- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Multiplication and Growth Inhibition Activity Assays for the Zoonotic Malaria Parasite, Plasmodium knowlesi
Published: Vol 10, Iss 17, Sep 5, 2020 DOI: 10.21769/BioProtoc.3743 Views: 6050
Reviewed by: Paurvi ShindeAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Malaria remains a major cause of morbidity and mortality globally. Clinical symptoms of the disease arise from the growth and multiplication of Plasmodium parasites within the blood of the host. Thus in vitro assays to determine how drug, antibody and genetic perturbations affect the growth rate of Plasmodium parasites are essential for the development of new therapeutics and improving our understanding of parasite biology. As both P. falciparum and P. knowlesi can be maintained in culture with human red blood cells, the effect of antimalarial drugs and inhibitory antibodies that target the invasion or growth capacity of Plasmodium parasites are routinely investigated by using multiplication assays or growth inhibition activity (GIA) assays against these two species. This protocol gives detailed step-by-step procedures to carry out flow cytometry-based multiplication assays and growth inhibition activity assays to test neutralizing antibodies based on the activity of the parasite enzyme lactate dehydrogenase of Plasmodium knowlesi adapted to human red blood cell culture. Whilst similar assays are well established for P. falciparum, P. knowlesi is more closely related to all other human infective species (Pacheco et al., 2018) and so can be used as a surrogate for testing drugs and vaccines for other malaria species such as P. vivax, which is the most widespread cause of malaria outside of Africa, but cannot yet be cultured under laboratory conditions.
Keywords: Plasmodium knowlesiBackground
Plasmodium blood stage parasites are responsible for the clinical symptoms of malaria, including high periodic fever and anemia. Plasmodium merozoites invade red blood cells, grow and multiply intracellularly until the blood cells burst and daughter merozoites are released to infect new red blood cells. The merozoite is briefly extracellular between egress and invasion of red blood cells, and this is a key target for blood stage vaccines as it is directly exposed to antibodies in the blood. Several invasion proteins are in the focus of vaccine development including Plasmodium falciparum merozoite surface protein 1 (MSP1), MSP2, apical membrane antigen 1 (AMA1), reticulocyte binding protein homologue 5 (RH5), erythrocyte binding antigen (EBA-175) and Plasmodium vivax Duffy binding protein (PvDBP) (Genton et al., 2003; Thera et al., 2011; Koram et al., 2016; Payne et al., 2017a and 2017b).
Investigation of antimalarial drugs or inhibitory antibodies that target the blood stages of Plasmodium parasites are carried out by using multiplication or growth inhibition activity (GIA) assays. Flow cytometry-based invasion/multiplication rate assays have previously been described for various Plasmodium species (Basco et al., 1995; Bhakdi et al., 2007; Xie et al., 2007; Izumiyama et al., 2009; Bei et al., 2010; Moon et al., 2013). The advantage of flow cytometry-based assays is that they provide absolute measurements of parasite numbers, so are particularly good for examining changes in parasitemia between different parasite lines. As parasitemia increase only occurs after the release of merozoites from schizonts resulting in invasion and formation of new ring stage parasites–the flow cytometry-based assays are particularly useful for examining this transition. They can be expressed either as % of control, or as an absolute value like fold-growth or parasite multiplication rate.
The activity of the enzyme lactate dehydrogenase (LDH) was first measured as a means to detect the presence of Plasmodium falciparum as a fast alternative to microscopy screening (Makler and Hinrichs, 1993). The LDH assay is based on the fact that Plasmodium LDH can rapidly convert lactate to pyruvate by employing the NAD analog 3-acetylpyridine NAD (APAD) as a coenzyme, whereas human erythrocyte LDH uses APAD instead of NAD at a much smaller rate (200-fold slower). Measuring the malarial LDH activity in the presence of APAD is a specific and sensitive method for the detection of Plasmodium parasites (Basco et al., 1995).
The standardized P. falciparum growth inhibition activity assays based on the LDH activity are used as standard to investigate vaccine candidate antigen activity (Kennedy et al., 2002) and routinely used for analysis of clinical trials with the international GIA Reference Laboratory at the NIH, USA (Malkin et al., 2005). LDH activity assays are often simpler than flow cytometry-based assays, but measure the presence of parasite derived LDH rather than directly measuring the number of infected cells. As such they can only provide relative growth rate values, so are best suited for comparison of multiple treatments of specific parasite lines e.g. for drug or antibody inhibition assays. Relative growth rates are normally expressed as a % of an untreated control.
This protocol describes the methodology for both multiplication assays based on flow cytometry and GIA assays based on LDH activity of Plasmodium blood stage parasites (Figure 1). Here, the focus is on the simian malaria parasite Plasmodium knowlesi, but they can easily be adapted to other Plasmodium species by adjusting incubation times based on the life cycle length and specific culture conditions. P. knowlesi has recently been adapted to grow in human Duffy positive blood (Moon et al., 2013) and is therefore the second human malaria parasite with a long-term in vitro culture system, next to P. falciparum. Due to its close ancestry and biology, it is a suitable model to study invasion genes of P. vivax, which lacks a long-term in vitro culture system.
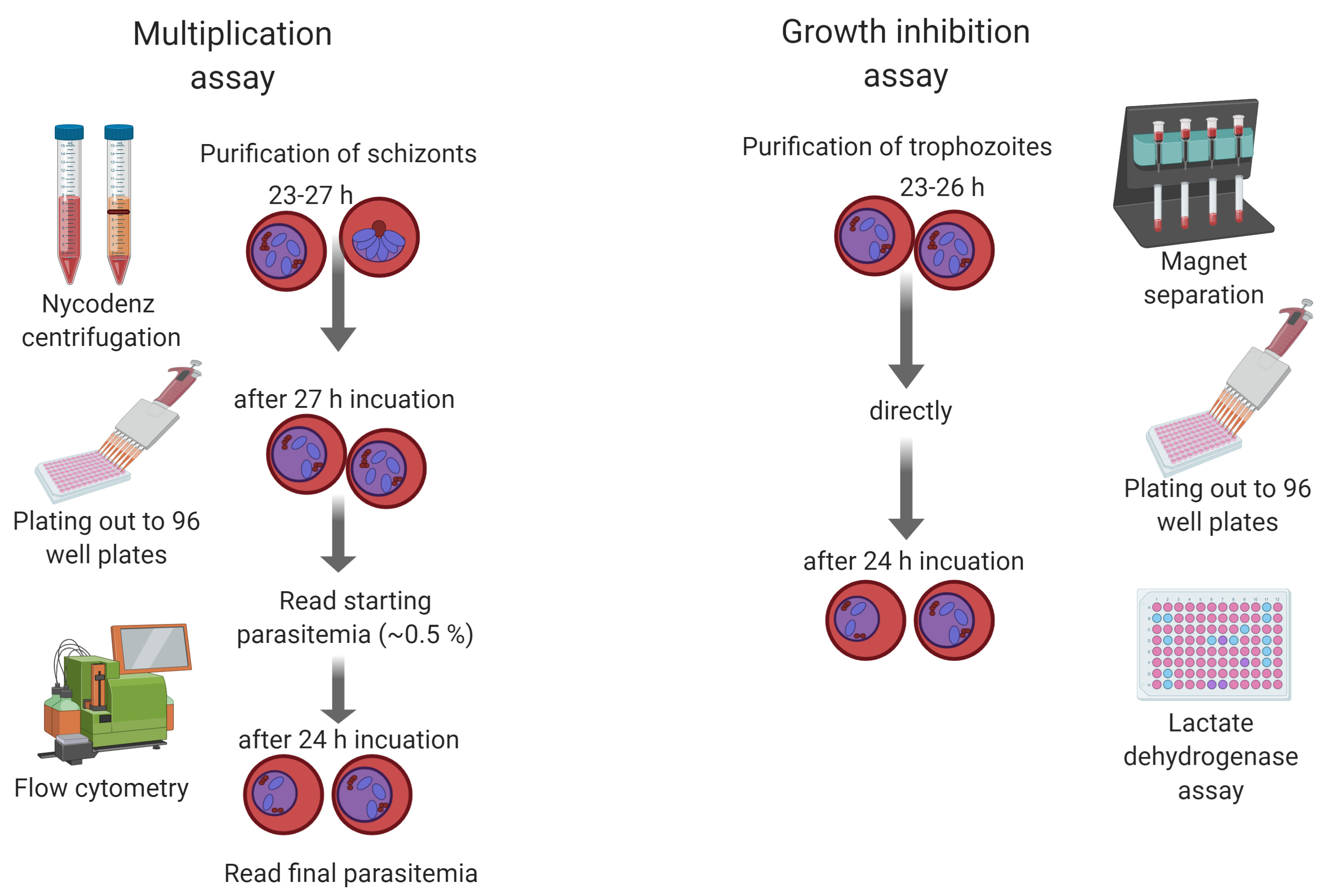
Figure 1. Schematic showing the procedures of multiplication and growth inhibition assays for Plasmodium knowlesi
Materials and Reagents
- 1.5 ml micro centrifuge tube (Eppendorf, catalog number: 0030120086)
- 15 ml centrifuge tube (Falcon, Corning, catalog number: 352196)
- 24-well plates (CytoOne, Starlab, catalog number: CC7672-7524)
- 96 flat-bottom plates (CytoOne, Starlab, catalog number: CC7672-7596)
- 96-well flat/half area tissue culture cluster plates (Corning, catalog number: 3697)
- Aluminium foil
- Plasmodium knowlesi A1-H.1 wild type (Mike Blackman, Francis Crick Institute London) (Moon et al., 2013)
- Duffy positive (Fy+) human red blood cells
- RPMI-1640 Media (Sigma-Aldrich, catalog number: R5886)
- L-glutamine (Sigma, catalog number: G7513-100ML), -20 °C
- Sodium bicarbonate (Sigma-Aldrich, catalog number: S5761)
- Dextrose (Sigma, catalog number: G7021)
- Hypoxanthine (Sigma-Aldrich, catalog number: H9636)
- Albumax II (Gibco, catalog number: 11560376)
- Horse serum (PAN BIOTECH, catalog number: P30-0711), -20 °C
- Nycodenz (Progen, catalog number: 1002424), room temperature
- Paraformaldehyde (Pierce 16% Formaldehyde [w/v]) (Thermo Fisher, catalog number: 28906) room temperature
- PBS tablets (MP BiomedicalsTM, catalog number: MP2810305), room temperature
- Glutaraldehyde Solution Grade I, 25% in water (Sigma-Aldrich, catalog number: G5882-10X1ML)
- Triton X-100 (Roche, Sigma, catalog number: 11332481001)
- Ribonuclease A (MP BiomedicalsTM, catalog number: 0219398050)
- SYBR® Green I nucleic acid gel stain (Life Technologies, Sigma-Aldrich, catalog number: S9430-5ML)
- Tris HCl, Trizma® hydrochloride solution 1 M, pH 8.0, (Sigma-Aldrich, catalog number T3038-1L)
- Sodium L-lactate (Sigma-Aldrich, catalog number: 71718)
- 3-acetylpyridine adenine dinucleotide (APAD) (Sigma-Aldrich, catalog number: A5251)
- Phenazine ethosulfate (Sigma-Aldrich, catalog number: P4544)
- Nitro Blue Tetrazolium tablets (Sigma-Aldrich, catalog number: N5514)
- 4-[7-[(dimethylamino)methyl]-2-(4-fluorphenyl)imidazo[1,2-a]pyridin-3-yl]pyrimidin-2-amine; compound 2 (Michael Blackman, Francis Crick Institute, London, UK)
- Diaphorase from Clostridium klyveri (Sigma-Aldrich, catalog number: D5540-1.5KU)
- Custom Modified RPMI media w/o glutamine (Life Technology Brand, see Recipes), 4 °C
- Fixative: 4% paraformaldehyde with 0.4% glutaraldehyde (see Recipes)
- LDH substrate buffer, pH 7.5 (see Recipes)
- Nitro Blue Tetrazolium (NBT) solution (see Recipes)
- 3-Acetylpyridine Adenine Dinucleotide (APAD) stock solution (10 mg/ml) (see Recipes)
- Diaphorase stock solution 50 units/ml (see Recipes)
Equipment
- Becton Dickenson LSR-II Flow Cytometer or equivalent
- Upright binocular compound light microscope with 100x oil objective
- Multichannel pipette (8-Channel Pipette, 30-300 μl) (ErgoOne®, catalog number: S7108-3300)
- Plate centrifuge (Eppendorf, model: 5810R, catalog number: 5811000660)
- Titramax 100 Flatbed shaker (Heidolph Instruments, catalog number: 544-11200-00)
- Microplate Spectrophotometer Spectra Max 340P
- Class II Microbiological Safety Cabinet
- -20 °C freezer
- -70 °C freezer
Software
- FACSDiva 6.1.3 software
- FlowJo, https://www.flowjo.com/
- Flowing software, http://flowingsoftware.btk.fi/
Procedure
The protocol of thawing Plasmodium knowlesi A1-H.1 parasite depends on the source of parasites and needs to be checked with the person that froze the sample. Parasites are maintained in a flask gassed with a mixture of 90% N2, 5% O2 and 5% CO2 at 37 °C, monitored by microscopy using Giemsa-stained thin films, and parasitaemia maintained at between 0.5% and 10%. All procedures need to be carried out with sterile equipment, materials and reagents with aseptic techniques. Local safety policies must be followed for all work involving human infectious agents.
- Parasite multiplication assay
- Synchronize P. knowlesi parasites via purification with Nycodenz
- Transfer 5 ml of 55% Nycodenz working solution to a 15 ml conical tube and warm up to room temperature (check Note 1).
- Centrifuge down a high parasitemia (4-10%) P. knowlesi 50 ml culture with 2% hematocrit at 900 x g for 4 min at high brake/acceleration at room temperature.
- Resuspend parasite pellet at 50% hematocrit in RPMI.
- Carefully lay 2 ml of this culture onto 5 ml Nycodenz in a 15 ml tube.
- Centrifuge at 900 x g for 12 min with low brake/acceleration (check Note 2).
- Transfer the brownish colored top layer schizonts to a new conical tube and wash with RPMI to remove Nycodenz (see Figure 2).
- Incubate schizonts in culture media with 1 μM Compound 2 for 2-3 h (check Note 3).
- Wash off Compound 2 with RPMI (centrifuge at 900 x g for 4 min at high brake/acceleration at room temperature) and transfer schizonts back to culture (with 2% hematocrit red blood cells). The number of harvested schizonts depends on the starting parasitemia and age of the parasites (check Note 4).
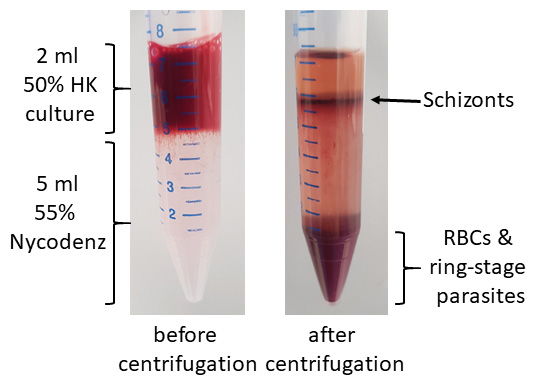
Figure 2. Schizont enrichment with Nycodenz
- In the following cycle, when parasites are again reaching the schizont stage, set up the multiplication assay in a 96-well plate (see Figure 3 for the plate layout).
- Fill out all outer wells in the 96-well plate with 100 μl of RPMI or sterile water. This ensures inner wells are not affected by evaporation.
- Plate out 75 μl complete growth media alone (or with vehicle control) or with 2x concentration of drug or antibody to be tested. Plate out 75 µl of the parasite culture per well as indicated in the plate layout. The plate layout can be customized depending on the individual experiment (see Figure 1 for plate layout).
- Prepare a culture with young schizonts at around 1% parasitemia and 4% hematocrit.
- Purify synchronous schizonts with Nycodenz as described in Step A1.
- Resuspend parasite pellet with 1 pellet-volume complete media to 50% hematocrit.
- Prepare complete media with 4% red blood cells (e.g., 5 ml media + 200 μl red blood cells).
- Transfer 1% schizonts to media + 4% red blood cells (around 4-6 μl of 50% schizonts to 5 ml, depending on the parasitemia of enriched schizonts that is usually between 80 and 90%) and mix well.
- Confirm the parasitemia by counting at least 400 cells of a blood smear.
- Add 75 μl of the culture to each well that contains 75 μl media and mix by pipetting.
- Plate out 150 μl of uninfected red blood cells (2% hematocrit) as a control.
- Mix by pipetting up and down and transfer 50 μl of each well to a new 96-well plate. Carry out Step A3. if the parasites can be measured with FACS within the next 30 min or with Step A4. if parasites are fixed and measured up to 1 week later (check Notes 5, 6, 7).
- Incubate the 96-well plate with 100 μl parasite cultures for one growth cycle (24 h) under standard parasite culture conditions (37 °C, 3% O2 and 3% CO2 and 94% Nitrogen).
- After 24 h incubation, mix each well and transfer 50 μl of the remaining 100 μl culture to a new 96-well plate. This is plate 2 (final parasitemia). Continue with Step A3. if the parasites can be measured with FACS within the next 30 min or with Step A4. if parasites are fixed and measured up to 1 week later (check Notes 5, 6, 7).
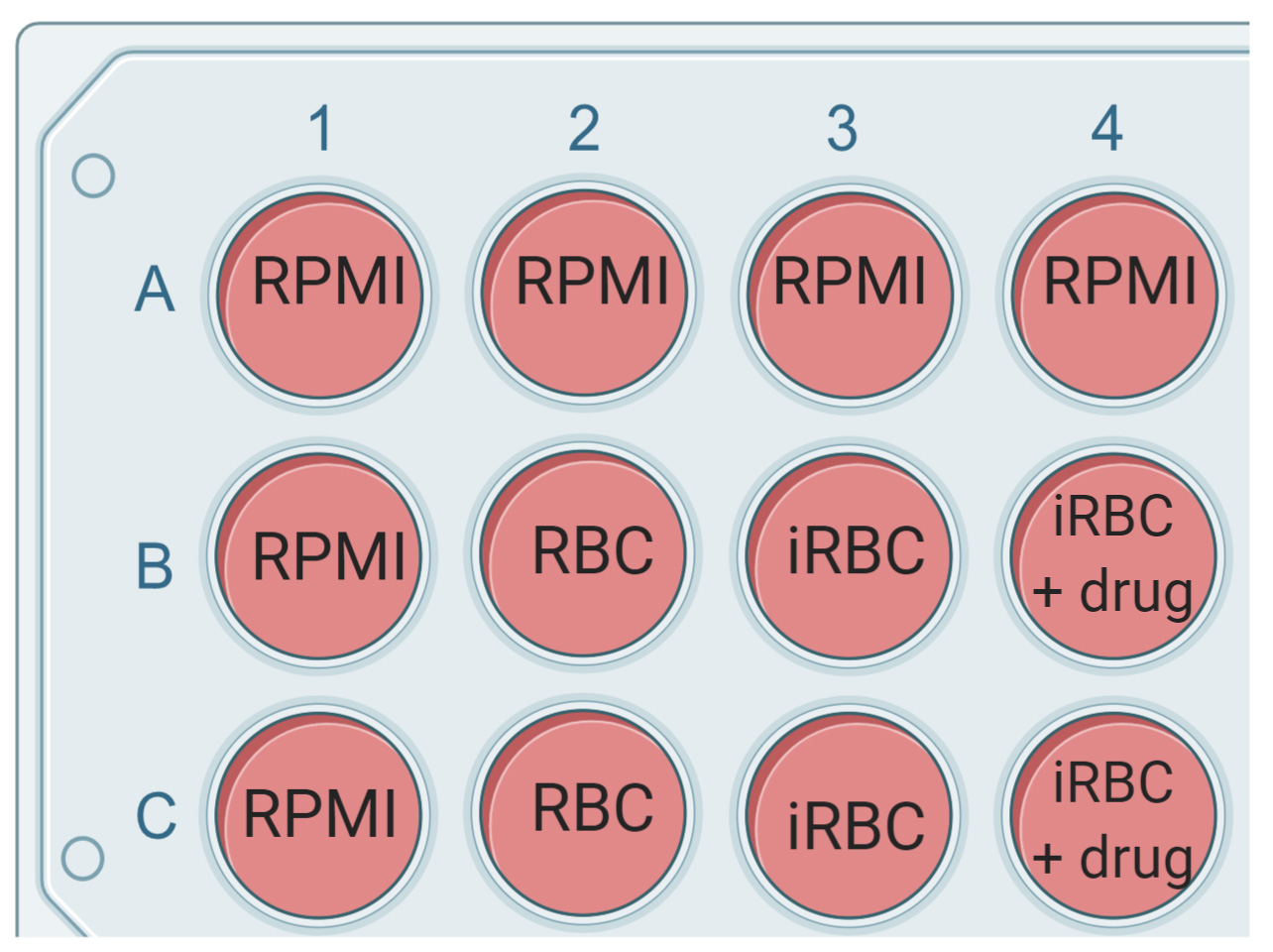
Figure 3. Example of the plate layout
- Prepare samples for flow cytometry for live cell flow cytometry
- Prepare filtered PBS and a 1:5,000 dilution of SYBR Green I in PBS.
- Add 50 μl of SYBR Green I dilution to the 50 μl of parasite culture and incubate for exactly 30 min at room temperature.
- Dilute stained cultures 1:5 in filtered PBS by adding 40 μl of stained cells to 160 μl of PBS and run on a FACS machine.
- Prepare samples for fixed cell flow cytometry
- Prepare all buffers in filtered PBS [0.3% (v/v) Triton X-100, 0.5 mg/ml ribonuclease A, 1:10,000 dilution of SYBR Green I)].
- Add 50 μl of fixative to 50 μl of parasite culture and incubate at 4 °C for at least 1 h or overnight.
- Centrifuge down plates for 4 min, at 4 °C and 763 x g in bench top centrifuge for plates with medium brake/acceleration settings.
- Remove the supernatant and resuspend the cell pellet with 100 μl PBS and spin down again.
- Remove the supernatant and resuspend the cell pellet with 100 μl of PBS with 0.3% (v/v) Triton X-100 and incubate for 10 min at room temperature.
- Wash twice with 100 μl PBS.
- Remove the supernatant and resuspend the pellet with 100 μl 0.5 mg/ml ribonuclease A in PBS and incubate for 1 h at 37 °C.
- Wash with 100 μl PBS.
- Remove as much PBS as possible and resuspend pellet with 100 μl 1:10,000 SYBR Green in PBS and incubate for exactly 30 min at room temperature (always keep SYBR Green tube and stained cells protected from light and keep SYBR GREEN I incubation consistent for all samples).
- Resuspend cells again and dilute 1:5 in PBS (Transfer 40 μl to a new 96-well plate with 160 μl PBS. No need to wash off SYBR Green I).
- Keep plates at 4 °C and protected from light. Measure parasitemia by FACS on the same day.
- Record events by flow cytometry
- For the Becton Dickenson LSR-II the following Voltages are used:
FSC 170 log
SSC 209 log
Green laser 530/30 488 F 360 log - Record 50,000-100,000 cells/treatment group.
- Export data to fsc file format.
- For the Becton Dickenson LSR-II the following Voltages are used:
- Analyze the flow cytometry data
- Upload the fsc files to FlowJo, Flowing or a comparable flow cytometry software.
- Open Dot Plots for each well of the 96-well plate.
- Within the Dot Plot of the RBC only control, generate a gate for RBCs (R1) that excludes other blood cells or broken red cells. X-axis is FSC-A and y-axis is SSC-A (see Figure 2).
- Open a Histogram for R1 and generate a gate for parasites (H-2). X-axis is SYBR-Green signal 530/30 488F-A and y-axis is number of cells. Compare RBC only control with a parasite containing well to find the right areas on the x-axis where RBCs and parasites need to be separated (Figure 4). Use the same gates for all wells.
- Save data as xls file and open it with excel.
- Calculate ratios (H-2/R1*100) and subtract the ratio of RBC only control. Calculate the ratio of End (24 h incubation, plate 2) and Start (0 h incubation, plate 1) to determine the multiplication rate (Example in Figure 5).
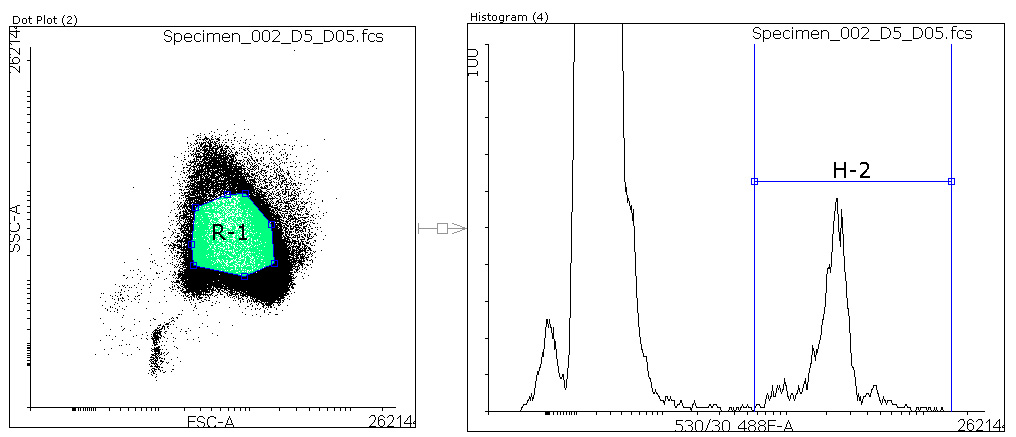
Figure 4. Gating of flow cytometry data. Within the Dot Plot a region (R1, green) defines red blood cells. All cells within that region are analyzed within a Histogram. In the Histogram a region (H-2) is defined to separate parasites from uninfected red blood cells (RBCs). The ratio of Parasite/total RBCs ((H-2/R1)*100) is calculated to give the parasitemia.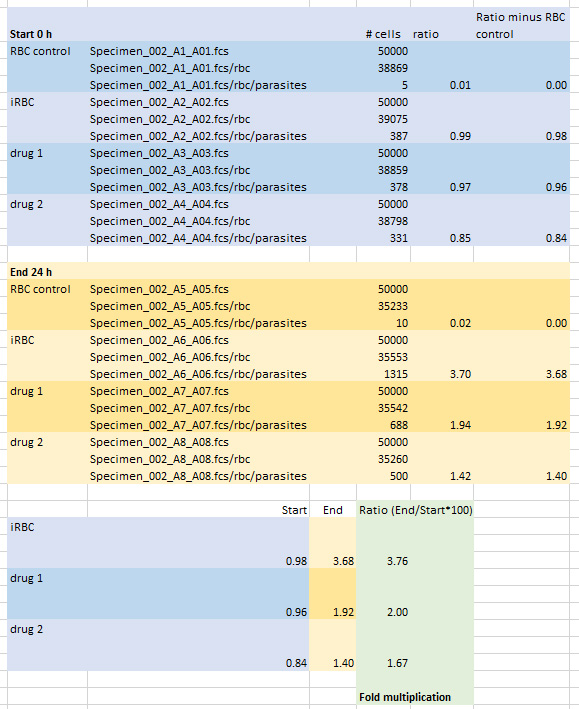
Figure 5. Calculation of Multiplication rates
- Synchronize P. knowlesi parasites via purification with Nycodenz
- Parasite growth inhibition activity (GIA) assay
- Set up GIA assay in 96-well plate flat/half area tissue cluster culture plates
- Plates should include: Triplicate wells of each test IgG concentration
- Plate out 20 μl of purified IgG (check Note 8) in 2x concentration in incomplete media to wells. Use serial 1:2 dilutions of immune serum or pre-immune serum of around eight final concentrations, depending on the EC50 value (for example 10, 5, 2.5, 1.25, 0.625, 0.312, 0.15 and 0.075 mg/ml). Set up each treatment in triplicate test wells.
- Enrich P. knowlesi parasites via Nycodenz purification (see Step A1) or by magnetic separation (Ribaut et al., 2008).
- Prepare 20 ml cell culture at 1.5 % parasitemia in 4% red blood cell suspension in 2 x warm complete media (e.g., 20 ml media + 800 μl red blood cells + 12 μl trophozoites (~1.2 x 109).
- Confirm parasitemia by counting of blood smear.
- Add 20 μl of culture with 4% hematocrit and 1.5% parasitemia trophozoites or only uninfected RBCs to the IgG plates using a multichannel pipette. Change tips for each set of triplicate samples.
- Incubate for one cycle (in P. knowlesi 26-27 h) in standard culture conditions, together with a tracking culture (5 ml cell culture + 5 ml media in culture flask) to check parasitemia and life cycle stage.
- Before harvest check parasitemia (~ 4%) and life cycle stage (trophozoites are ideal, young parasites don’t have adequate LDH levels) of tracking culture.
- GIA assay harvest
- Add 100 μl of cold PBS to each well.
- Spin down plates at 1,300 x g for 4 min with maximum acceleration/break setting.
- Aspirate 100 μl of supernatant from all wells. Do not aspirate any RBCs. Tilting the plate away from the RBC pellet will help.
- Repeat the Steps a to conce.
- Freeze at -80 °C if the assay is not going to be performed immediately–but this is not necessary otherwise.
- Measure parasitemia with the lactate dehydrogenase(LDH) activity assay
- Have NBT solution in LDH buffer ready and warm to room temperature.
- If plates have been frozen, thaw them at room temperature for at least 30 min. Plates need to be uniformly warmed to room temperature. If the plates have not been frozen, it is important that RBCs are uniformly resuspended prior to assay e.g. shake plate on Titramax at max settings for 1 min and check if RBC pellets have fully disappeared (check Note 9).
- Prepare complete LDH substrate by adding 50 μl of 3-Acetylpyridine Adenine Dinucleotide (APAD) stock and 200 μl of Diaphorase stock to every 10 ml of NBT solution. After all reagents are combined, use prepared LDH substrate immediately.
- Start the timer and add 120 μl of complete LDH substrate to all wells. Avoid bubbles (fully depress the plunger of the multichannel pipette before picking up the first aliquot of substrate, pipette should draw up extra substrate and no air will be ejected. Pipette down at the edge of the wells).
- Incubate plate covered with aluminium foil to protect from light and measure absorbance at 650 nm with a 96-well Microplate Spectrophotometer every 5 to 10 min until OD reaches 0.4-0.6 in iRBC and ICM control (0% GIA).
- Calculate percent inhibition: 100 – [(A650 of immune sample – A650 of RBCs only)/(A650 of pre-immune control – A650 of RBCs only) x 100].
Triplicate wells of positive control
6 x wells with infected erythrocytes and media only (0% GIA control)
6 x wells with uninfected erythrocytes at 2% hematocrit or iRBCs + 5 mM EDTA (100% GIA control / background)
3 x wells with non-malarial human antibody as a negative control
Notes
- 5 ml Nycodenz tube is required to purify parasites from 1 ml packed red blood cells at 4-10% parasitemia (e.g., 50 ml culture maintained at 2% hematocrit and 4-10% parasitemia will yield 1 ml of packed red blood cells after centrifugation). Add 1 ml of medium to 1 ml of packed red blood cells to make a 2 ml culture with 50% hematocrit.
- Uninfected red cells and ring-stage parasites will sink to the bottom and schizonts form a layer on top of the Nycodenz.
- Compound 2 is a PKG inhibitor that reversibly blocks merozoite egress. This step is optional but will help to maximize yield of late schizonts and also provides the user with some flexibility in timing for subsequent steps. Viability of parasites will dramatically decline for incubations longer than 3 h. As an alternative to Compound 2 a highly specific and potent derivative of Compound 2, referred to as ML10, can be used as well, which is available from LifeArc (Ressurreição et al., 2020).
- To get parasites even more synchronized let them invade red blood cells for 30 to 60 min and purify with Nycodenz again, only this time keep the ring stage parasite pellet and remove the schizonts. You can slow down the aging of the parasites by leaving them at room temperature for several h in order to get them to the schizont stage at a convenient time for the next purification.
- Set up all experiments in three biological replicates (different days, parasite preparations, and RBCs).
- For parasite multiplication assays, setting up with purified schizonts is critical, as this removes residual uninfected RBCs from the parasites and enables us to examine differences in multiplication in different host RBCs (e.g., comparing human and macaque RBC invasion).
- Whilst the initial starting parasitemia should be fixed for all samples, it is still critical to obtain a timepoint 0 for parasitemia (i.e., plate 1 for multiplication assay). This is because small variations in starting parasitemia can have a significant effect on the calculated growth rate–using different RBCs between samples can also alter the precise starting parasitemia.
Whilst here we use a second time point of 24 h, various other timepoints could be used depending on experimental aims. A timepoint of 24 h works well because all parasites would have undergone reinvasion and progressed to schizonts again, which are very easily identified using the flow cytometry assay. The downside of this is that both invasion efficiency and viability of developing rings and trophozoites are measured. Shorter timepoints (e.g., 2-6 h) can be used to more specifically look at invasion and early ring formation, but these require very synchronous and late stage schizonts, as any schizonts that have not yet progressed to form new rings at the specified timepoint will affect interpretation of the data.
- Parasites that are fixed become aggregated and settle more quickly to the bottom of the plate, otherwise there are no noticeable effects between fixed and unfixed samples.
- Assays can also be set up in 24-well plates with final volume of 1 ml.
- For polyclonal antibodies; purified IgG from serum is typically used and no background inhibition is usually seen from negative control samples (N.B. mouse IgG can sometimes be problematic in P. falciparum GIA assays; but all other species tested have been OK), purified IgG from plasma cannot typically be used as the anticoagulant (EDTA or heparin) cannot be fully removed even with IgG purification, leading to background GIA. P. falciparum GIA assays using diluted serum are reported, but are less common. Monoclonal antibodies work well when purified.
- If necessary, use a pipette to resuspend, but this poses the risk of introducing bubbles. Do not vortex).
Recipes
- Fixative: 4% paraformaldehyde with 0.4% glutaraldehyde
10x PBS 5 ml
16% Formaldehyde (w/v) from Pierce 12.5 ml
Glutaraldehyde 125 μl
H2O 32.5 ml
Final volume 50 ml - LDH substrate buffer, pH 7.5
- To prepare 500 ml of the buffer, mix 50 ml of 1 M Tris HCI (pH 8.0) and 450 ml H2O
- Add 2.8 g sodium L-Lactate
- Add 1.25 ml Triton X-100
- Mix on a magnetic stirrer at room temperature for at least 30 min
- Make 50 ml aliquots and freeze at -20 °C
- Nitro Blue Tetrazolium (NBT) solution
- Remove one 50 ml aliquot of LDH buffer from the freezer
- Warm up to room temperature.
- In a 50 ml tube, add 50 ml of LDH buffer to one NBT tablet (10 mg)
- Cover the tube with aluminium foil and leave in fridge overnight to dissolve
- Do not shake! Mix gently
- Prepared solution can be kept at 4 °C up to 3 weeks in the dark (covered with aluminium foil)
- 3-Acetylpyridine Adenine Dinucleotide (APAD) stock solution (10 mg/ml)
- To prepare 10 ml of APAD stock solution, dissolve 100 mg of APAD in 10 ml of distilled water
- Store the APAD stock solution in 50 μl aliquots in PCR tubes at -20 °C
- Diaphorase stock solution 50 units/ml
To prepare Diaphorase stock solution, dissolve the 1,500 units Diaphorase vial contents in 30 ml of distilled water. Store 200 μl aliquots at -20 °C - Complete media
RPMI-1640 (HEPES Modification, 25 mM HEPES, without L-glutamine, Merck) with the following additions:
2.3 g/L sodium bicarbonate (Sigma)
2 g/L dextrose (Sigma)
0.05 g/L Hypoxanthine (Sigma)
5 g/L Albumax II (Gibco)
0.3 g/L L-glutamine (10 ml of 200 mM solution pro 1 L media, Merck)
10% (vol/vol) horse serum
Sterile filter and store at 4 °C
Acknowledgments
This work is supported by an MRC Career Development Award (MR/M021157/1) jointly funded by the UK Medical Research Council and Department for International Development (R.W.M, F.M), (Basco et al., 1995; Mohring et al., 2019). This work was supported in-part by funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement for MultiViVax [number 733073]. T.A.R. held a Wellcome Trust Research Training Fellowship [108734/Z/15/Z]; and S.J.D. is a Jenner Investigator; a Lister Institute Research Prize Fellow and a Wellcome Trust Senior Fellow [106917/Z/15/Z]. We thank Amelia Lias and Don Van Schalkwyk for their advice on the manuscript.
Competing interests
The authors declare no competing financial interests.
Ethics
The project, consent and protocol were approved by the LSHTM Observational Research Ethics Committee under project reference 5520-1.
References
- Basco, L. K., Marquet, F., Makler, M. M. and Le Bras, J. (1995). Plasmodium falciparum and Plasmodium vivax: lactate dehydrogenase activity and its application for in vitro drug susceptibility assay. Exp Parasitol 80(2): 260-271.
- Bei, A. K., Desimone, T. M., Badiane, A. S., Ahouidi, A. D., Dieye, T., Ndiaye, D., Sarr, O., Ndir, O., Mboup, S. and Duraisingh, M. T. (2010). A flow cytometry-based assay for measuring invasion of red blood cells by Plasmodium falciparum. Am J Hematol 85(4): 234-237.
- Bhakdi, S. C., Sratongno, P., Chimma, P., Rungruang, T., Chuncharunee, A., Neumann, H. P., Malasit, P. and Pattanapanyasat, K. (2007). Re-evaluating acridine orange for rapid flow cytometric enumeration of parasitemia in malaria-infected rodents. Cytometry A 71(9): 662-667.
- Genton, B., Al-Yaman, F., Betuela, I., Anders, R. F., Saul, A., Baea, K., Mellombo, M., Taraika, J., Brown, G. V., Pye, D., Irving, D. O., Felger, I., Beck, H. P., Smith, T. A. and Alpers, M. P. (2003). Safety and immunogenicity of a three-component blood-stage malaria vaccine (MSP1, MSP2, RESA) against Plasmodium falciparum in Papua New Guinean children. Vaccine 22(1): 30-41.
- Izumiyama, S., Omura, M., Takasaki, T., Ohmae, H. and Asahi, H. (2009). Plasmodium falciparum: development and validation of a measure of intraerythrocytic growth using SYBR Green I in a flow cytometer. Exp Parasitol 121(2): 144-150.
- Kennedy, M. C., Wang, J., Zhang, Y., Miles, A. P., Chitsaz, F., Saul, A., Long, C. A., Miller, L. H. and Stowers, A. W. (2002). In vitro studies with recombinant Plasmodium falciparum apical membrane antigen 1 (AMA1): production and activity of an AMA1 vaccine and generation of a multiallelic response. Infect Immun 70(12): 6948-6960.
- Koram, K. A., Adu, B., Ocran, J., Karikari, Y. S., Adu-Amankwah, S., Ntiri, M., Abuaku, B., Dodoo, D., Gyan, B., Kronmann, K. C. and Nkrumah, F. (2016). Safety and immunogenicity of EBA-175 RII-NG malaria vaccine administered intramuscularly in semi-immune adults: A Phase 1, double-blinded placebo controlled dosage escalation study. PLoS One 11(9): e0163066.
- Makler, M. T. and Hinrichs, D. J. (1993). Measurement of the lactate dehydrogenase activity of Plasmodium falciparum as an assessment of parasitemia. Am J Trop Med Hyg 48(2): 205-210.
- Malkin, E. M, Diemert, D. J. McArthur, J. H., Perreault, J. R., Miles, A. P., Giersing, B. K., Mullen, G. E., Orcutt, A., Muratova, O., Awkal, M., Zhou, H., Wang, J., Stowers, A., Long, C.A., Mahanty S., Miller, L.H., Saul, A. and Durbin, A.P. (2005). Phase 1 clinical trial of apical membrane antigen 1: an asexual blood-stage vaccine for Plasmodium falciparum malaria. Infect Immun 73(6):3677-85.
- Mohring, F., Hart, M. N., Rawlinson, T. A., Henrici, R., Charleston, J. A., Diez Benavente, E., Patel, A., Hall, J., Almond, N., Campino, S., Clark, T. G., Sutherland, C. J., Baker, D. A., Draper, S. J. and Moon, R. W. (2019). Rapid and iterative genome editing in the malaria parasite Plasmodium knowlesi provides new tools for P. vivax research. Elife 8: 45829.
- Moon, R. W., Hall, J., Rangkuti, F., Ho, Y. S., Almond, N., Mitchell, G. H., Pain, A., Holder, A. A. and Blackman, M. J. (2013). Adaptation of the genetically tractable malaria pathogen Plasmodium knowlesi to continuous culture in human erythrocytes. Proc Natl Acad Sci U S A 110(2): 531-536.
- Pacheco, M. A., Matta, N. E., Valkiunas, G., Parker, P. G., Mello, B., Stanley, C. E., Lentino, M., Garcia-Amado, M. A., Cranfield, M., Kosakovsky Pond, S. L. and Escalante, A. A. (2018). Mode and rate of evolution of haemosporidian mitochondrial genomes: timing the radiation of avian parasites. Mol Biol Evol 35(2):383-403.
- Payne, R. O., Silk, S. E., Elias, S. C., Miura, K., Diouf, A., Alanine, D., Jin, J., Labbe, G., Brian, I., Poulton, I., Griffiths, O., Edwards, N., Berrie, E., Siani, L., Douglas, A., Roberts, R., Vekemans, J., Nugent, F., Hill, A. V., Long, C., Lawrie, A. M. and Draper, S. J. (2017a). Safety and immunogenicity of the novel Plasmodium falciparum blood-stage vaccine chad63-mva rh5 in a phase ia clinical trial. Am J Trop Med Hyg 95(5): 313-313.
- Payne, R. O., Silk, S. E., Elias, S. C., Milne, K. H., Rawlinson, T. A., Llewellyn, D., Shakri, A. R., Jin, J., Labbe, G. M., Edwards, N. J., Poulton, I. D., Roberts, R., Farid, R., Jorgensen, T., Alanine, D. G., de Cassan, S. C., Higgins, M. K., Otto, T. D., McCarthy, J. S., de Jongh, W. A., Nicosia, A., Moyle, S., Hill, A. V., Berrie, E., Chitnis, C. E., Lawrie, A. M. and Draper, S. J. (2017b). Human vaccination against Plasmodium vivax Duffy-binding protein induces strain-transcending antibodies. JCI Insight 2(12): e93683.
- Ressurreição, M., Thomas J.A., Nofal S.D., Flueck, C., Moon, R.W., Baker, D.A., van Ooij, C. (2020). Use of a highly specific kinase inhibitor for rapid, simple and precise synchronization of Plasmodium falciparum and Plasmodium knowlesi asexual stage parasites. bioRxiv. doi.org/10.1101/2020.04.24.059493.
- Ribaut, C., Berry, A., Chevalley, S., Reybier, K., Morlais, I., Parzy, D., Nepveu, F., Benoit-Vical, F. and Valentin, A. (2008). Concentration and purification by magnetic separation of the erythrocytic stages of all human Plasmodium species. Malar J 7: 45.
- Thera, M. A., Doumbo, O. K., Coulibaly, D., Laurens, M. B., Ouattara, A., Kone, A. K., Guindo, A. B., Traore, K., Traore, I., Kouriba, B., Diallo, D. A., Diarra, I., Daou, M., Dolo, A., Tolo, Y., Sissoko, M. S., Niangaly, A., Sissoko, M., Takala-Harrison, S., Lyke, K. E., Wu, Y., Blackwelder, W. C., Godeaux, O., Vekemans, J., Dubois, M. C., Ballou, W. R., Cohen, J., Thompson, D., Dube, T., Soisson, L., Diggs, C. L., House, B., Lanar, D. E., Dutta, S., Heppner, D. G., Jr. and Plowe, C. V. (2011). A field trial to assess a blood-stage malaria vaccine. N Engl J Med 365(11): 1004-1013.
- Xie, L., Li, Q., Johnson, J., Zhang, J., Milhous, W. and Kyle, D. (2007). Development and validation of flow cytometric measurement for parasitaemia using autofluorescence and YOYO-1 in rodent malaria. Parasitology 134(Pt 9): 1151-1162.
Article Information
Copyright
![]() Mohring et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Mohring et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Mohring, F., Rawlinson, T. A., Draper, S. J. and Moon, R. W. (2020). Multiplication and Growth Inhibition Activity Assays for the Zoonotic Malaria Parasite, Plasmodium knowlesi. Bio-protocol 10(17): e3743. DOI: 10.21769/BioProtoc.3743.
- Mohring, F., Hart, M. N., Rawlinson, T. A., Henrici, R., Charleston, J. A., Diez Benavente, E., Patel, A., Hall, J., Almond, N., Campino, S., Clark, T. G., Sutherland, C. J., Baker, D. A., Draper, S. J. and Moon, R. W. (2019). Rapid and iterative genome editing in the malaria parasite Plasmodium knowlesi provides new tools for P. vivax research. Elife 8: 45829.
Category
Immunology > Antibody analysis > Antibody function
Cell Biology > Cell-based analysis > Flow cytometry
Cell Biology > Cell viability > Cell proliferation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.