- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Screening Method for CRISPR/Cas9 Inhibition of a Human DNA Virus: Herpes Simplex Virus
(*contributed equally to this work) Published: Vol 10, Iss 17, Sep 5, 2020 DOI: 10.21769/BioProtoc.3748 Views: 6965
Reviewed by: Sylvain Baron Thirupugal GovindarajanAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Dec 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
The efficiency of cleavage of individual CRISPR/Cas9-sgRNAs remains difficult to predict based on the CRISPR target sequence alone. Different intracellular environments (dependent on cell type or cell cycle state for example) may affect sgRNA efficiency by altering accessibility of genomic DNA through DNA modifications such as epigenetic marks and DNA-binding proteins (e.g., histones) as well as alteration of the chromatin state of genomic DNA within the nucleus.
We recently reported a multi-step screening method for the identification of efficient sgRNAs targeting the Herpes simplex virus (HSV-1) genome and reported a differential mechanism for viral inhibition by CRISPR-Cas9 in the latent versus lytic phase. The screening platform detailed in this protocol allows step-by-step testing of the efficiency of cleavage in a cell-free system and in the context of viral target cells such as human foreskin fibroblasts followed by functional testing of the effects of CRISPR/sgRNA on viral protein expression, replication, and reactivation. This strategy could be readily applied to other target cells such as pluripotent stem cell-derived human sensory neurons or other human DNA viruses.
Background
Herpes simplex virus (HSV) is a neurotropic DNA virus of the Herpesviridae family that causes lifelong and incurable infection in the majority of the human population and can lead to significant morbidity and mortality (Liesegang et al., 1989; Roizman et al., 2013). After the initial acute (primary) infection, HSV establishes lifelong latent infection within sensory ganglia. Latent HSV can reactivate periodically and cause serious illness such as HSV keratitis, the most common cause of corneal blindness (Liesegang et al., 1989), and recurrent genital herpes, which carries high morbidity. In the fetus, neonate and the immunosuppressed, herpesviruses such as HSV-1 and -2, human cytomegalovirus (HCMV), and Varicella-Zoster Virus (VZV) can cause disseminated disease with high mortality including meningitis and encephalitis. In the US, HSV infection occurs in 1 in 3,200 neonates (Thompson and Whitley, 2011), and half of these infants have disseminated disease or encephalitis (Kimberlin, 2004), which, despite antiviral treatment, carries 25% mortality and long-term neurologic morbidity in more than two-thirds of survivors (Kimberlin et al., 2001). In addition, HSV1 infection has been linked with Alzheimer's disease risk in APOE-E4 carriers (Itzhaki, 2018).
Current antiviral treatments (nucleoside analogues) do not prevent establishment of latency or episodes of reactivation and are not effective for clearing latent HSV from infected individuals. Thus, there is a critical need for novel, specific, and efficacious therapeutic and prophylactic avenues that directly cleave and/or edit viral genomes and permanently eliminate latent virus or disable its reactivation and effectively suppress viral replication in disseminated acute HSV infections.
CRISPR/Cas9 systems have evolved in bacteria as a form of adaptive immunity to provide resistance against bacteriophage infection and plasmid transformation (Bhaya et al., 2011; Doudna and Charpentier, 2014; Gaj et al., 2013; Hsu et al., 2014). This system has been successfully transported into human cells (Cong et al., 2013; Mali et al., 2013). However, sgRNAs with comparable cutting efficiencies in a cell-free system can differ significantly when used in living cells (Oh et al., 2019), indicating that the intracellular environment may alter the accessibility of genomic DNA for cleavage by CRISPR/Cas9. Possible mechanisms are the covalent modification of genomic DNA in the form of epigenetic marks, differential density of DNA binding proteins (such as histones), and alteration of the chromatin state of genomic DNA in different cell types and/or cellular states (Yarrington et al., 2018; Liu et al., 2020).
The screening platform described in this protocol allows identification of sgRNAs that efficiently cleave the HSV genome and inhibit lytic viral replications as well as reactivation of quiescent virus in the chosen type of target cells. We used SaCas9 to allow for further testing of the antiviral activity of individual SaCas9/sgRNAs in vivo using recombinant AAV-based delivery systems. This screening strategy can be extended to other HSV target cells such as human sensory neurons which can be differentiated from human pluripotent stem cells (Chambers et al., 2012; Young et al., 2014). In addition, this screening strategy can be applied to clinically important members of the Herpesviridae family such as HSV-2, HCMV, VZV and KSHV as well as other human DNA viruses lacking effective therapies.
Materials and Reagents
- Eppendorf Safe-Lock Tubes, 1.5 ml (Thermo Fisher Scientific, Eppendorf, catalog number: 0 22363212 )
- PCR tubes (any kind)
- Pipette tips 10 μl, 200 μl, 1,000 μl (Thermo Fisher Scientific, Thermo ScientificTM, catalog numbers: 2140-05 , 2160P , 2079 )
- Tissue culture flasks T-75 filtered flasks (Corning, Falcon®, catalog number: 353824 )
- 50 ml Conical tube (Corning, Falcon®, catalog number: 352098 )
- 0.45 µm syringe filter (Pall, catalog number: 4508 )
- 6-well plates (Corning, catalog number: CLS3516 )
- AmpliTaq GoldTM 360 Master Mix (Thermo Fisher Scientific, Applied Biosystems, 4398901)
- Wizard® SV Gel and PCR Clean-Up System (Promega, catalog number: A9281 )
- PlatinumTM II Taq Hot-Start DNA Polymerase (Thermo Fisher Scientific, Invitrogen, catalog number: 14966001 )
- MAXIscriptTM T7 Transcription Kit (Thermo Fisher Scientific, Invitrogen, catalog number: AM1314 )
- Phusion® High-Fidelity DNA Polymerase (Thermo Fisher Scientific, New England Biolabs, catalog number: M0530S )
- Gene SnipperTM SaCas9 Protein (CRISPR-associated endonuclease Cas9 from Staphylococcus aureus) with SaCas9 Buffer: 200 mM HEPES, 50 mM MgCl2, 1 M NaCl,1 mM EDTA, pH 6.5 (Biovision, catalog number: M1280 ) or SpCas9 Protein (CRISPR-associated endonuclease Cas9 from S. pyogenes) with 1x NEBuffer 3.1: 100 mM NaCl, 50 mM Tris-HCl, 10 mM MgCl2, 100 µg/ml BSA, pH 7.9 (New England Biolabs, catalog number: M0386 )
- E.Z.N.A.® Micro RNA Kit (Omega Bio-Tek, catalog number: R7034-01 )
- QIAfilter Plasmid Midi Kit (Qiagen, catalog number: 12245 )
- Lenti-X qRT-PCR Titration Kit (Takara, catalog number: 631235 )
- BsmBI restriction enzyme (Thermo Fisher Scientific, New England Biolabs, catalog number: R0580 )
- DNeasy Blood & Tissue kit (Qiagen, catalog number: 69504 )
- Agarose (Sigma-Aldrich, catalog number: A9539 )
- Polyethyleneimine (Polysciences, catalog number: 23966 , average Mw ~25,000)
- Effectene (Qiagen, catalog number: 301425 )
- NuPAGETM LDS Sample Buffer (4x) (Thermo Fisher Scientific, Invitrogen, catalog number: NP0008 )
- NuPAGE 4-12% Bis-Tris Gels (Thermo Fisher Scientific, Invitrogen, catalog number: NP0321PK2 )
- NuPAGE MOPS SDS Running Buffer (Thermo Fisher Scientific, Invitrogen, catalog number: NP0001 )
- Nitrocellulose membrane (Bio-Rad, catalog number: 1620112 )
- Odyssey Blocking Buffer (TBS) (LI-COR, catalog number: 927-50000 )
- IRDye 800CW (P/N: 926-32211) or IRDye 680RD (P/N: 926-68070) secondary antibody (LI-COR)
- Giemsa stain (Sigma-Aldrich, catalog number: G4507 or GS500 )
- Assay buffer (see Recipes)
- KCl
- MgCl2
- Tris
- Glycerol
- EDTA
- SDS
- Bromophenol blue
- Reaction Stop Buffer (see Recipes)
Media
- DMEM (Thermo Fisher Scientific, Gibco, catalog number: 11960051 )
- Opti-MEMTM I (Thermo Fisher Scientific, Gibco, catalog number: 31985062 )
- Fetal Bovine Serum (Thermo Fisher Scientific, Gibco, catalog number: 26140079 )
- Bovine calf serum (Thermo Fisher Scientific, catalog number: 16030074 )
- L-Glutamine (Thermo Fisher Scientific, Gibco, catalog number: 25030164 )
- G418 Geneticin (Thermo Fisher Scientific, Gibco, catalog number: 10131035 )
- Puromycin Dihydrochloride (Thermo Fisher Scientific, Gibco, catalog number: A1113802 or Santa Cruz, catalog number: sc-134220 )
- Polybrene Infection reagent (Millipore Sigma, catalog number: TR-1003-G or Santa Cruz: sc-134220 )
- GAMMAGARD LIQUID Immune Globulin Infusion (Takeda, Shire, catalog number: LE1500190 , NDC: 0944 2700 02)
Plasmids
- lenti-SaCas9-sgRNA-Puro
Can be obtained by cloning SaCas9 and its trans-activating sgRNA into the lentiCRISPRv2 plasmid (addgene # 52961 ). lentiCRISPRv2 is digested using BsmBI (NEB) and BamHI (NEB) and purified using a DNA purification kit (Zymo). DNA assembly can be performed using NEBuilder® HiFi DNA Assembly (NEB) according to the manufacturer’s protocol with three DNA fragments, the purified linear lentiCRISPRv2, a double-stranded DNA gBlock (IDT) containing the sgRNA sequence (IDT, 5’ CTTTATATATCTTGTGGAAAGGA CGAAACACCGGAGACGtGATATCaCGTCTCAGTTTTAGTACTCTGGAAACAGAATCTACTAAAACAAGGCAAAATGCCGTGTTTATCTCGTCAACTTGTTGGCGAGATTTTTGAATTCGTAGACTCGAGGCGTTGACAT TG 3’) and a PCR fragment containing SaCas9.
- psPAX2 (Addgene plasmid # 12260 )
- pVSV-G (Addgene plasmid # 8454)
Cell lines and media
Human Foreskin Fibroblasts (HFF, Hs27, ATCC, catalog number: CRL-1634), U2OS (known as U-2 OS, ATCC, catalog number: HTB-96), 293T (ATCC, catalog number: CRL-3216) and Vero (ATCC, catalog number: CCL-81) cells can be obtained from the American Type Culture Collection.
- HFF and 293T cells: Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% (v/v) Fetal Bovine Serum (FBS)
- V27 cells: DMEM supplemented with 5% (v/v) FBS, 5% (v/v) bovine calf serum (BCS), 2 mM L-glutamine, and 500 μg/ml of G418 in 5% CO2 (Rice et al., 1989)
- U2OS and Vero cells: DMEM supplemented with 5% (v/v) FBS, 5% (v/v) bovine calf serum (BCS), and 2 mM L-glutamine in 5% CO2
- FO6 cells: are maintained in DMEM supplemented with 5% (v/v) FBS, 5% (v/v) BCS, and 2 mM L-glutamine with 500 μg/ml of G418 (Gibco) and 300 μg/ml of hygromycin B (Invitrogen) in 5% CO2 (Samaniego et al., 1997)
Viruses
The KOS strain is a commonly used wild-type HSV-1 strain (Schaffer et al., 1970). HSV-1 d109 is a KOS strain-derived mutant that is deleted for all five immediate early (IE) genes and contains the green fluorescent protein (GFP) gene under the control of the human cytomegalovirus (HCMV) IE promoter in the HSV-1 UL54 gene locus. d109 can be grown and titrated on U2OS ICP4/27 and FO6 cells (Samaniego et al., 1998).
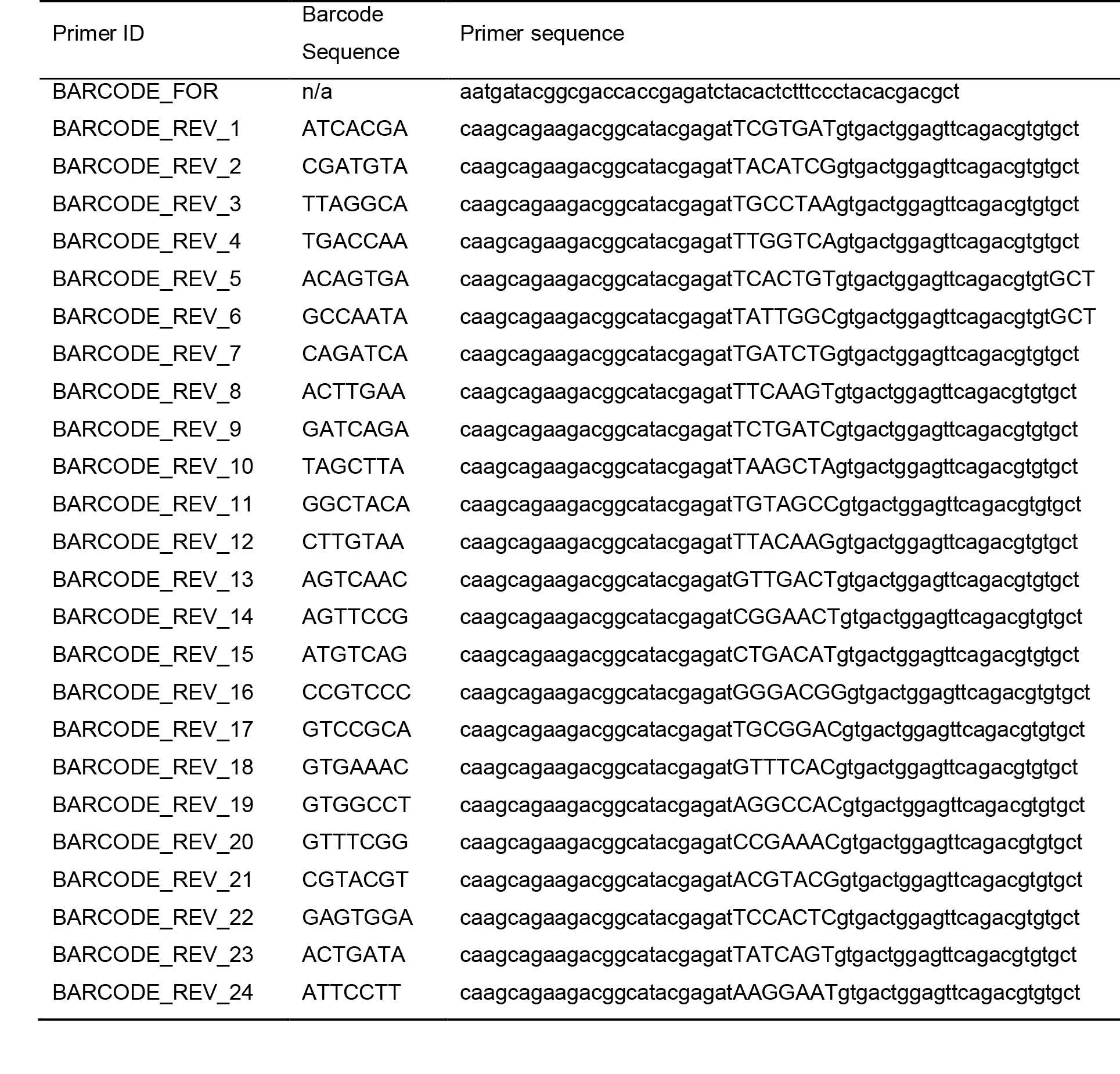
MiSeq adapter primers (Table 1)
Forward adapter primer: 5′-TCTTTCCCTACACGACGCTCTTCCGATCT-CRISPR site forward primer-3′
Reverse adapter primer: 5′-TGGAGTTCAGACGTGTGCTCTTCCGATCT-CRISPR site reverse primer-3′
HSV-1 gene specific primers
UL30-5_rev: 5′-CTCCACGTTCTCCAGGATGT-3′
UL29-3_for: 5′-ATAGACTCGAGGGCCAGGG-3′
UL29-3_rev: 5′-TGACGAAAACCACGAGGGC-3′
Table 1. MiSeq barcode primers
Equipment
- Pipettes (capable of accurately pipetting from 1 μl-10 ml) (any source)
- Thermocycler (Bio-Rad, model: C1000 )
- Vortex (any source)
- Micro centrifuge capable of 14 kG (Eppendorf)
- Incubators (37 °C)
- DNA gel electrophoresis apparatus
- Odyssey CLx Imaging System (LI-COR)
- UV transilluminator (or handheld UV lamp)
- Spectrophotometer (Thermo Fisher Scientific, model: NanoDrop 2000)
Software
- Outknocker (http://www.outknocker.org/)
- Cas-OFFinder (http://www.rgenome.net/cas-offinder/)
- mutect2 (https://gatk.broadinstitute.org/hc/en-us/articles/360037593851-Mutect2)
- Prism 6 (Version 6.01) software
Procedure
- Cell-free CRISPR/Cas9 cutting assay
- Design sgRNAs
- Identify essential genes (e.g., UL29, UL30, UL54, a4 for HSV-1) or DNA regions within the viral genome.
- Design appropriate sgRNA target sequences within ~1 kbp downstream of the start codon. The requirement for SaCas9 cleavage is the presence of a PAM sequence (5’-NNGRRT-3’) immediately downstream of the ~21 base pair target. Select multiple independent CRISPR sgRNAs for each viral gene target for efficiency testing.
- Assess off-target activity of sgRNAs: The sgRNA sequences should be unique to the viral genome to prevent off-target cleavage of human DNA. Useful web sites for sgRNA identification and ranking of off-target activity are http://chopchop.cbu.uib.no/ [CHOP CHOPv2 (Labun et al., 2016) and crispr.mit.edu (Zhang lab, MIT) (Hsu et al., 2013)].
- In vitro sgRNA synthesis
- Generate templates for T7 In vitro transcription of sgRNAs
- Design and order oligos for sgRNA synthesis: For each sgRNA design a 60 nt forward oligo (target specific oligo), containing the T7 promoter (5’-TAATACGACTCACTATA) followed by 21 nt of the specific SaCas9 sgRNA DNA binding sequence with the first 2 nucleotides replaced by GG (GGNNNNNNNNNNNNNNNNNNN) for T7 initiation and a constant 23 nt tail of sgRNA scaffold sequence for annealing (GTTTTAGTACTCTGGAAACAGAA-3’). These forward oligos are used in combination with a constant 82 nt reverse oligo (5’- AAAAAATCTCGCCAACAAGTTGACGAGATAAAC ACGGCATTTTGCCTTGTTTTAGTAGATTCTGTTTCCAGAGTACTAAAAC-3’) encoding the reverse-complement of the tracrRNA tail to add the sgRNA invariable 3’ end (SaCas9 scaffold oligo, Figure 1).
- Anneal sgRNA primers with the tail primer: For each sgRNA assemble the following reaction in PCR tubes:
Reagent [μl]
Oligo 1 (variable sgRNA oligo), 100 μM 1
Oligo 2 (constant tail oligo), 100 μM 1
H2O 8
Total 10
Anneal protocol:
Hold at 95 °C for 5 min.
Ramp from 95 °C -> 85 °C at 2 °C/s
Ramp from 85 °C -> 25 °C at 0.1 °C/s
Hold at 4 °C. - Fill in single-stranded DNA (ssDNA) overhangs to generate double- stranded DNA (dsDNA) templates for T7 transcription by assembling the following reaction for each sgRNA in PCR tubes (Figure 1):
Reagent [μl]
AmpliTaq GoldTM 360 Master Mix 10
Taq polymerase (5 U) 1
Anneal Rx from ii. 9
Total 20
Incubate at 72 °C for a total of 1.5 h and add an additional 1 μl Taq after 45 min. - Purify the dsDNA using the Promega Wizard Clean up kit and elute in 20 μl H2O.
- (Optional) Run dsDNA templates on an agarose gel to check the size.
- sgRNA in vitro transcription (IVT):
- For each sgRNA assemble the following reaction using the MAXIscriptTM T7 Transcription Kit and approximately 150 ng of dsDNA template and incubate overnight at 37 °C:
Reagent [μl]
ATP solution 2
CTP solution 2
GTP solution 2
UTP solution 2
10X Reaction Buffer 2
Template DNA (150 ng) ~4
T7 Enzyme Mix 2
H2O 4
Total 20
Note: Assemble transcription reaction at room temp (The spermidine in the 10x Reaction Buffer can coprecipitate the template DNA if the reaction is assembled on ice). Add the 10x Reaction Buffer after the water and the ribonucleotides are already in the tube. - Remove template DNA: Add 56 μl H2O and 4μl DNAse turbo to each IVT reaction and incubate at 37 °C for 15 min.
- sgRNA purification: Purify the sgRNAs using the Omega EZNA PF kit and measure the concentration of the purified sgRNAs using a NanoDrop 2000. Elute in H2O or assay buffer). After purification, dilute the sgRNAs to 200 ng/µl each and store at -80 °C.
- (Optional) Confirm the expected size (~97bp) of the sgRNA on an RNA gel.
- For each sgRNA assemble the following reaction using the MAXIscriptTM T7 Transcription Kit and approximately 150 ng of dsDNA template and incubate overnight at 37 °C:
- Generate dsDNA substrate for the In vitro cutting assay
- For each sgRNA amplify a 500-2,000 bp segment around the sgRNA sequence to be tested from HSV-1 genomic DNA using PCR. If the amplification is difficult, dsDNA for the cleavage assay can also be obtained as g-blocks (IDT).
- Purify the PCR product using the Promega Wizard Clean up kit.
- In vitro cutting assay with CRISPR sgRNA and SaCas9 protein
- Reagent setup:
- Dilute Cas9 protein in assay buffer to 500 nM.
- Dilute purified PCR product to 50-100 ng/μl in H2O.
- Confirm each sgRNA is at 200 ng/μl or higher.
- Assembly of cleavage assay: Each guide requires four wells of a PCR strip. Cleavage assays are performed in a reaction volume of 10 μl with final SaCas9 concentrations of 300 nM, 100 nM, 33 nM, and 0 nM, 400ng sgRNA, 200 ng dsDNA substrate in 1x SaCas9 nuclease reaction buffer.
- Add 6 μl of assay buffer to wells 2-4 of a PCR strip and 6 μl of 500 nM SaCas9 protein to well 1. Then add 2 μl of 500 nM SaCas9 protein to well 2, mix, transfer 2 μl of this to well 3, mix and remove and discard 2 μl of this mix. Since the final volume of the reaction is 10 μl, the final Cas9 concentrations are 300 nM, 100 nM, 33 nM, and 0 nM.
- Add 2 μl of sgRNA to each of the 4 wells.
- Incubate for 10 min at 37 °C to allow SaCas9-sgRNA complexes to form.
- Add 2 μl purified PCR product to each of the 4 wells.
- Incubate for 30 min at 37 °C.
- Add 4 μl of reaction stop buffer to each well and incubate at 80 °C for 10 min.
- Run all 10 μl of all reactions on a 2% agarose gel and image.
- Analysis: Because the assay provides sgRNA in excess, SaCas9 protein is the limiting factor. Cutting efficiency is constrained by the total amount of template provided, but in general, sgRNAs that work well show robust cutting (larger band at 0-50% intensity of no SaCas9 control) and there will be evidence of graded activity at lower SaCas9 concentrations. Figure 2 shows the results of the in vitro cleavage assay for three sgRNA targeting the essential HSV UL30 gene (encoding the viral polymerase).
- Cell-based CRISPR/SaCas9 cutting assay using next generation sequencing (MiSeq)
- Cloning of sgRNAs into SaCas9 lentivirus expression vector
- Clone the sgRNA CRISPR sequence into lentiCRISPR SaCas9 after BsmBI digestion using oligos as described previously (Sanjana et al., 2014; Shalem et al., 2014) to generate a lenti vector expressing SaCas9 land a sgRNA as well as the selection marker puro.
- Purify plasmid DNAs using the endotoxin-free Midiprep kit (Qiagen) according to the manufacturer’s protocol.
- Lentivirus prep
- Plate 3 x 106 human 293T cells in a 100 mm dish 20-24 h before transfection.
- Transfect the 293T cells with lenti-SaCas9-sgRNA-Puro (5 µg), psPAX2 (4 µg), and pVSV-G (1 µg) using Effectene according to the manufacturer’s protocol or using polyethyleneimine (PEI).
- For PEI transfection, mix a total of 10 µg of DNAs in 500 µl of Opti-MEMI (Gibco) and 30 µg of PEI in 500 µl of Opti-MEMI.
- Mix DNA and PEI (total 1 ml) and incubate at ambient temperature for 20 min.
- Add mixture directly to 293T cells containing fresh 5 ml of DMEM (supplemented with 10% (vol/vol) FBS and 2 mM glutamine).
- Incubate the cells at 37 °C for 8-12 h, then replace media with 10 ml of fresh DMEM supplemented with 10-30% (vol/vol) FBS and 2 mM glutamine, and continue incubation at 37 °C.
- At every 12-24 h for 48-60 h, harvest media and replace with fresh DMEM (supplemented with 10-30% (vol/vol) FBS and 2 mM glutamine).
- Pool the harvested media on ice, filter the pooled media using 0.45 µm syringe filter (Pall).
- Optional: To concentrate, centrifuge the virus for 1.5 h at 25,000 rpm (SW28 rotor of Beckman, ~120,000 x g) at 4 °C.
- Titration of lentivirus: use the Lenti-X qRT-PCR Titration Kit (Takarabio) or qPCR Lentivirus Complete Titration Kit (abm) according to the manufacturer’s protocol.
- Keep lentivirus at 0-4 °C until transduction of target cells (within one day) or freeze at -80 °C for long-term storage.
- For quiescent HSV infection:
- Establish quiescent HSV-1 infection in HFF cells:
- Infect HFF cells with the HSV-1 mutant d109 (which is missing all 5 immediate early genes) at an MOI of 10 in PBS containing 0.1% glucose (wt/vol) and 0.1% BCS (v/v) for 1 h with shaking at 37 °C (Ferenczy and DeLuca, 2011).
- Replace virus inoculum with DMEM supplemented with 10% (v/v) FBS and incubate at 37 °C for 7-10 days.
- Transduction of HFFs containing quiescent HSV-1 viral genomes with SaCas9/sgRNA
- Transduce the d109 infected cells with 2 or 10 ml of lentivirus (or at an MOI of 5) containing 3-4 µg/ml of polybrene in 6-well plates or in T150 flasks respectively (optional: repeat the lentivirus transduction next day).
- The next day, replace the transduction medium with fresh medium and incubate cells for 2 days at 37 °C followed by puromycin treatment (1 µg/ml) for 7-10 days.
- Isolation of HSV genomic DNA: Harvest cells and isolate genomic DNA using the DNeasy Blood & Tissue kit (Qiagen) according to the manufacturer’s protocol.
- For lytic HSV infection:
- Transduction of HFFs with SaCas9/sgRNA:
- To transduce HFF cells with lentivirus for the lytic infection assay, plate HFF cells at a density of 2 x 105/well in a T25 flask one day prior to transduction.
- Transduce with 2-5 ml of lentivirus (or at an MOI of 5) containing 3-4 µg/ml of polybrene (optional: repeat the lentivirus transduction next day).
- The next day, replace the transduction medium with fresh medium and incubate cells for 2 days at 37 °C followed by puromycin treatment (1 µg/ml) for 7-10 days.
- Lytic infection with wild-type HSV-1
- Infect the transduced cells with wild-type HSV-1 at an MOI of 0.1 or 5 in phosphate-buffered saline (PBS) containing 0.1% glucose (wt/vol), 0.1% BCS (v/v) for 1 h with shaking at 37 °C.
- Replace virus inoculum with DMEM containing 1% BCS and incubate at 37 °C.
- Harvest cells at various time points for viral replication by qPCR and titration.
- Isolation of genomic and viral DNA: isolate genomic DNA using the DNeasy Blood & Tissue kit according to the manufacturer’s protocol.
- Deep sequencing (MiSeq): The process of deep sequencing involves an initial PCR to generate a ~200 bp product around the region of interest followed by a second PCR to add barcodes to the amplicons. Following this, each of the products is run on an agarose gel to determine the relative amount of each amplicon. Based on the brightness of the amplicon band on the gel, the PCR products of a particular loci are pooled such that each amplicon is ~equally represented in the final pool. The pooled product is then run on an agarose gel and gel-purified before being submitted for sequencing. With a set of 24 unique barcoding primer pairs a total of 24 sgRNAs can be analyzed in parallel with one deep sequencing sample.
- Adapter (first round) PCR with target gene-specific primers
- Design a primer set generating a ~200 bp product around the CRISPR site and add the adapter sequences to forward and reverse primer (see Figure 3). The CRISPR site should be in the center 1/3 of the amplicon to obtain good sequencing across the targeted site. The adapter sequences are used in the second round (barcoding) PCR for primer annealing.
- Perform a Phusion touch-down PCR, which increases the specificity of the reaction, according to the following protocol. Use the NEB calculator to determine the annealing temperature (http://tmcalculator.neb.com/#!/).
Reagent [μl]
5x GC buffer 2
dNTPs (10 mM) 0.2
Forward primer (25 μM) 0.05
Reverse primer (25 μM) 0.05
Template (~1-10 ng gDNA from B4b-iii) 1
Phusion polymerase 0.1
DMSO 0.3
H2O 6.3
Total 10
Cycle protocol:
Initial denaturation 98 °C 30 s
15x
Denaturation 98 °C 10 s
Annealing 72 °C (-0.5 °C/cycle) 30 s
Elongation 72 °C 2 min
15x
Denaturation 98 °C 10 s
Annealing Tm 30 s
Elongation 72 °C 2 min
Final elongation 72 °C 5 min
Hold at 4 °C - Run a small amount (2 μl) on a 1.5% agarose gel to ensure that the PCR worked. If so, move on to the second round (barcoding) PCR.
- Barcode (second round) PCR with barcoding primers: This PCR uses universal primers (not gene-specific, see Table 1) to attach the sequencing adaptors and barcode to the amplicon. The forward primer is the same, but there are 24 reverse primers containing different barcodes (for testing of up to 24 different sgRNAs in parallel).
- Set up a standard 10 μl PCR mix using 1 μl of the first round PCR product as the template as follows:
PCR Rx [μl]
5x GC buffer 2
dNTPs (10 mM) 0.2
Forward primer (10 μM) 0.5
Reverse primer (5 μM) 1
Adapter PCR (template) 1.0
Phusion 0.1
DMSO 0.3
H2O 4.9
Total 10
Cycle protocol:
Initial denaturation 98 °C 30 s
20x
Denaturation 98 °C10 s
Annealing 66 °C20 s
Elongation 72 °C20 s
Final elongation 72 °C 3 min
Hold at 4 °C - Run a small amount (2 μl) on a 1.5% agarose gel. This allows you to ensure that the PCR worked and to normalize the amount of each amplicon when PCR products are pooled together for MiSeq. Of note, in some cases, one may see a large primer dimer amplicon around 175 bp in addition to the correct band.
- Pool amplicons and perform gel extraction:
- Submit purified/pooled amplicons for IlluminaMiSeq using the 16S Metagenomic Sequencing Library Preparation kit (Illumina). Determine DNA concentrations using the Qubit fluorometer 2.0 (Life Technologies, USA) with the Qubit dsDNA High Specificity assay kit. Sequence DNA libraries by IlluminaMiSeq (150 bp paired-end [Gagnon et al., 2014]).
1)Pool (up to 24) amplicons for different sgRNAs–do not pool across amplicons from different targeted viral genes as this will make the gel extraction more difficult. From the previous gel, estimate equal molarities when pooling (i.e., compensate for weaker looking amplicons by adding more to the pool). This will even out the read counts across different amplicons during MiSeq.2)Run pooled amplicons on a 1.5% agarose gel and gel extract the DNA using the Promega Gel Extraction Kit. - Set up a standard 10 μl PCR mix using 1 μl of the first round PCR product as the template as follows:
- Indel analysis: Determine indel frequencies for each sgRNA by quantifying aligned reads containing insertions or deletions 1bp or larger using (http://www.outknocker.org/). Analyze indel size using the ICE analysis toolbox (https://www.synthego.com/products/bioinformatics/crispr-analysis). Figure 4 shows the analysis of indel mutations in the HSV-1 genome during the quiescent infection for sgRNAs targeting the essential HSV-1 genes UL29 and UL30.
- Analysis of off-target effects in the human an viral genome
- Transduce HFFs with lentivirus expressing SaCas9/sgRNA as above and SaCas9 without sgRNA as control.
- Isolate genomic DNA using the DNeasy Blood & Tissue kit according to the manufacturer’s protocol.
- Perform 30x whole genome sequencing (WGS) of SaCas9/sgRNA treated sample and control.
- To detect off-target effects in the human genome: Map WGS reads to the human genome (hg38) and use mutect2's tumor-normal calling treating the CRISPRed sample as a ‘tumor’ to identify possible variants within the human genome in the sgRNA treated HFFs relative to the control. Use Cas-OFFinder (http://www.rgenome.net/cas-offinder/) in offline mode to look for all predicted off-target sites with 6 or fewer mismatches within hg38 for each sgRNA and determine the overlap between predicted variants and off-target sites.
- To detect off-target effects in the viral (HSV) genome: Align the unmapped reads to the HSV-1 KOS genomic sequence (GenBank: KT899744) and call variants using mutect2's tumor-normal calling. One can then determine the overlap between predicted variants and off-target sites within HSV-1 KOS.
- Functional testing of CRISPR/SaCas9 antiviral activity (see Figure 5A)
- Inhibition of viral protein expression: SDS-PAGE and immunoblotting
- Lyse HFF cells in 1x NuPAGE sample buffer (Life Technologies) with protease inhibitors (cOmplete Protease Inhibitor Cocktail, Millipore Sigma).
- Resolve samples in NuPAGE 4-12% Bis-Tris Gels (Life Technologies).
- Transfer proteins to a nitrocellulose membrane (Bio-Rad).
- Block the membrane in Odyssey Blocking Buffer (LI-COR) for 1 h.
- Incubate the membrane with antibodies specific for individual proteins [anti-ICP4 (1:3,000), anti-ICP8 (1:5,000), anti-ICP27 (1:5,000), anti-UL30 (1:2,000), anti-GAPDH (1:10,000), and anti-HA (1:5,000)] for 1 h at room temperature or overnight at 4 °C.
- Wash membrane 5 x 5 min with PBS-T (PBS with 0.1% Tween 20).
- Incubate the membranes with secondary antibodies, IRDye 680RD (1:15,000, LI-COR) or IRDye 800CW (1:15,000, LI-COR), for 45 min.
- Wash membrane 5 x 5 min with PBS-T (PBS with 0.1% Tween 20).
- Detect near-infrared fluorescence using Odyssey (LI-COR) and quantify protein expression levels using Image J or ImageStudio V4 (LI-COR) (see Figure 5D).
- Inhibition of lytic replication
- Transduction of HFFs with SaCas9/sgRNA:
- Prepare HFF cells at a density of 2 x 105/well in a T25 flask one day prior to transduction
- Transduce with 2-5 ml of lentivirus (or at an MOI of 5) and 3-4 µg/ml of polybrene.
- The next day, replace the transduction medium with fresh medium and incubate cells for additional 2 days at 37 °C followed by puromycin treatment (1 µg/ml) for 7-10 days.
- Lytic infection with wild-type HSV-1
- Infect the transduced cells with wild-type HSV-1 at an MOI of 0.1 or 5 in phosphate-buffered saline (PBS) containing 0.1% glucose (wt/vol), 0.1% heat-inactivated BCS (v/v) for 1 h with shaking at 37 °C.
- Replace virus inoculum with DMEM containing 1% BCS and incubate at 37 °C.
- Harvest cells at various time points for viral replication by qPCR and titer.
- To determine viral titer, harvest media and cells and mix with the same volume of autoclaved powder milk and freeze down at -80 °C (addition of sterilized milk to regular media can stabilize HSV (Blaho et al., 2005).
- To isolate whole DNA (cellular and viral DNAs), isolate genomic DNA using the DNeasy Blood & Tissue kit according to the manufacturer’s protocol.
- Titration of viral yield:
- Thaw harvested cells in media/milk and repeat freeze-thaw cycle two times (3 cycles total).
- Dilute the thawed samples in PBS containing 0.1% glucose (wt/vol), 0.1% BCS (v/v) (10 fold serially up to 6-8 logs) and incubate the serially diluted virus inoculum with the appropriate cell type monolayer, which is prepared one day prior, for 1 h with shaking at 37 °C.
- Replace the virus inoculum with DMEM containing 1% BCS and 0.17% human IgG and incubate at 37 °C for 2-3 days until plaques appear.
- Remove media and fix cells with 100% cold methanol for 10 min.
- Stain monolayer with GIEMSA stain and count plaques (see Figures 5B and 5C)
- Inhibition of reactivation of quiescent virus (see Figure 6A)
- Establish quiescent HSV-1 infection in HFF cells (Ferenczy and DeLuca, 2011):
- Infect HFF cells with the HSV-1 d109 mutant virus at an MOI of 10 in PBS containing 0.1% glucose (wt/vol) and 0.1% BCS (v/v) for 1 h with shaking at 37 °C.
- Incubate at 37 °C for 7-10 days in DMEM supplemented with 10% (v/v) FBS.
- Transduction of HFFs containing quiescent HSV-1 viral genomes with SaCas9/sgRNA
- Transduce the d109 virus-infected cells with 2 or 10 ml of lentivirus (or at a MOI of 5) and 3-4 µg/ml of polybrene in 6-well plates or in T150 flasks.
- The next day, replace the transduction medium with fresh medium and incubate cells for 2 days at 37 °C followed by puromycin treatment (1 µg/ml) for 7-10 days.
- To reactivate quiescent d109 virus, superinfect HFF cells with wild-type HSV-1 at an MOI of 5 in PBS containing 0.1% glucose (wt/vol) and 0.1% BCS (v/v) for 1 h with shaking at 37 °C.
- Replace virus inoculum to DMEM containing 1% BCS for 24 h.
- Harvest cells at 24 hpi and follow the protocol for titration of viral yield in Step C2c above using FO6 and V27 cells.
- To count the number of green plaques, fix the cells with 1% formaldehyde in PBS for 10 min.
- To calculate reactivated d109, count green plaques and subtract the number of GFP-positive plaques on V27 cells from the number of GFP-positive plaques on FO6 cells.
- Reactivation analysis: For the reactivation assay, it is important to include a control for recombination between WT HSV-1 and d109 which could transfer the GFP sequence to WT HSV-1 and result in production of false GFP-positive plaques. We quantify d109 viral reactivation using plaque assays by counting GFP-positive plaques on complementing FO6 cells. FO6 is a Vero-derived cell line expressing ICP4, ICP27, and ICP0 upon HSV-1 infection, thereby complementing replication of d109 virus (Samaniego et al., 1998). To measure these recombinant viruses, we count GFP-positive plaques formed on V27 cells, which express ICP27 (but not ICP4 and ICP0) upon infection with HSV. Because ICP27 is replaced with GFP in HSV-1 d109, any recombinant GFP-positive but ICP27-negative HSV mutants that arise (but not HSV-1 d109), can replicate in V27 cells. To calculate the number of plaques originating from reactivated d109 genomes, we subtract the number of GFP-positive plaques on V27 cells from the number of GFP-positive plaques on FO6 cells (see Figures 6B and 6C).

Figure 1. T7 in vitro transcription of sgRNAs. The 60 nt target-specific oligo contains the T7 promoter sequence followed by 21 nt of the specific S. aureus Cas9 sgRNA DNA binding sequence with the first 2 nucleotides replaced by GG and a 23 nucleotide overlap sequence of the SaCas9 scaffold. The overlap sequence is complementary to the first 23 nt of the (constant) SaCas9 scaffold oligo. The two oligos are annealed and both oligos extended from their 3′ ends by DNA polymerase resulting in a double-stranded DNA template for T7 transcription. RNA polymerase recognizes the double-stranded DNA of the T7 promoter and initiates transcription at the ‘GG’. The resulting sgRNA contains the target-specific CRISPR sequence as well as the RNA scaffold.
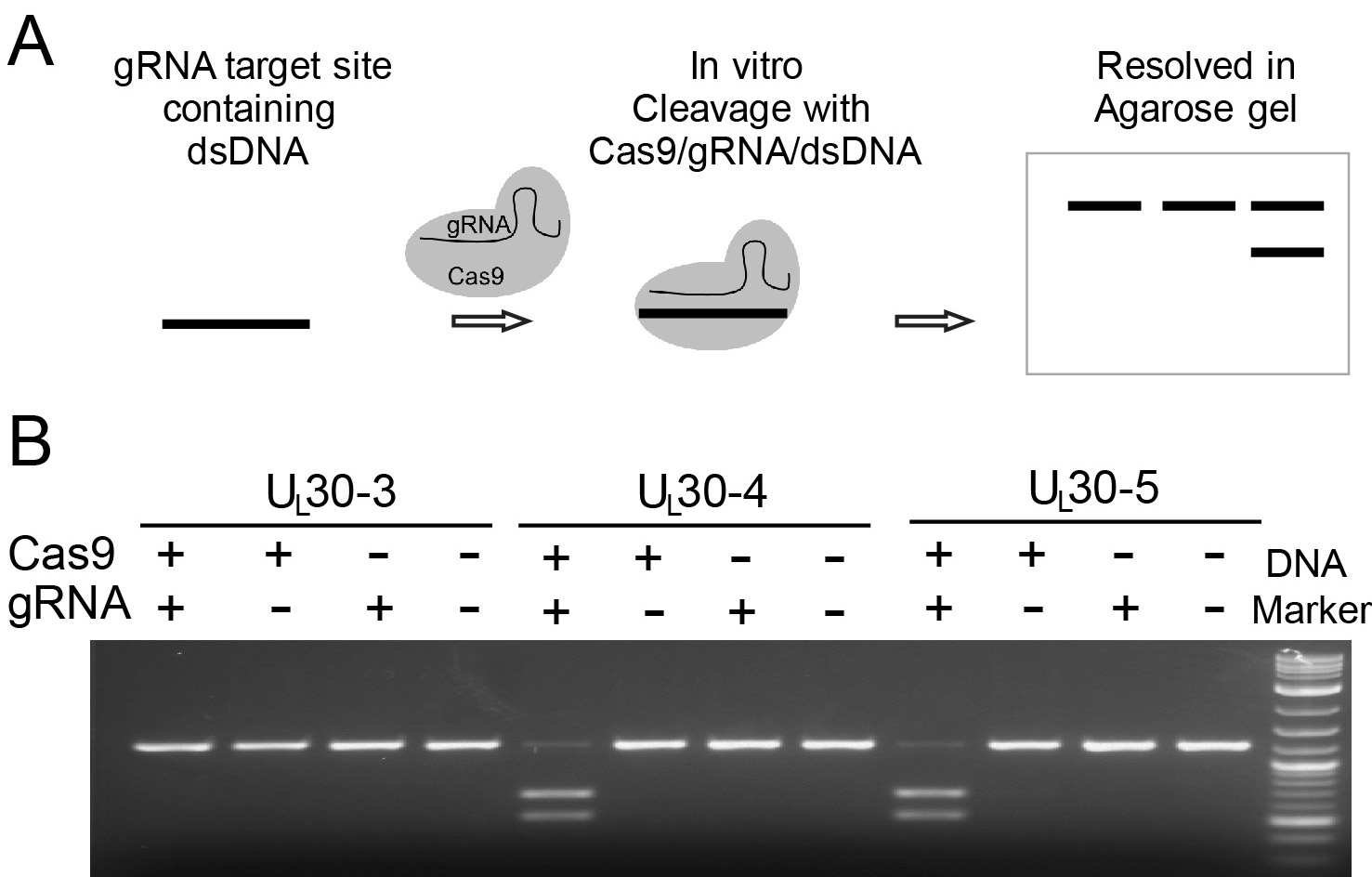
Figure 2. In vitro cleavage assay for the essential HSV-1 gene UL30 (Oh et al., 2019). A. Schematic diagram of in vitro cleavage assay; B. Results are shown for three sgRNAs targeting UL30 (UL30-3, -4, and -5). T7 in vitro transcribed sgRNA was combined with SpCas9 protein and a PCR template containing the CRISPR sequence, incubated 1 h at 37 °C and run on an agarose gel. Lane (1) SpCas9+sgRNA, lane (2) Cas9 only, lane (3) sgRNA only, lane (4) no Cas9/sgRNA. Efficient cutting is seen for UL30-4 and -5 but not UL30-3.

Figure 3. Deep sequencing (MiSeq) process. A diagram of the two step PCR (adapter PCR, barcode PCR). The first step (adapter PCR) uses gene specific primers (black) with extensions (orange) for the second step (barcode PCR). The following sequences are the adaptors that should be added to the gene-specific primers: Adapter sequence for forward primer: 5’-TCTTTCCCTACACGACGCTCTTCCGATCT-target specific sequence-3’, Adapter sequence for reverse primer: 5’-TGGAGTTCAGACGTGTGCTCTTCCGATCT-target specific sequence-3’. The barcode PCR primers anneal to the adapter sequences and contain a barcode (blue) as well as P5/P7 sequences for binding to the flow cell. The three sequencing reads cover the amplicon/target sequence and read the barcode.
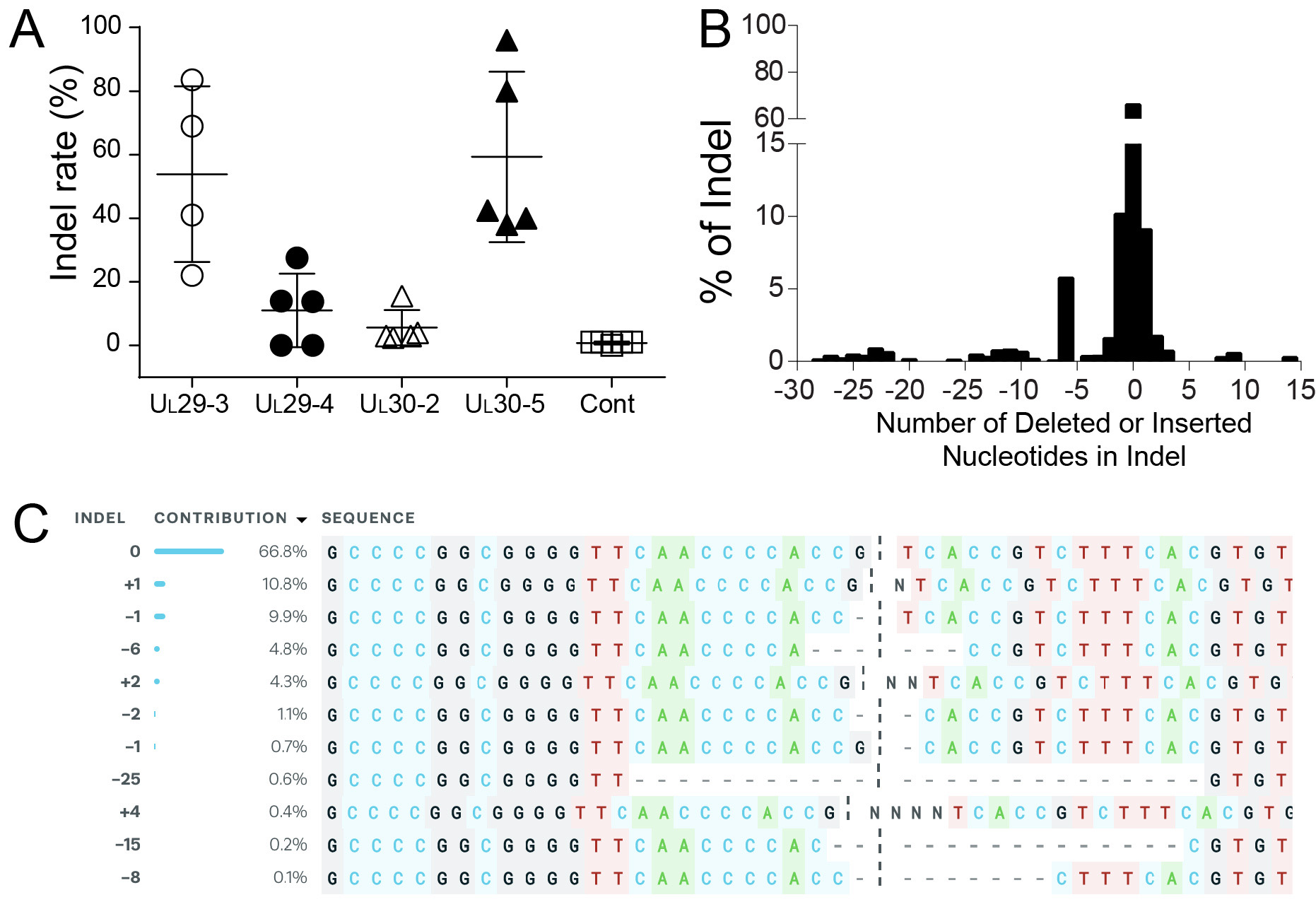
Figure 4. Indel mutations in the HSV-1 genome during the quiescent infection (Oh et al., 2019). A. Indel mutation frequencies of quiescent d109 genomes are shown at the indicated sgRNA target sites. B. Histogram representing the frequency (count) of indel lengths induced by SaCas9/UL30-5 in quiescent d109 genomes. (-) deletions (+) insertions. C. Examples of sequences that show mutations induced by SaCas9/UL30-5 in quiescent d109 viral genomes.
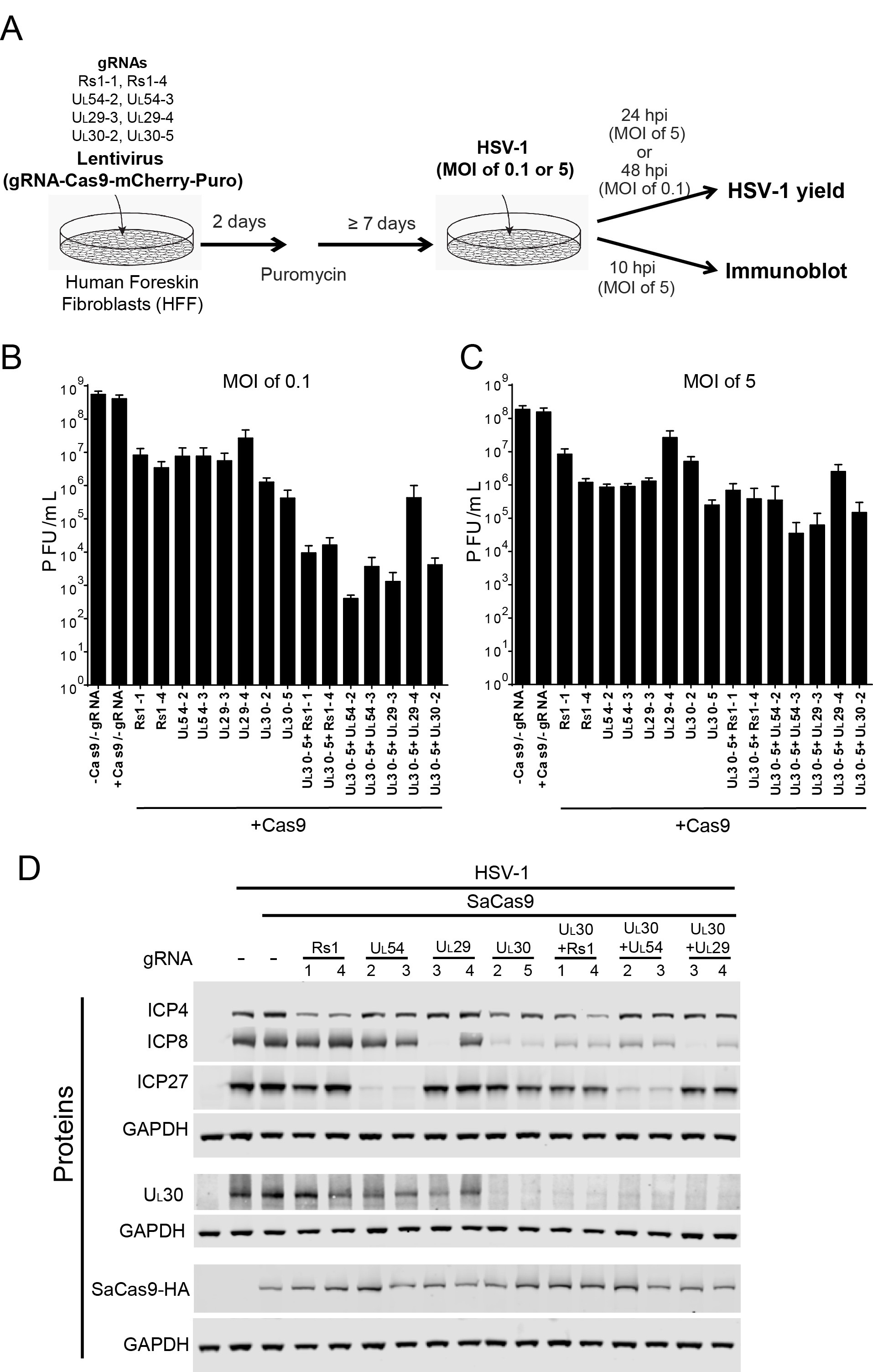
Figure 5. Effect of CRISPR-Cas9 on HSV-1 lytic infection (Oh et al., 2019). A. Experimental scheme of SaCas9/sgRNA-mediated inhibition of HSV lytic infection. B. and C. HFFs transduced with lentivirus expressing SaCas9 and sgRNAs were infected with HSV-1 at an MOI of 0.1 (C) or 5 (D) and harvested at 48 hpi or 24 hpi, respectively. Viral yields were determined by plaque assays. The histogram shows the mean values and standard deviations of biological replicates at an MOI of 0.1 (N = 3) or at an MOI of 5 (N = 4). All the sgRNA added conditions showed statistical significance compared to +Cas9/-gRNA (t-test, *P < 0.05). D. HFFs transduced with lentivirus expressing SaCas9 and sgRNA were infected with HSV-1 at a MOI of 5 and harvested at 10 hpi. Proteins were detected using immunoblotting with antibodies specific for the indicated proteins. Three SDS-PAGE gels loaded with the same amount of proteins were used to detect multiple proteins. Immunoblots of GAPDH were shown as a control under the individual immunoblots.
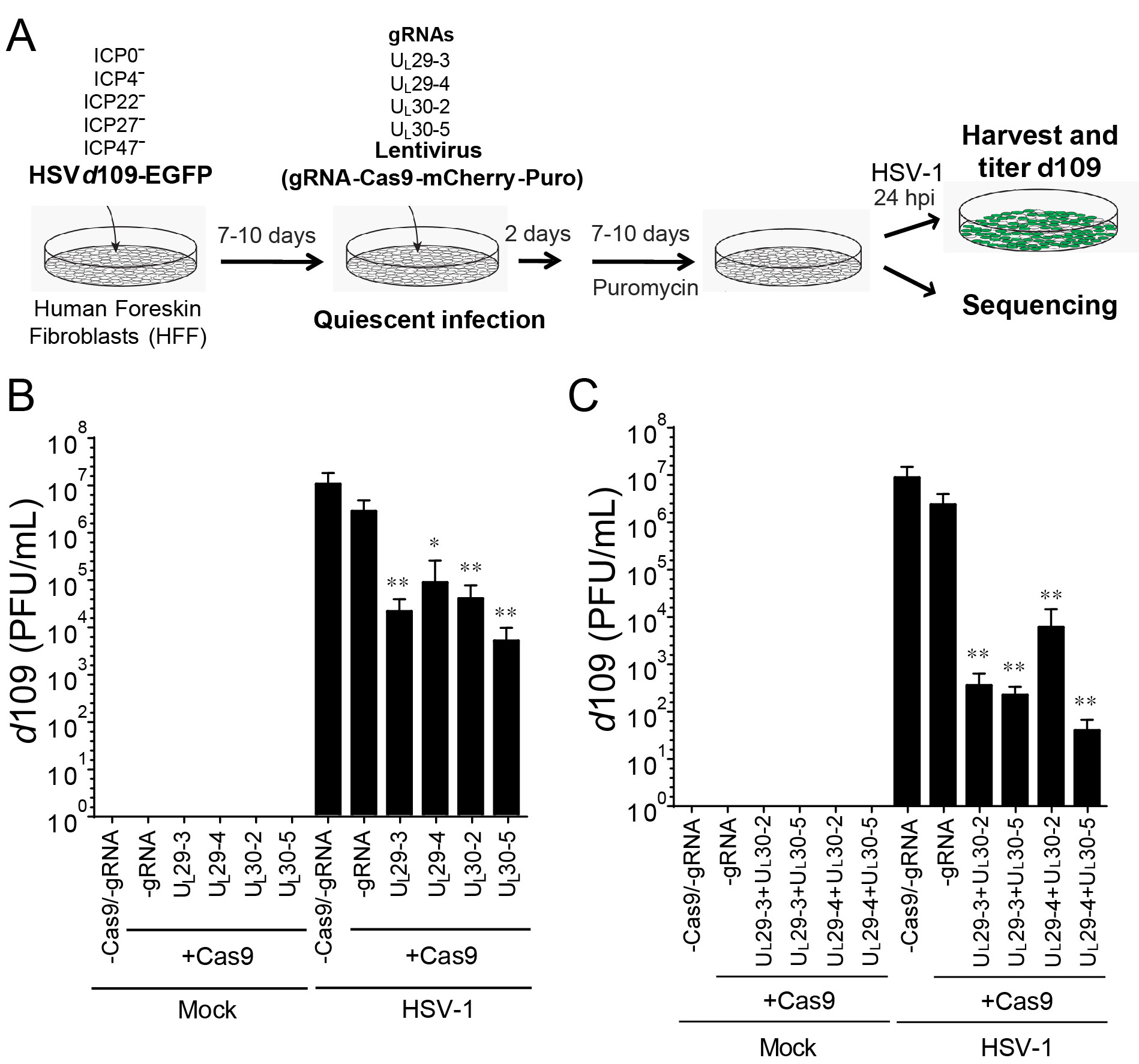
Figure 6. CRISPR/Cas9-induced mutagenesis of quiescent d109 genomes and effect on reactivation (Oh et al., 2019). A. Experimental scheme of SaCas9/sgRNA-mediated inhibition of reactivation of quiescent d109 genomes in HFFs. HFFs were infected with HSV-1 d109 virus to establish quiescent infection for 7-10 days and transduced with lentivirus expressing SaCas9 and sgRNAs for 7-10 days. B. and C. HFF were infected with d109 to establish quiescent infection for 7-10 days and transduced with lentivirus expressing SaCas9 and sgRNAs for 7-10 days. To reactivate quiescent d109 genomes, HFFs were superinfected with WT HSV-1 at an MOI of 5 and harvested at 24 hpi. GFP-positive viral yields were determined by plaque assays on FO6 and V27 cells. The histogram shows the mean values and standard deviations of biological replicates (B and C: N = 5 and N = 7 respectively). All the sgRNA added conditions showed statistical significance compared to +Cas9/-gRNA (Ratio paired t test, *P < 0.05 and **P < 0.01).
Data analysis
Controls: We include a control without sgRNA (SaCas9 only) for all experiments.
Statistical analysis: We use a minimum of three biological replicates (independent experiments that are performed using the same test at different times) to determine statistical significance. We use Prism 6 (Version 6.01) software (GraphPad Software) for statistical analysis. To determine statistical significance, we use Student’s t-test, ratio paired t-test, or one-way ANOVA with Dunnett’s multiple comparison test with two-sided (95% confidence level) (Oh et al., 2019).
Recipes
- Assay buffer (concentrations given are final concentration) 200 mM KCl
- Reaction Stop Buffer (Enzyme Assay) 30% glycerol by weight 15 g
10 mM MgCl2
20 mM Tris pH 8
Store at RT
0.5 M EDTA (6 ml)
2% SDS by weight 1 g
Small amount Bromophenol blue
Bring volume to 50 ml with water and vortex
to mix thoroughly
Store at RT
Acknowledgments
This research was supported by NIH grants AI135423 (KE and DMK) and AI106934 (DK) and a Q-FASTR Award from Harvard University (DMK). This protocol and results are derived from the following original research paper (Oh et al., 2019).
Competing interests
The authors declare no conflicts of interest or competing interests.
References
- Bhaya, D., Davison, M. and Barrangou, R. (2011). CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet 45: 273-297.
- Blaho, J. A., Morton, E. R. and Yedowitz, J. C. (2005). Herpes simplex virus: propagation, quantification, and storage. Curr Protoc Microbiol Chapter 14: Unit 14E 11.
- Chambers, S. M., Qi, Y., Mica, Y., Lee, G., Zhang, X. J., Niu, L., Bilsland, J., Cao, L., Stevens, E., Whiting, P., Shi, S. H. and Studer, L. (2012). Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat Biotechnol 30(7): 715-720.
- Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A. and Zhang, F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121): 819-823.
- Doudna, J. A. and Charpentier, E. (2014). Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346(6213): 1258096.
- Ferenczy, M. W. and DeLuca, N. A. (2011). Reversal of heterochromatic silencing of quiescent herpes simplex virus type 1 by ICP0. J Virol 85(7): 3424-3435.
- Gagnon, J. A., Valen, E., Thyme, S. B., Huang, P., Akhmetova, L., Pauli, A., Montague, T. G., Zimmerman, S., Richter, C. and Schier, A. F. (2014). Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS One 9, e98186.
- Gaj, T., Gersbach, C. A. and Barbas, C. F., 3rd (2013). ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31(7): 397-405.
- Hsu, P. D., Lander, E. S. and Zhang, F. (2014). Development and applications of CRISPR-Cas9 for genome engineering. Cell 157(6): 1262-1278.
- Hsu, P. D., Scott, D. A., Weinstein, J. A., Ran, F. A., Konermann, S., Agarwala, V., Li, Y., Fine, E. J., Wu, X., Shalem, O., Cradick, T. J., Marraffini, L. A., Bao, G. and Zhang, F. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31(9): 827-832.
- Itzhaki, R. F. (2018). Corroboration of a Major Role for Herpes Simplex Virus Type 1 in Alzheimer's Disease. Front Aging Neurosci 10: 324.
- Kimberlin, D. W. (2004). Neonatal herpes simplex infection. Clin Microbiol Rev 17(1): 1-13.
- Kimberlin, D. W., Lin, C. Y., Jacobs, R. F., Powell, D. A., L, F., Gruber, W., Rathore, M., Bradley, J., Diaz, P. S., Kumar, M., Arvin, A. M., Gutierrez, K., Shelton, M., Weiner, L. B., Sleasman, J. W., de Sierra, T. M., Soong, S. J., Lakeman, F. D., Whitley, R. J. and the National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group (2001). Natural history of neonatal herpes simplex virus infections in the acyclovir era. Pediatrics 108: 223-229.
- Labun, K., Montague, T. G., Gagnon, J. A., Thyme, S. B. and Valen, E. (2016). CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res 44(W1): W272-276.
- Liesegang, T. J., Melton, L. J., 3rd, Daly, P. J. and Ilstrup, D. M. (1989). Epidemiology of ocular herpes simplex. Incidence in Rochester, Minn, 1950 through 1982. Arch Ophthalmol 107(8): 1155-1159.
- Liu, B., Chen, S., Rose, A., Chen, D., Cao, F., Zwinderman, M., Kiemel, D., Aissi, M., Dekker, F. J. and Haisma, H. J. (2020). Inhibition of histone deacetylase 1 (HDAC1) and HDAC2 enhances CRISPR/Cas9 genome editing. Nucleic Acids Res 48(2): 517-532.
- Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., Norville, J. E. and Church, G. M. (2013). RNA-guided human genome engineering via Cas9. Science 339(6121): 823-826.
- Oh, H. S., Neuhausser, W. M., Eggan, P., Angelova, M., Kirchner, R., Eggan, K. C. and Knipe, D. M. (2019). Herpesviral lytic gene functions render the viral genome susceptible to novel editing by CRISPR/Cas9. Elife 8: e51662.
- Rice, S. A., Su, L. S. and Knipe, D. M. (1989). Herpes simplex virus alpha protein ICP27 possesses separable positive and negative regulatory activities. J Virol 63(8): 3399-3407.
- Roizman, B., Knipe, D. M. and Whitley, R. J. (2013). Herpes Simplex Viruses. Fields Virology. D. M. Knipe and P. M. Howley. Philadelphia, Lippincott Williams & Wilkins: 1823-1897.
- Samaniego, L. A., Neiderhiser, L. and DeLuca, N. A. (1998). Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J Virol 72(4): 3307-3320.
- Samaniego, L. A., Wu, N. and DeLuca, N. A. (1997). The herpes simplex virus immediate-early protein ICP0 affects transcription from the viral genome and infected-cell survival in the absence of ICP4 and ICP27. J Virol 71(6): 4614-4625.
- Sanjana, N. E., Shalem, O. and Zhang, F. (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11(8): 783-784.
- Schaffer, P., Vonka, V., Lewis, R. and Benyesh-Melnick, M. (1970). Temperature-sensitive mutants of herpes simplex virus. Virology 42(4): 1144-1146.
- Shalem, O., Sanjana, N. E., Hartenian, E., Shi, X., Scott, D. A., Mikkelson, T., Heckl, D., Ebert, B. L., Root, D. E., Doench, J. G. and Zhang, F. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343(6166): 84-87.
- Thompson, C. and Whitley, R. (2011). Neonatal herpes simplex virus infections: where are we now? Adv Exp Med Biol 697: 221-230.
- Yarrington, R. M., Verma, S., Schwartz, S., Trautman, J. K. and Carroll, D. (2018). Nucleosomes inhibit target cleavage by CRISPR-Cas9 in vivo. Proc Natl Acad Sci U S A 115(38): 9351-9358.
- Young, G. T., Gutteridge, A., Fox, H. D., Wilbrey, A. L., Cao, L., Cho, L. T., Brown, A. R., Benn, C. L., Kammonen, L. R., Friedman, J. H., Bictash, M., Whiting, P., Bilsland, J. G. and Stevens, E. B. (2014). Characterizing human stem cell-derived sensory neurons at the single-cell level reveals their ion channel expression and utility in pain research. Mol Ther 22(8): 1530-1543.
Article Information
Copyright
![]() Neuhausser et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Neuhausser et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Neuhausser, W. M., Oh, H. S., Eggan, P., Angelova, M., Kirchner, R., Eggan, K. C. and Knipe, D. M. (2020). Screening Method for CRISPR/Cas9 Inhibition of a Human DNA Virus: Herpes Simplex Virus. Bio-protocol 10(17): e3748. DOI: 10.21769/BioProtoc.3748.
- Oh, H. S., Neuhausser, W. M., Eggan, P., Angelova, M., Kirchner, R., Eggan, K. C. and Knipe, D. M. (2019). Herpesviral lytic gene functions render the viral genome susceptible to novel editing by CRISPR/Cas9. Elife 8: e51662.
Category
Microbiology > Pathogen detection > PCR
Molecular Biology > RNA > RNA detection
Molecular Biology > DNA > Mutagenesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.