round-bottom polysterene test tube with 35 µm cell strainer cap (Corning, catalog number: 352235 ). Store at RT
Depending on the tissue and cell type of interest, other strainer sizes are available (i.e., 70 µm). - Oligo probes and hairpin (see section on “Oligo and hairpin ordering”)
- Fluorescent oligos (see section on “Probe Hybridization Recipes”)
- Permeabilization Buffer (see Recipes)
- FACS Collection Solution (see Recipes)
- 40% wHyb (see Recipes)
- Hyb1 (see Recipes)
- Probe Mix (see Recipes)
- Fluorescent Oligo Mix (see Recipes)
Reagents for Library Preparation
- RecoverAll Total Nucleic Acid Isolation Kit for FFPE (Thermo Fisher Scientific, catalog number: AM1975 ). Store components as indicated in the kit user manual
- DNA LoBind microcentrifuge tube (Eppendorf, catalog number: Z666548 ). Store at RT
- SMART-Seq v.4 Ultra Low Input RNA Kit (Takara Bio, catalog number: 634889 ). Store components as indicated in the kit user manual
- Nextera XT DNA Library Preparation Kit (Illumina, catalog number: FC-131-1024 ). Store at -20 °C
- Nextera XT Index Kit (Illlumina, catalog number: FC-131-1001 ). Store at -20 °C
- Ampure XP Beads (Beckman Coulter, catalog number: A63880 ). Store at 4 °C Necessary for cDNA purification during RNA-Seq library preparation
- High Sensitivity DNA Analysis Kit (Agilent, catalog number: 5067-4626 ). Store at 4 °C
Reagents for Nuclei Isolation (optional)
- Sucrose (MilliporeSigma, catalog number: S9378 ). Store at RT
- KCl (MilliporeSigma, catalog number: P9541 ). Store at RT
- MgCl2 (MilliporeSigma, catalog number: M8266 ). Store at RT
- 1 M Tris Buffer pH 8.0 (Thermo Fisher Scientific, catalog number: AM9855G ). Store at RT
- Protease Inhibitor Cocktail (50x) (Promega, catalog number: G6521 ) Reconstitute in DMSO. Store at 4 °C It will be frozen at 4 °C
- DTT (Millipore Sigma, catalog number: 10197777001 ). Store at RT
- Isopentane (Millipore Sigma, catalog number: 32361 ). Store at RT
- Buffer 1 (see Recipes)
- Homogenization Buffer (see Recipes)
- Sucrose Buffer (see Recipes)
- Sucrose Bed (see Recipes)
Equipment
- P200 multichannel pipette
- P1000 pipette
- Hybridization Oven (Bellco, catalog number: 7930-01230 )
Any other hybridization oven that can be set at 37 °C and 43 °C will likely work. Make sure to measure the temperature with a thermometer before starting. If a hybridization oven is not available, a water bath should work, although it has not been tested. Ideally, 2 ovens should be used. - Centrifuge (Eppendorf, model: 5804 )
- Centrifuge rotor (Eppendorf, model: A-4-44 )
Any other centrifuge/rotor that can hold 5-ml polypropylene tubes should suffice. Ideally, a swinging bucket rotor so that the pellet settles at the bottom of the tube. - FACS Aria with 405 nm, 488 nm, 561 nm, and 633 nm lasers (BD Biosciences)
Other FACS machines can be used. We have also tested MoFlo Astrios EQ with success. The important parameter to take note of is the availability of lasers. If using a 633 nm laser, make sure to use fluorescent oligos with 633 nm excitation fluorophore. - BioAnalyzer 2100 (Agilent)
- Thermal cycler (Bio-Rad, model: C1000 ). Any other thermal cycler should suffice
- Dounce homogenizer (Thomas Scientific, catalog number: 1234F37 ). Only for nuclei isolation
Software
- OligoMiner (Brian Beliveau, GitHub, https://github.com/beliveau-lab/OligoMiner)
- UCSC Genome Browser (UCSC, https://genome.ucsc.edu)
- Brackets (Adobe, https://brackets.io)
Procedure
Gene-Specific Probe Set Synthesis
Note: For optimal isolation of cell types of interest using Probe-Seq, the marker used is the most important parameter. Based on other methods such as single cell RNA sequencing, it should be verified that the marker is expressed highly and specifically in the cell type of interest. Then, check the length of the transcript. Ideally, the transcript is at least 2 kb to ensure that enough oligo sequences are generated in the BED file. It is possible, however, that shorter transcript (i.e., 1 kb) will produce enough oligo sequences (> 20 oligos).
Oligo Design
Adapted from the original protocols on Primer Exchange Reaction and SABER (Kishi et al., 2018 and 2019).
Notes:
- Brian Beliveau has previously published Genome-wide probe sets for C. elegans, D. melanogaster, H. sapiens, A. thaliana, M. musculus, and S. danios. If you would like to use other species, please refer to OligoMiner (Beliveau et al., 2018) to generate your genome-wide probe set.
- A user-friendly web-version of probe design software called PaintSHOP has been recently created (Hershberg et al., 2020). The website can be accessed here: https://oligo.shinyapps.io/paintshop/.
The manual probe design protocol described below uses genome-wide probe sets with a length window of 35-41 nucleotides while the new Balanced probe sets for PaintSHOP has a length window of 30-37 nucleotides. Therefore, more oligos are expected using the PaintSHOP software. - Another probe design software is available (Passaro et al., 2020). The website can be accessed here: http://oligominerapp.org/home.
However, probe design for Probe-Seq did not use these websitse because they were not available at the time. Therefore, we have not tested the probes generated by these software.
Before you start:
•Get access to your institution’s computing cluster.
•Create a folder on the cluster (i.e., “SABER_Probe_Design”).
•Download Brian Beliveau’s OligoMiner-master folder from Github: https://github.com/brianbeliveau/OligoMiner.
•Transfer the OligoMiner-master folder in the SABER_Probe_Design folder on the cluster. For example, use the rsync command in the Terminal (Mac) to transfer files between the cluster and the local computer (example shown below).
•Download the Genome-wide probe set for your favorite organism from Ting Wu’s OligoPaint website: https://oligopaints.hms.harvard.edu/genome-files.
•Download the “Complete Genome” with the “Balance” setting. Unzip the file and transfer the folder in the SABER_Probe_Design folder on the cluster.
Obtaining the gene-specific BED file (30 min)
- Go to the UCSC Genome Browser: https://genome.ucsc.edu/.
- Click on Genomes and go to the genome browser of your organism (i.e., GRCm38/mm10 for mouse).
- Enter the gene of your choice in the search box (i.e., Grik1), click on the correct mRNA transcript in the drop-down menu [i.e., Grik1 (Mus musculus glutamate receptor, ionotropic, kainate 1 (Grik1), transcript variant 1, mRNA (from RefSeq NM_146072)], and hit Enter.
- With the correct mRNA in the viewer, hover the cursor over Tools, and click on Table Browser in the drop-down menu.
- Fill in as follows:
Group: Genes and Gene Predictions
Track: NCBI RefSeq
Table: UCSC RefSeq (refGene)
Region: position
Output format: BED
File type returned: plain text - Name the output file (i.e., Grik1_Mm).
- Click on Get Output.
- Click on “Exons plus 0 bases at each end”.
Note: If the target RNA is short and does not produce sufficient number of oligos, including introns may help. This is especially true if using nuclei as most of the RNA will be nascent. - Click “get BED”. This will download your BED file.
- Open the BED file. We use a software called Brackets, which is free to download at: http://brackets.io/.
- If there are multiple isoforms listed (i.e., multiple NM_# with different #), choose one of the isoforms to keep and manually delete the others. Save the file.
- Note the 2 features in the BED file as you will need them later:
a.The chromosome number.
b.Strand orientation “+” or “-“. - Transfer the BED file to the SABER_Probe_Design folder on the cluster [i.e., type “rsync Grik1_Mm user@o2.hms.harvard.edu:/home/user/SABER_Probe_Design” in the Terminal (Mac)].
- Log into the cluster.
Note: How you log into your cluster will be different institution to institution. - Run an interactive mode (i.e., “srun --pty -p interactive --mem 500M -t 0-06:00 /bin/bash”).
- Load these modules:
a.conda2 (i.e., type “module load conda2”).
b.bedtools (i.e., type “module load bedtools/2.27.1”).
c.Any other dependencies (i.e., gcc).
Note: The versions will be different for every cluster. - Run intersectBed between the genome-wide probe set (using the correct chromosome BED file) and your BED file with the option of–f 1. (i.e., Type in “intersectBed –a mm10b/mm10_chr21b.bed –b Grik1_Mm –f 1 > Grik1_Mm+_probes.bed”)
Notes:
a.Make sure you’re running this command in the folder with the genome-wide probe set (i.e., mm10b) and your gene-specific BED file (i.e., Grik1_Mm).
b.If you have enough computing power, it’s also possible to run this command locally. - If your gene-specific BED file had a “+” orientation, you need to reverse complement. Use the probeRC.py file from the OligoMiner-master folder (i.e., Type in “python OligoMiner-master/probeRC.py –f Grik1_Mm_probes.bed –o Grik1_Mm-_probes”)
- Transfer the finalized gene-specific probe set file to the local computer (i.e., using the Terminal, be in a folder on the local computer, and type “rsync user@o2.hms.harvard.edu:/home/ra108/SABER_Probe_Design/Grik1_Mm-_probes.bed.”
- Open the gene-specific probe set file (i.e., using Brackets) and see how many oligos you have.
Note: Sufficient number of oligos is crucial for successful Probe-Seq. We have used as few as 12 oligos for high expressing transcripts and saw a nice separation by FACS. However, in general, more is better. For low expressing transcripts, or if fewer than < 20 oligos, Probe-Seq may not work well.Oligo and hairpin ordering (10 min + wait time for synthesis and delivery)
Note: The oligos without the primer sequence contain the sequences for hybridization to the RNA, but not the hairpin concatemers that are hybridized by fluorescent oligos. Specific hairpin primer sequences are appended to the oligo sequences to attach hairpin concatemers during the probe set synthesis step (see below). Therefore, at this step, determine which hairpin will be used for which gene. If using two genes for one sort, two different hairpin primer sequences should be added because fluorescent oligos will hybridize to the hairpin region. For example, in order to sort two populations using Vsx2 and Grik1, for Vsx2, add the 25.25 hairpin primer sequence (“CCAATAATA”) to each oligo sequence and extend Vsx2 oligos with the 25 hairpin during the probe set synthesis step, while for Grik1, add the 27.27 hairpin primer sequence (“CATCATCAT”) to each oligo sequence and extend Grik1 oligos with the 27 hairpin during the probe set synthesis step. Then, hybridize 25.488 nm fluorescent oligo and 27.565 nm fluorescent oligo to detect Vsx2 and Grik1, respectively. - For ordering the oligos, we use IDT. Oligos from other companies have not been tested.
- Go to www.idtdna.com and under “Products & Services” click on “Custom DNA oligos.”
- Under “DNA oligos,” click on “Order Now” and “All ordering options.”
- Under “Ordering,” click on “Plates” and order 25 nmole DNA Plate oligo in a 96 well format.
Note: For most experiments, 25 nmole permits thousands of Probe-Seq experiments, which is likely excessive. If possible, order less DNA (i.e., 10 nmole) in order to decrease cost. - Click on “Upload Plates” and download the sample ordering template.
- Open the template with Excel.
- Copy and paste the contents of the gene-specific probe set file into a region of the Excel template that is not being used.
- Copy the oligo sequences into the column under “Sequences.” Delete the other pasted information.
- For each oligo sequence, add “TTT” and a primer sequence. From our experience, the hairpin primer sequences that have worked well are:
25.25 CCAATAATA
27.27 CATCATCAT
28.28 CAACTTAAC
36.36 AACTAATCT
Note: Other hairpins have been used by our group and others. However, these are the hairpins that have worked most consistently. For other hairpins, please see the Oligonucleotide_List file. - For each line, the sequence should read “(oligo sequence)TTT(primer sequence)”.
Note: You can find examples of sequences in the Oligonucleotide_List file. - Name each line with the name of the gene, hairpin used, and a number (i.e., Grik1_25_1, Grik1_25_2...).
- Many genes can fit on one plate, but it’s advisable that a new gene starts in a new row every time (i.e., If Grik1 oligos use 30 wells, and thus 3 rows: 2 full rows + 6 wells, then the Grm6 oligos should start on the 4th row).
- Upload the Excel file on IDT.
- Under Plate Specification, use the following:
Scale: 10 nmole (or 25 nmole)
Purification: Standard Desalting
Plate Type: V-Bottom
Ship Option: Wet
Buffer: IDTE 7.5 pH
Normalization Type: Normalized Yield
Quantity: 5 nmol
Concentration: 100 µM
Volume: 50 µl - Order plate.
- When the plate arrives, thaw the oligos, and use a P200 multichannel pipette to pipette the oligos into a sterile reservoir (i.e., MilliporeSigma) to pool the oligos. The pooled oligos can be stored at -20 °C.
- Make a working stock of the pooled oligos (10 µM) by diluting the 100 µM stock with IDTE pH 7.5 (IDT).
Notes:
a.Do not use other TE buffers.
b.The working stock can undergo freeze-thaw multiple times. - Order hairpins as written in the Oligonucleotide_List or Supplementary Files of the SABER (Kishi et al., 2019) or Probe-Seq (Amamoto et al., 2019) paper. They can be ordered dry and resuspended in IDTE pH 7.5 as 100 µM stock solution. Working stock (5 µM) can be made using IDTE pH 7.5 and aliquoted for further use.
Note: The 25 hairpin requires a InvdT at the 3’ end and HPLC purification.
Probe set synthesis (2-3 h)
- Add the following regents in a PCR tube on ice:
UltraPure Water 44.5 µl
10x PBS 10 µl
100mM MgSO4 (NEB) 10 µl
dNTP(A, C, T 6 mM each, NEB) 5 µl
Clean.G (1 µM) 10 µl
BST enzyme (McLab) 0.5 µl
Hairpin (5 µM) 10 µl
Notes:- 6 mM A, C, T dNTP is made by mixing dATP, dCTP, and dTTP to a final concentration of 6 mM.
- Clean.G is ordered through IDT with the following sequence (standard desalting, diluted with UltraPure water): CCCCGAAAGTGGCCTCGGGCCTTTTGGCCCGAGGCCACTTTCG.
- In a thermal cycler, incubate for 15 min at 37 °C.
- Add 10 µl of the oligo pool (10 µM) and mix.
- Incubate for 100 min at 37 °C, incubate for 20 min at 80 °C, incubate indefinitely at 4 °C. You can store this unpurified reaction at -20 °C.
- Check the length of the extended probe set by taking 8 µl of the unpurified probe set and mix it with 1.6 µl of 6x Loading Dye (or any other Loading Dye should suffice). Run a 1.25% ethidium bromide agarose gel (2-3 µl of EtBr for 50 ml volume) at 150 V for 8 min. The band should be between 300-700 bp. See Figure 2 for an example gel.
Notes:- Longer running time will lead to fainter band. Two bands are expected–the top band is your extended probe and the bottom band contains catalytic hairpins and the Clean.G oligo. Even after purification, this bottom band remains and may even help stabilize the long ssDNA probes during storage.
- If the probe set is too short or too long, you can change the extension time from 100 min to 80 min (if too long) or 120 min (it too short). The hairpin amount can also be changed. For example, if you used 10 µl of hairpin and your probe runs at ~250 bp, you can try doubling the hairpin to 20 µl (and decrease the amount of water in the reaction) to theoretically double the length to 500 bp.

Figure 2. Example DNA gel of three gene-specific probe sets. The left lane is the DNA ladder with the 500 bp band indicated in red. For each lane, the bottom band is the unextended hairpin and other small DNA pieces. The top band is the extended probe set. The bands are always fuzzy because the probes are ssDNA. - Purify the remaining 92 µl of probe set using a MinElute PCR Purification Kit (Qiagen). We use 7x PB buffer (instead of 5x) to increase binding efficiency.
- Elute in 25 µl of UltraPure water.
- Determine the concentration by NanoDrop using the ssDNA setting. The concentration should be ~200-400 ng/µl.
- Store purified probe set at -20 °C. Freeze-thaw is tolerated.
Probe-Seq: Day 1
Notes:
- If Probe-Seq is not successful for any reason, please check the Troubleshooting Guide at the end of the document.
- If you are interested in using nuclei from frozen tissue, please refer to the Appendix.
Tissue Dissociation (30 min)
Notes:
Dissociation may differ for different tissues. This dissociation protocol is for the adult mouse retina.
- If starting with already dissociated cells, start with Probe Hybridization (Step 12).
- Prepare all solutions.
Note: Be sure that RNasin has been added to all solutions. Papain mix should be moved to 37 °C to warm for ~15 min. HBSS/FBS and DMEM/BSA should be at room temperature. 4% PFA and Permeablization Buffer should be placed on ice. 40% wHyb (warm 500 µl per sample) and Probe Mix should be warmed to 43 °C. - Dissect retinas in HBSS or PBS at RT.
- Place retina in a microcentrifuge tube and remove most of the PBS/HBSS without disturbing the retina tissue.
- Add 400 µl of the pre-warmed (37 °C) Papain Mix.
- Incubate for 7 min at 37 °C with no agitation.
- Spin 600 x g (rcf) for 2.5 min at RT.
- Remove supernatant and add 1 ml of HBSS/FBS without agitation.
- Spin 600 x g for 2.5 min at RT.
- Remove supernatant and add 600 µl of DMEM/BSA.
- Dissociate by triturating the tissue with a P1000 pipette. ~15 times in/out pipetting should be sufficient to dissociate into single cells. The solution should turn homogenous with no visible clumps.
Note: Avoid bubbles. If large chunks of debris or clumped cells are present at this stage, carefully remove them using a pipette or forceps. - Transfer homogenate to a 5 ml polypropylene tube (Thermo Fisher Scientific). Make sure to seal the cap by pushing in.
Probe Hybridization (~1 h hands on time + overnight incubation)
- Spin 600 x g for 5 min at 4 °C.
Note: If a temperature-controlled centrifuge is not available, RT spin is also acceptable. However, there may be slightly more RNA degradation. - Remove supernatant and add 1 ml of 4% PFA. Resuspend. Incubate for 15 min at 4 °C with rocking.
Notes:- To minimize cell loss, ~50-100 µl of liquid can be left at the bottom after each spin step.
- For tissues with a high amount of debris (i.e., mouse or human brain), an extra filtering with a FACS cell strainer tube can be done after the 15 min incubation. This may improve the staining index.
- Spin 2,000 x g for 5 min at 4 °C.
Note: The centrifugation speed has changed to 2,000 x g after fixation. - Remove supernatant and add 1 ml of Permeabilization Buffer. Resuspend. Incubate for 10 min at 4 °C with rocking.
- Spin 2,000 x g for 5 min at 4 °C.
- Remove supernatant and add 500 µl of pre-warmed (43 °C) 40% wHyb. Resuspend. Incubate for at least 30 min at 43 °C in the oven.
Note: If an in situ hybridization oven is not available (i.e., Bellco), a water bath should suffice although it has not been tested in our lab. - Spin 2,000 x g for 5 min at RT.
Notes:- When changing liquids, it is critical to perform every step quickly to minimize change in temperature. Ideally, the oven and the centrifuge are close together.
- It is crucial that the Probe Mix and the sample are at 43 °C when it is added to the sample. If the Probe Mix is added at a lower temperature, significant off-target binding of probes can occur causing unwanted background. This is also important for subsequent addition of wHyb solution for washes.
- From here, resuspension is not necessary until FACS. Resuspension will cause cells to get stuck in the pipette tip.
- Remove supernatant and add 100 µl of pre-warmed (43 °C) Probe Mix. Incubate for at least 16 h at 43 °C in the oven.
Probe-Seq: Day 2
Fluorescent oligo hybridization (~2 h)
Note: If RNA sequencing is not going to be performed for your samples and the RNA quality is not important after FACS, you can perform a longer incubation. Increasing the incubation time to 2 overnight incubations (~48 h) has increased the staining index. However, the RNA quality after such long incubation has not been tested.
- Prepare all solutions (see Recipes).
Notes:- Be sure that RNasin has been added to all solutions.
- Approximately 30 min before starting, you should warm the 40% wHyb (1 ml/sample) and 2x SSC (1 ml/sample) to 43 °C. It is critical that they reach 43 °C.
- At this point, another oven should be set to 37 °C for the fluorescent oligo hybridization and washes. You should warm the Fluorescent Oligo Mix and PBS (1.5 ml/sample) to 37 °C.
- If you are working with a single oven, you can pause after Step 24 below. Remove the supernatant from the 2x SSC wash (leaving ~100 μl and tightly cap to prevent desiccation) and switch the oven temperature to 37 °C. Put the sample in the oven at 37 °C and wait ~10 min for equilibration. Then, resume at Step 25 below by adding pre-warmed 37 °C PBS.
- Add 500 µl of pre-warmed (43 °C) 40% wHyb and spin 2,000 x g for 5 min at RT.
Note: The pellet might become transparent after incubation in the 40% wHyb. Continue to leave ~100 μl in the tube when removing supernatant to avoid disturbing the pellet. - Remove supernatant and add 500 µl of pre-warmed (43 °C) 40% wHyb. Incubate for 15 min at 43 °C.
- Spin 2,000 x g for 5 min at RT.
- Remove supernatant and add 1 ml of pre-warmed (43 °C) 2x SSC. Incubate for 5 min at 43 °C.
- Spin 2,000 x g for 5 min at RT.
- Remove supernatant and add 500 µl of pre-warmed (37 °C) PBS.
- Spin 2,000 x g for 5 min at RT.
- Remove supernatant and add 100 µl of pre-warmed (37 °C) Fluorescent Oligo Mix. Incubate for 10 min at 37 °C.
- Add 500 µl of pre-warmed (37 °C) PBS and spin 2,000 x g for 5 min at RT.
- Remove supernatant and add 500 µl of pre-warmed (37 °C) PBS. Incubate for 5 min at 37 °C.
- Spin 2,000 x g for 5 min at RT.
Note: Before resuspending, pipette up and down with FACS Collection Solution (BSA/PBS) to coat the inside of the pipette tip with BSA to help minimize cells sticking inside the pipette tip. - Resuspend in 500-1,000 µl of PBS depending on the number of cells. Keep tubes on ice.
- Filter into FACS cell strainer tube and proceed to FACS.
Notes:
a. Make sure to bring collection tubes (microcentrifuge tubes) with 500 µl of FACS Collection Solution per population.
b. To visualize the cells before FACS, pipette 10 µl of the suspension onto a glass slide and air dry. Coverslip with mounting media.
FACS (~30 min for 1 million cells)
- Gate first based on Hoechst histogram (Figure 3). This step will ensure that you get 2N cells. Make sure to NOT include doublets. You can also gate based on FSC/SSC for cells vs. debris, but this step is likely unnecessary as debris will be negative for Hoechst.
Note: If your tissue contains polyploid cells (4N+ cells), it will be difficult to distinguish them from doublets or other cell clumps. In this case, it may be best to use the nuclei Probe-Seq protocol. - Gate using a 2D plot with appropriate wavelengths. The negative population will likely run diagonal. The positive population will be left- or right-shifted compared to the diagonal events (i.e., the Vsx2+ cells). Often, the separation will not be as obvious as GFP or well characterized cell surface markers. Thus, it’s a good idea to run a negative control (fluorescent oligo only control).
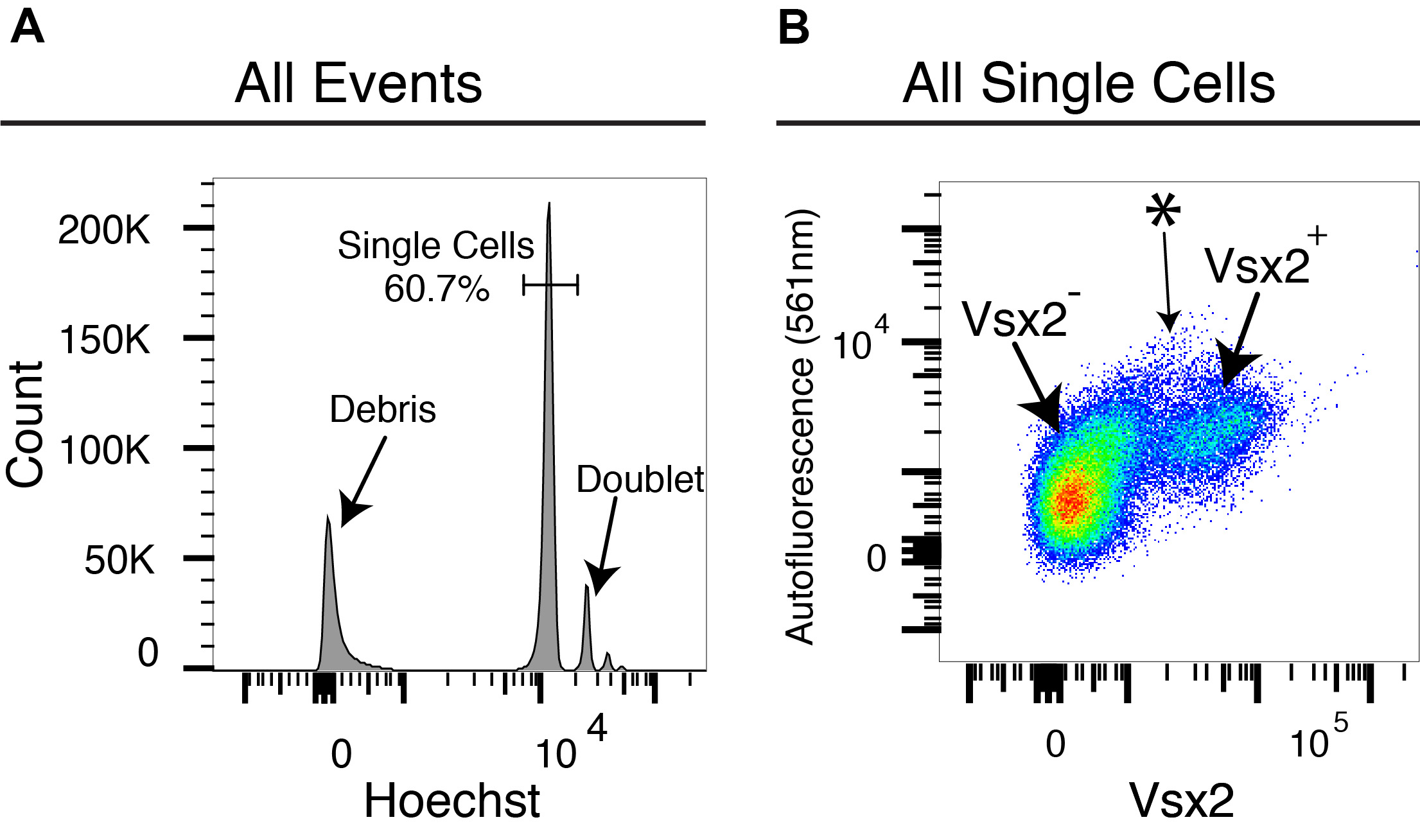
Figure 3. FACS gating strategy for Probe-Seq. A. Using a Hoeschst histogram, single cell peak is gated first. B. The percentage of single cell events will depend on the tissue. For example, the mouse retina will have a higher percentage of single cell events compared to the mouse brain because less debris is generated during the dissociation protocol for the retina. Out of the single cell events, the population of interest is identified. Here, the Vsx2 fluorescence is indicated on the x-axis, and the Vsx2+ population is right-shifted from the negative population. It is advisable to use an empty autofluorescence channel as the opposing axis as this will help to determine the events that have high fluorescence in all channels. * Do not collect these events even if they have a high fluorescence intensity in the Vsx2 channel. These events are also highly fluorescent for the empty autofluorescence (561 nm) channel. If using a histogram or a plot without an autofluorescence channel, these events will look like they are Vsx2+ even though they are unlikely to be the population of interest.
Note: It is advisable to keep one laser channel unused as this can be used to determine autofluorescence (i.e., the 561 nm channel in Figure 3). If there are events that are highly fluorescent in your channel of interest (i.e., the Vsx2 channel) AND the autofluorescence channel (i.e., 561 nm), these events are likely autofluorescent in all channels (i.e., the cells indicated by * in the FACS plot). For cells that are true positive, they generally form a cluster of events as opposed to a stream. Thus, these events should not be collected. - FACS isolated cells are sorted into FACS Collection Solution in a microcentrifuge tube (start with 500 µl per population already in the tube) and kept at 4 °C. This is NOT a stopping point. Go to the decrosslinking step ASAP.
Note: The BSA in the FACS Collection Solution helps pellet the cells in the subsequent steps.
Decrosslinking and RNA isolation (~4 h)
- Transfer the collected cells to a 5-ml polypropylene tube (with white caps).
- Spin at 3,000 x g for 7 min at 4 °C (or at RT if temp-controlled centrifuge is not available)
Note: Centrifugation at RT is also acceptable. Note the change in spin speed. - Remove as much supernatant as possible. Usually, there is ~40-50 µl left. Depending on the number of cells, the pellet may not be visible (i.e., not visible with 10,000 cells).
- From the Recoverall Total Nucleic Acid Isolation Kit for FFPE (Thermo Fisher Scientific), mix 100 µl of Digestion Buffer and 4 µl of protease for each sample.
- Add the Digestion Buffer/Protease Mix (104 µl) to the pellet. Resuspend.
- Transfer the resuspended cells to a DNA LoBind microcentrifuge tube (Eppendorf) if available. Wrap the lid with parafilm.
- Incubate at 50 °C for 3 h.
Note: This incubation time differs from the manufacturer’s protocol. - After 3 hours, the samples can be stored at -80 °C indefinitely or proceed to next steps according to the kit protocol.
STOPPING POINT
- Prep Isolation Additive/Ethanol mix (for 100 µl digest, 120 µl isolation + 275 µl 100% Ethanol. Usually, there is ~140 µl so adjust accordingly).
- Add Isolation/Ethanol Mix and pipette to mix.
- Add onto filter and spin 10,000 x g for 30 s at RT.
- Discard flow-through. Wash with 700 µl of Wash 1. Spin 10,000 x g for 30 s at RT.
- Discard flow-through. Wash with 500 µl of Wash 2. Spin 10,000 x g for 30 s at RT.
- Discard flow-through. Spin 10,000 x g for 30 s at RT.
- Prep DNase Mix (6 µl 10x DNase Buffer, 4 µl DNase, 50 µl UltraPure Water).
- Add 60 µl of DNase Mix to center of cartridge. Close cap and incubate for 30 min at RT.
- Add 700 µl of Wash 1, incubate 1 min at RT. Spin 10,000 x g for 30 s at RT.
- Discard flow-through. Wash twice with 500 µl of Wash 2.
- Spin 10,000 x g for 1 min to remove residual fluid.
- Elute in ~17 µl of UltraPure water (Use more water if lower concentration desired. Eluting with 17 µl will leave you with ~12 µl of RNA, which is enough for SMART-Seq v.4, Qubit, and/or BioAnalyzer).
- Store RNA at -80 °C.
STOPPING POINT
Note: Qubit or BioAnalyzer can be used to estimate RNA concentration. If less than 10,000 cells, concentration may not be available by these methods. In this case, proceed directly to SMART-Seq v.4 using 9.5 µl of RNA as the starting volume. - Proceed to the SMART-Seq v.4 protocol per manufacturer’s protocol for cDNA synthesis and amplification. Depending on the number of cells, you will use 1-9.5 µl of RNA for cDNA synthesis.
Notes:- The number of cycles will depend on the cell type, RNA concentration, RNA integrity. The goal is to use the least number of cycles to obtain ~500 pg/µl of cDNA. This will ensure as little amplification bias as possible while having enough cDNA for the subsequent steps. One strategy would be to perform SMART-Seq with 20-50% of your purified RNA with a high number of cycles (i.e., 18 cycles) and then calculate the number of cycles needed from there.
- If you collected the gene-negative and gene-positive populations and you would like to know if you collected the correct cell types, you can run qPCR or ddPCR with cell type specific markers at this stage.
- Other Library prepration kits can be used. However, they have not been tested.
- Use 150 pg of cDNA for Nextera XT library kit.
Note: HS DNA BioAnalyzer chip should be run on the BioAnalyzer 2100 after the SMART-Seq v.4 protocol and after Nextera XT indexing to ensure proper library construction.
Appendix: Nuclei Isolation (30 min)
Notes:
- All of the FISH reagents remain the same as the whole cell protocol. Before beginning, make sure to add RNasin to every solution. Also, before beginning the nuclei isolation, make sure to pre-warm the reagents. 40% wHyb (warm 500 µl per sample) and Probe Mix should be warmed to 43 °C.
- Because you are using only nuclear RNA as the marker, the signal intensity is likely to decrease compared to whole cell. Therefore, the expression level and transcript length of the marker gene will be more critical.
- If not enough signal is detected, you can design the oligos with both Exon and Intron sequences, which should be present in nascent nuclear RNA.
- The total extracted RNA is less abundant compared to whole cell. Therefore, when performing SMART-Seq v.4, more cycles will be needed to amplify your cDNA.
- This nuclei isolation method works for mouse and human retina and brain. It has not been tested for other tissues in other organisms.
- This nuclei isolation is meant for frozen tissue. If you have fresh tissue, flash-freeze the tissue in isopentane/dry ice slurry. This nuclei isolation protocol has not been tested with fixed-frozen tissue. Keep the collection tubes on dry ice before starting. The tissue goes directly into the slurry, without too much residual liquid around the tissue. Quickly transfer the frozen tissue into the collection tube. Keep at -80 °C.
- Prepare all solutions and keep on ice with the Dounce homogenizer (i.e., Thomas Scientific).
- Fill the glass Dounce homogenizer with 1 ml of cold homogenization buffer. Keep on ice.
- Mince the tissue into little pieces and place in 1% PFA for 5 min on ice.
- Transfer the tissue pieces into the homogenizer with the buffer. With the tight pestle, homogenize with 10-15 strokes on ice. Avoid foaming.
- Transfer the homogenate into a 5-ml polypropylene tube (Thermo Fisher Scientific). Make sure to seal the cap by pushing in.
- Carefully add 2 ml of Sucrose Bed solution to the bottom of the tube so that the homogenate is above the sucrose solution.
- Spin at 500 x g, 12 min, 4 °C.
- Remove supernatant. Add 1 ml of 4% PFA solution and resuspend pellet. Incubate for 15 min at 4 °C with rocking.
Note: Remove the top layer supernatant first. Then, remove the rest of the supernatant. - Centrifuge at 2,000 x g for 5 min at 4 °C.
- Go to Step 17 under Probe Hybridization of the Probe-Seq protocol.
Notes
Troubleshooting Guide
Table 1. Troubleshooting Guide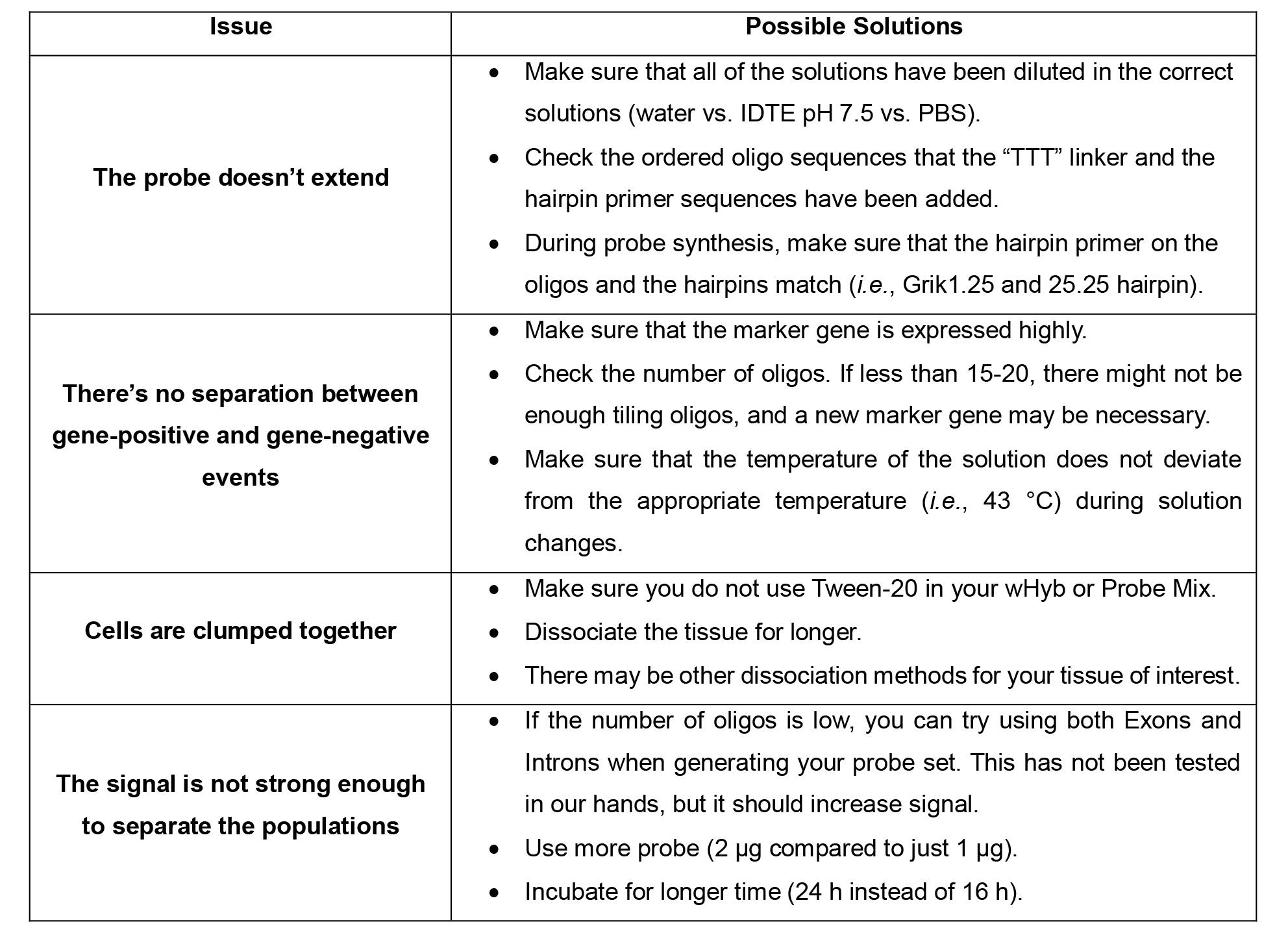
Recipes
- Retina Dissociation Recipes (optional if using other tissue)
Notes:
a.Especially for the Probe Hybridization/Probe Synthesis Reagents, use only the reagents with the catalog numbers listed below. We have not tested a wide range of reagents made by other companies.
b.These volumes are per sample except the DMEM/BSA, which is for 5 samples. If performing Probe-Seq for more than one sample, adjust volume accordingly.
1.HBSS/FBS
HBSS 900 µl
FBS 100 µl
Total Volume 1,000 µl
2.DMEM/BSA
DMEM 3,000 µl
BSA 12 mg
Total Volume 3,000 µl
3.Papain Mix
HBSS 315 µl
1 M HEPES pH 7.0 35 µl
50 mM L-Cysteine 20 µl
0.5 M EDTA pH 8.0 0.4 µl
UltraPure Water 19.6 µl
Papain 10 µl
Total Volume 400 µl
Notes:- Make the above solutions fresh before the experiment.
- Keep HBSS/FBS and DMEM/BSA at RT.
- The papain mix should be incubated at 37 °C for 10 min before use. It will be cloudy initially and become clear after warming.
- Add 1 µl of RNasin Plus (Promega) for every 1 ml of every solution used. Incubate for at least 10 min with the RNasin before use.
- Probe Hybridization Recipes
The details of the SABER FISH reagents can be found in the original SABER FISH paper (Kishi et al., 2019). For detailed protocol for gene-specific probe set design, see the saber.fish website. Here are the reagents that you will need for Probe-Seq:- Permeabilization Buffer
PBS 989 µl
Hoechst 33342 (10 mg/ml) 1 µl
10% Triton X-100 10 µl
Total Volume 1,000 µl
Make fresh before experiment - FACS Collection Solution (1% BSA/PBS)
BSA 10 mg
PBS 1,000 µl
Total Volume 1,000 µl
Make fresh before experiment - 40% wHyb
20x SSC 5 ml
UltraPure Water 25 ml
Deionized formamide 20 ml
Total Volume 50 ml
Store at -20 °C. This solution should not freeze at -20 °C
Pre-warm to 4 °C before use
Note: Compared to the original SABER-FISH paper (Kishi et al., 2019), these solutions do not contain Tween-20. This is because we have found that Tween-20 causes clumping in dissociated cells. - Hyb1
20x SSC 1 ml
UltraPure Water 1 ml
Deionized formamide 4 ml
50% Dextran sulfate 2 ml
Total Volume 8 ml
Store aliquots of 120 µl at -20 °C. This solution should not freeze at -20 °C - Probe Mix
Hyb1 96 µl
Purified Probe set 1 µg
UltraPure Water up to 120 µl
Pre-warm to 43 °C before use
Notes:- Increasing the amount of purified probe set to 2 µg has shown to increase fluorescence intensity. It is unclear if, with 2 µg, a point of saturation has been achieved. Therefore, more probe set may improve fluorescence intensity.
- The total volume of purified probe sets cannot exceed 24 µl.
- Fluorescent Oligo Mix
PBS 100 µl
10 µM Fluorescent oligo 2 µl (each)
Pre-warm to 37 °C before use
Notes:- Fluorescent oligos are ordered from IDT. The sequences and the fluorescent tags (on the 5’ end) can be found in the Oligonucleotide_List file. Listed are 2 different fluorophores per hairpin, but other fluorophores can be used as well. They are resuspended in UltraPure water as 100 µM stocks, and 10 µM working stocks (diluted in water) can be aliquoted and stored at -20 °C.
- Add 1 µl of RNasin Plus (Promega) for every 1 ml of every solution used. Incubate for at least 10 min with the RNasin before use.
- Permeabilization Buffer
- Nuclei Isolation Recipes
1.Buffer 1
1.5 M Sucrose 2,500 µl
1 M KCl 375 µl
1 M MgCl2 75 µl
1 M Tris Buffer pH 8.0 150 µl
UltraPure Water 11,900 µl
Total Volume 15,000 µl
Buffer 1 can be stored at 4 °C for up to 6 months
2.Homogenization Buffer
Buffer 1 968 µl
10% Triton X-100 10 µl
Protease Inhibitor (50x) 20 µl
1 mM DTT 1 µl
Hoechst 33342 1 µl
Total Volume 1,000 µl
Homogenization Buffer should be made fresh
Note: 1 ml of Homogenization Buffer is necessary for each sample.
3.Sucrose Buffer
1 M KCl 2,250 µl
1 M MgCl2 450 µl
1 M Tris Buffer pH 8.0 900 µl
UltraPure Water 11,400 µl
Total Volume 15,000 µl
Sucrose Buffer can be stored at 4 °C for up to 6 months
4.Sucrose Bed
Sucrose Buffer 2,500 µl
24% Sucrose 12,500 µl
Total Volume 15,000 µl
Sucrose Bed can be stored at 4 °C for up to 6 months
Note: Add 1 µl of RNasin Plus (Promega) for every 1 ml of every solution used. Incubate for at least 10 min with the RNasin before use.
Acknowledgments
We would like to thank former and current members of the Cepko and Tabin Labs for the insightful discussion and feedback. We thank C. Araneo, F. Lopez, and the Flow Cytometry Core Facility for their assistance with flow cytometry. This work was supported by the Howard Hughes Medical Institute (J.C., C.L.C. and N.P.), Edward R. and Anne G. Lefler Postdoctoral Fellowship (R.A.), HSCI Internship Program (M.D.G), and NIH NEI K99/R00 Grant 5K99EY028215 (S.W.L.). This protocol was derived from the original Probe-Seq and SABER manuscripts (Amamoto et al., 2019; Kishi et al., 2019).
Competing interests
The authors declare no conflict of interest.
Ethics
All animals were handled according to protocols approved by the Institutional Animal Care and Use Committee (IACUC) of Harvard University (protocol number: 1695).
References
- Amamoto, R., Garcia, M. D., West, E. R., Choi, J., Lapan, S. W., Lane, E. A., Perrimon, N. and Cepko, C. L. (2019). Probe-Seq enables transcriptional profiling of specific cell types from heterogeneous tissue by RNA-based isolation. Elife 8: e51452.
- Beliveau, B. J., Kishi, J. Y., Nir, G., Sasaki, H. M., Saka, S. K., Nguyen, S. C., Wu, C. T. and Yin, P. (2018). OligoMiner provides a rapid, flexible environment for the design of genome-scale oligonucleotide in situ hybridization probes. Proc Natl Acad Sci U S A 115(10): E2183-E2192.
- Jaitin, D. A., Kenigsberg, E., Keren-Shaul, H., Elefant, N., Paul, F., Zaretsky, I., Mildner, A., Cohen, N., Jung, S., Tanay, A. and Amit, I. (2014). Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 343(6172): 776-779.
- Hershberg, E. A., Close, J. L., Camplisson, C. K., Attar, S., Chern, R., Liu, Y., Akilesh, S., Nicovich, P. R. and Beliveau, B. J. (2020). PaintSHOP enables the interactive design of transcriptome- and genome-scale oligonucleotide FISH experiments. bioRxiv doi: https://doi.org/10.1101/2020.07.05.188797.
- Kishi, J. Y., Lapan, S. W., Beliveau, B. J., West, E. R., Zhu, A., Sasaki, H. M., Saka, S. K., Wang, Y., Cepko, C. L. and Yin, P. (2019). SABER amplifies FISH: enhanced multiplexed imaging of RNA and DNA in cells and tissues. Nat Methods 16(6): 533-544.
- Kishi, J. Y., Schaus, T. E., Gopalkrishnan, N., Xuan, F. and Yin, P. (2018). Programmable autonomous synthesis of single-stranded DNA. Nat Chem 10(2): 155-164.
- Klein, A. M., Mazutis, L., Akartuna, I., Tallapragada, N., Veres, A., Li, V., Peshkin, L., Weitz, D. A. and Kirschner, M. W. (2015). Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161(5): 1187-1201.
- Klemm, S., Semrau, S., Wiebrands, K., Mooijman, D., Faddah, D. A., Jaenisch, R. and van Oudenaarden, A. (2014). Transcriptional profiling of cells sorted by RNA abundance. Nat Methods 11(5): 549-551.
- Macosko, E. Z., Basu, A., Satija, R., Nemesh, J., Shekhar, K., Goldman, M., Tirosh, I., Bialas, A. R., Kamitaki, N., Martersteck, E. M., Trombetta, J. J., Weitz, D. A., Sanes, J. R., Shalek, A. K., Regev, A. and McCarroll, S. A. (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161(5): 1202-1214.
- Passaro, M., Martinovic, M., Bevilacqua, V., Hershberg, E. A., Rossetti, G., Beliveau, B. J, Bonnal, R. J. P. and Pagani, M. (2020). OligoMinerApp: a web-server application for the design of genome-scale oligonucleotide in situ hybridization probes through the flexible OligoMiner environment. Nucleic Acid Res 48(W1): w332-w339.
- Picelli, S., Bjorklund, A. K., Faridani, O. R., Sagasser, S., Winberg, G. and Sandberg, R. (2013). Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods 10(11): 1096-1098.
Article Information
Copyright
![]() Amamoto et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Amamoto et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Amamoto, R., Garcia, M. D., West, E. R., Choi, J., Lapan, S. W., Lane, E. A., Perrimon, N. and Cepko, C. L. (2020). Probe-Seq: Method for RNA Sequencing of Specific Cell Types from Animal Tissue. Bio-protocol 10(18): e3749. DOI: 10.21769/BioProtoc.3749.
- Amamoto, R., Garcia, M. D., West, E. R., Choi, J., Lapan, S. W., Lane, E. A., Perrimon, N. and Cepko, C. L. (2019). Probe-Seq enables transcriptional profiling of specific cell types from heterogeneous tissue by RNA-based isolation. Elife 8: e51452.
Category
Neuroscience > Sensory and motor systems > Retina
Developmental Biology > Cell growth and fate > Degeneration
Cell Biology > Cell isolation and culture > Cell isolation > Flow cytometry
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.