- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

An in vitro DNA Sensor-based Assay to Measure Receptor-specific Adhesion Forces of Eukaryotic Cells and Pathogens
(*contributed equally to this work) Published: Vol 10, Iss 17, Sep 5, 2020 DOI: 10.21769/BioProtoc.3733 Views: 5869
Reviewed by: Amberley D. StephensYINNIAN FENGVikash Verma
Original research article
The authors used this protocol in:

Jan 2020
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Motility of eukaryotic cells or pathogens within tissues is mediated by the turnover of specific interactions with other cells or with the extracellular matrix. Biophysical characterization of these ligand-receptor adhesions helps to unravel the molecular mechanisms driving migration. Traction force microscopy or optical tweezers are typically used to measure the cellular forces exerted by cells on a substrate. However, the spatial resolution of traction force microscopy is limited to ~2 µm and performing experiments with optical traps is very time-consuming.
Here we present the production of biomimetic surfaces that enable specific cell adhesion via synthetic ligands and at the same time monitor the transmitted forces by using molecular tension sensors. The ligands were coupled to double-stranded DNA probes with defined force thresholds for DNA unzipping. Receptor-mediated forces in the pN range are thereby semi-quantitatively converted into fluorescence signals, which can be detected by standard fluorescence microscopy at the resolution limit (~0.2 µm).
The modular design of the assay allows to vary the presented ligands and the mechanical strength of the DNA probes, which provides a number of possibilities to probe the adhesion of different eukaryotic cell types and pathogens and is exemplified here with osteosarcoma cells and Plasmodium berghei Sporozoites.
Background
Motile cells and pathogens interact in many different ways with their environment (Parsons et al., 2010; Nan, 2017; Muthinja et al., 2018). For example, transmembrane receptors anchor single cells in their environment and allow them to interact with other cells (Hynes, 1992). Integrins, which are the main class of receptors connecting cells to the extracellular matrix, transmit forces in a bidirectional manner (Schoen et al., 2013). On the one hand, extracellular mechanical signals are transduced to the cytosol, where they trigger signaling cascades and thereby control various cellular functions like proliferation, differentiation and migration (Wang et al., 1993; Janmey and Miller, 2011; Totaro et al., 2017). On the other hand, forces generated by the cytoskeleton can be applied to the extracellular environment, causing tissue shape changes and modulate cellular motility in specialized cells (Iskratsch et al., 2014).
To test for integrin-specific adhesion of cells, here, we first describe a method “option I” to prepare substrates presenting cyclic Arginin-Glycin-Aspartic acid (cRGD) ligands while non-specific surface interactions being prevented by a polyethylene glycol (PEG)-based passivation layer (Figure 1A). Of note, this method can be easily adopted to other receptors by varying the ligand.
To characterize the different aspects of cellular force transmission like absolute tension values, direction or the spatiotemporal distribution of forces several methods exist, which are based on the deformation of hydrogels, micropillars or unfolding of molecules, which differ in their spatial resolution and force range (Polacheck and Chen, 2016). One of the latest approaches visualizes traction forces exerted on single ligands via fluorescent DNA-based tension sensors (Wang and Ha, 2013; Blakely et al., 2014; Zhang et al., 2014). These double-stranded (ds) DNA or hairpin (particularly self-annealing single stranded (ss) DNA structures) sensors are immobilized on an in vitro surface and carry a ligand (e.g., cRGD) that enables for receptor (integrin)-specific cell interactions. Upon force, the dsDNA regions can be unfolded, which is detected with a fluorescent sensor (Figure 1B). In combination with standard wide-field or total internal reflection microscopy the spatiotemporal distribution of forces can be determined at high resolution, which is only limited by the optical resolution (~0.2 µm). The mechanical stability of dsDNA depends on its length and the GC content (Zhang et al., 2015). The force F1/2 that is required to unfold 50% of the DNA sensors, has been calibrated in previous single-molecule studies between ~4-60 pN for varying DNA sequences (Wang and Ha, 2013; Zhang et al., 2014).
The herein presented method “option II” builds on DNA hairpins, which allow to quantify single-molecular, force transmission in the range of 4-19 pN (Zhang et al., 2014; Liu et al., 2016). We modified the DNA strands to enable coupling of various ligands in a modular manner depending on the receptors of interest. In the closed configuration, the fluorescent tag is in close proximity to a quencher and thus dark. Above a specific threshold force the DNA-hairpins unzip and elongate, which lowers the quenching efficiency and gives rise to a digital ON/OFF signal (Figure 1B) (Woodside et al., 2006). They can refold if the force no longer persists (Bonnet et al., 1998; Woodside et al., 2006), which is accompanied by a decrease in fluorescence. This is especially important for the characterization of cells with high turnover of adhesion formations like motile pathogens. Here, DNA hairpin sensors were immobilized via gold nanoparticles, which serve as additional fluorescence quenchers. This reduces the background signals induced by thermodynamic fluctuations as well as the photobleaching in comparison to other sensor surfaces (Lang et al., 2004; Liu et al., 2016). If the range of forces a certain receptor can transmit is not previously known, it is advisable to start with the lowest force probe as it quantitively detects forces above its threshold. Next, the assay can be expanded by multiplexing sensors with different force thresholds and distinct fluorescent dyes to obtain semi-quantitative information on the distribution of cellular forces (Zhang et al., 2014).
Finally, we show how to combine substrate specific adhesion as established in “option I” with the molecular tension sensors “option II” allowing for traction measurements while assuring proper cell adhesion to the surface. This method “option III” can prevent frequent detaching of small and motile cells like Plasmodium sporozoites, even if they show low binding affinities for the ligands coupled to the force sensors.
By immobilizing the molecular tension sensors on such adaptable substrates, they can be used in a wide range of biophysical studies of cell adhesion to extracellular ligands. We recently employed molecular tension sensors to study the specific adhesion between pathogenic agents like viral particles and mammalian cells (Wiegand et al., 2020). Here we provide a protocol to study the interaction of different cell types, including Plasmodium sporozoites, with specific matrix ligands. The rapid movement of the Malaria parasites is a crucial part in the infection cycle (Frischknecht and Matuschewski, 2017) and has been investigated with traction force microscopy (Münter et al., 2009) and laser tweezers (Quadt et al., 2016). Note that the molecular sensors semi-quantitatively detect distribution of forces transduced via single receptors, while they do not report on the overall forces transmitted per cell (Goktas and Blank, 2017) nor the direction of force without integration of further fluorescence polarization techniques (Brockman et al., 2018). Since the methods are very sensitive to changes in the experimental procedure, we recommend to carefully control each step of the protocol.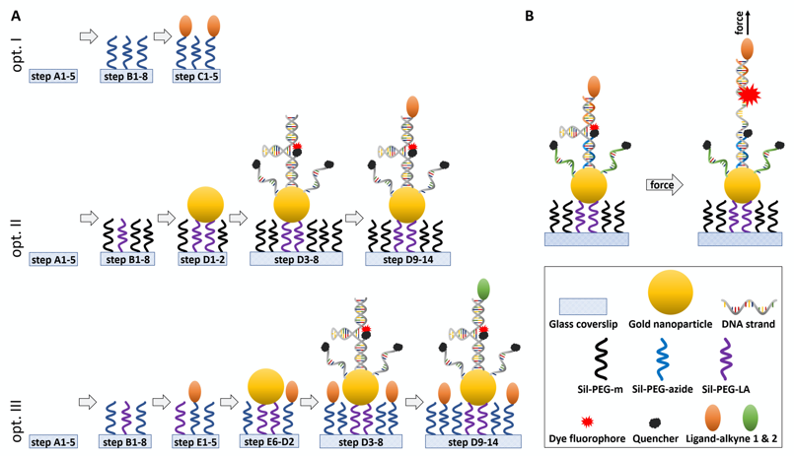
Figure 1. Schematic of the experimental design and working principle of the in vitro DNA-sensor force assay. A. Step-by-step representation of the individual steps in this protocol to generate ligand-presenting surfaces (Opt. I), surfaces with DNA-based force sensors (Opt. II) and combination of cell adhesion ligands presented in the background of the surface and DNA-based force sensor bearing another type of ligand (Opt. III). Briefly, glass coverslips are cleaned, passivated with a PEG-layer and cellular ligands, gold nanoparticles and DNA strands are immobilized on top. For details, the indicated steps lead to the corresponding sections in the procedure chapter. B. Design of the DNA hairpin force sensors in relaxed conformation and under load. ‘Hairpin’ ssDNA (grey) forms the backbone of the sensors and is linked to the Gold nanoparticle at the bottom and a ligand at the top. Top ssDNA (orange) is linked to the fluorescent dye, bottom ssDNA (blue) is linked to a quencher. Additional bottom ssDNA (green) increases the quenching of background fluorescence. Annealing procedure of the DNA hairpin probe is described in Recipe 8. When a force is applied the partially self-annealing ‘hairpin’ strand unfolds by rupturing the hydrogen bonds between nucleotides, lowering the quenching efficiency between the two dyes, and thus local increase in the fluorescent signal. Figure is not drawn to scale.
Materials and Reagents
Standard
- 1.5 ml plastic reaction tube (e.g., Eppendorf, catalog number: 00 30125150 )
- Aluminum foil
- Cell hood
- Erlenmeyer flask of different sizes
- Flasks for buffers
- Glass beaker of different sizes
- Heat-resistant glass tub for oil bath
- Pipettes of different sizes with tips
- Schlenk flasks of different sizes
- Fine-tipped metallic tweezers, e.g., #4, #7 (Sigma, catalog number: T6165 )
- Ice
- Ultra-pure deionized water max. resistance of 18.2 MΩ·cm, e.g., MilliQ purified
- Inert gas (Ar)
- Nitrogen gas
- Schlenk line (Schlenk flask with wide opening, lid for Schlenk flask, connection tubing for Vaccuum and Nitrogen gas)
- Deionised (DI) water
Consumables
- 20 x 20 mm coverslips #1 (Carl Roth, catalog number: H873.2 )
- DWK Life Sciences DURANTM Filter Funnels (No. 3) (Fisher Scientific, catalog number: 09-841-090 )
- GE Healthcare illustraTM, NAP-5 column (Fisher Scientific, catalog number: 10054394 )
- Glass coverslip holder (home-built)
- Kimwipe tissues (Kimberly-Clark Worldwide, catalog number: 0 5517 )
- Molecular sieve 3 Å (Carl Roth, catalog number: 8487 )
- Pasteur pipettes ISO 7712 (230 mm) (Karl Hecht, catalog number: 40567002 )
- Petri dish, PS, 35/10 mm with vents (Greiner Bio-One, catalog number: 627102 )
- Rotilabo-syringe filter, CME, sterile (Carl Roth, catalog number: P818.1 )
- Silicon chamber (Ibidi GmbH, catalog number: 80841 )
- Teflon coverslip holder (home-built, commercially available, e.g., at Thermo Fisher Scientific, catalog number: C14784 )
- Silicon glue‚ Twinsil 22, Addition-curing duplucating silicone (Picodent GmbH, catalog number: 13001000 )
- Silicon oil for oil bath (Sigma-Aldrich, catalog number: 85409 )
Chemicals
- Gold Nanospheres, 9 nm (nanoComposix, custom size, 9 ± 2 nm, 0.05 mg ml−1 in tannic acid)
- (+)-Sodium L-ascorbate (Sigma-Aldrich, catalog number: A7631 )
- 3-(triethoxysilyl)propylisocyanate (Sigma-Aldrich, catalog number: 413364 )
- Acetic acid ethyl ester (Carl Roth, catalog number: 6784 )
- Acetonitrile (CHROMASOLV for HPLC) (Honeywell International, catalog number: 60-002-31 )
- Aminoguanidine hydrochloride (Sigma-Aldrich, catalog number: A8835 )
- ɑ-Silane-ω-alkyne PEG, (CH3O)3-Si-PEG(3000)-C≡CH (Rapp Polymere, catalog number: 133000-70-71 )
- ɑ-Silane-ω-azido PEG, (CH3O)3-Si-PEG(3000)-N3 (Rapp Polymere, catalog number: 133000-5-71 )
- Copper sulfate (Sigma-Aldrich, catalog number: C1297 )
- Diethyl ether (Sigma-Aldrich, catalog number: 309966 )
- Dimethyl sulfoxide (for spectroscopy) (DMSO) (Merck, catalog number: 102950 )
- cRGD-alkyne (Biotrend Chemicals, custom synthesis: Cyclo(Arg-Gly-Asp-D-Phe-Pra))
- Hydrogen peroxide 30% (Merck, catalog number: 107209 )
- LA-PEG(3400)-NH2 (Biochempeg Scientific, catalog number: HE039005-3.4K )
- Methanol (Carl Roth, catalog number: 00 82 )
- N,N-Dimethylformamide (anhydrous, 99.8%) (DMF) (Sigma-Aldrich, catalog number: 227056 )
- Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P9333 )
- Potassium hydroxide (for cleaning bath) (Sigma-Aldrich, catalog number: P5958 )
- Potassium phosphate monobasic (KH2PO4) (Sigma-Aldrich, catalog number: P5655 )
- QSY21 (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q20131 )
- Silane-PEG-methoxy, Sil-PEG(2000)-m (Biochempeg Scientific, catalog number: MF001020-2K )
- Sodium carbonate (Na2CO3) (Carl Roth, catalog number: A135 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S7653 )
- Sodium phosphate dibasic (Na2HPO4) (Sigma-Aldrich, catalog number: 255793 )
- StarRed-NHS (Abberior) (Abberior, catalog number: 1-0101-011-3 )
- Sulfuric acid 96% (VWR CHEMICALS, catalog number: 1.08131.1000 )
- Toluene (Merck, catalog number: 108325 )
- Triethylamine (Sigma-Aldrich, catalog number: 471283 )
- Triethylammonium acetate (TEAA) (Sigma-Aldrich, catalog number: 69372 )
- Tris(3-hydroxypropyltriazolylmethyl)amine (THPTA) (Sigma-Aldrich, catalog number: 762342 )
- Click reaction mixture (CuAAC) (see Recipes)
- Dried toluene (see Recipes)
- Sil-PEG(3500)-LA (see Recipes)
DNA strands (custom synthesis by Integrated DNA Technologies, Inc.)
Note: Self-annealing sections underlined, top strand annealing dotted, bottom strand annealing dashed; a list of the hairpins used in literature and their calibration can be found in Zhang et al., 2014 and Liu et al., 2016.
- Top: 5′- /5AmMC6/ CGC ATC TGT GCG GTA TTT CAC -3′
- Bottom NH2-quencher:
5′- /5ThioMC6-D/TTT GCT GGG CTA CGT GGC GCT CTT/3AmMO/ -3′ - Hairpin 4.2 pN:
5'- /5AzideN/GTG AAA TAC CGC ACA GAT GCG TTT - GTA TAA ATG TTT TTT TCA TTT ATA C - TTT AAG AGC GCC ACG TAG CCC AGC AAA/3ThioMC3-D/ -3’ - Hairpin 12 pN:
5'- /5AzideN/GTG AAA TAC CGC ACA GAT GCG TTT - GGG TTA ACA TCT AGA TTC TAT TTT TAG AAT CTA GAT GTT AAC CC - TTT AAG AGC GCC ACG TAG CCC AGC AAA/3ThioMC3-D/ -3′ - Hairpin 19 pN:
5'- /5AzideN/GTG AAA TAC CGC ACA GAT GCG TTT - CGC CGC GGG CCG GCG CGC GGT TTT CCG CGC GCC GGC CCG CGG CG - TTT AAG AGC GCC ACG TAG CCC AGC AAA/3ThioMC3-D/ -3′ - Labeled DNA strands (see Recipes)
- Annealed DNA strands (see Recipes)
Enzymes
- StemPro Accutase Cell Dissociation Reagent (Thermo Fisher Scientific, Gibco®, catalog number: A1110501 )
Medium
- Albumin Fraction V (Carl Roth, catalog number: 8076 )
- Fetal Bovine Serum (USA) (Sigma-Aldrich, catalog number: F2442 )
- MEM α (Thermo Fisher Scientific, Gibco®, catalog number: 12561 )
- Penicillin-Streptomycin (10,000 U/ml) (Thermo Fisher Scientific, Gibco®, catalog number: 15140148 )
- RPMI 1640 Medium (Thermo Fisher Scientific, Gibco®, catalog number: 11-875 )
- Sodium Pyruvate 100 mM (100x) (Thermo Fisher Scientific, Gibco®, catalog number: 11360070 )
- Microscopy U2OS medium (see Recipes)
- Microscopy sporozoite gliding medium (see Recipes)
- PBS buffer (see Recipes)
Cell lines
- Plasmodium berghei ANKA strain (BEI resources (ATCC), catalog number: MRA-871 )
- Plasmodium berghei NK65 strain, CS-GFP line (optional usage, see Step H4) (not commercially available, requestable from cooperating parasitological institutes; further information can be found in Natarajan et al., 2001)
- U2OS, osteosarcoma cells (ATCC, catalog number: HTB-96 )
Equipment
- 300 °C Oven
- Freezer (-20 °C and -80 °C)
- Fridge (4 °C)
- Fume hood
- Centrifuge (Thermo Fisher Scientific, model: Heraeus Biofuge Pico )
- Centrifuge (Thermo Fisher Scientific, model: Heraeus Multifuge 1S-R ; catalog number: 75004331 )
- Hot Plates Accessories (IKA, model: ETS-D5 , catalog number: 0003378000 )
- HPLC (GE Comp Healthcare Life Sciences, model: ÄKTA Pure Chromatography System )
- HPLC column (Phenomenex, model: Luna 5u C18(2)-RP-HPLC , catalog number: 00G-4252-N0 )
- Lyophilisator (Labconco, model: FreeZone Plus 2.5 Liter Cascade Benchtop Freeze Dry System , catalog number: 7670020 )
- Magnetic Stirrers (IKA, model: C-MAG HS 7 , catalog number: 0003581200 )
- Wide-field, inverted fluorescence microscope (Applied Precision, model: DeltaVisionTM Elite Microscope System, based on an Olympus IX71 body) with home-built environmental chamber (commercially available incubators e.g., at OKOLAB)
- Microscope filter set (Applied Precision, model: DeltaVisionTM Elite Filter Sets)
- Microscope objective (Olympus, model: Objective 60x/1.4 NA PlanApo IX70 )
- Sonicator (EMAG Technologies, model: Emmi-H22 , catalog number: 9 82 17-DE)
- Spectrophotometer (NanoDrop Technologies, model: PEQLAB Nano Drop ND-1000 )
- Thermal cycler (Bio-Rad Laboratories, model: Bio-Rad T100 , catalog number: 1861096 )
Software
- Fiji is just ImageJ (Version 1.52e; https://fiji.sc/) (Schindelin et al., 2012)
- Stack contrast adjustment, plugin for Fiji (Michálek et al., 2007)
Procedure

Figure 2. Overview of the experimental procedure. Glass coverslips are handled with a pair of tweezers and inserted in a Teflon holder (white), which is transferred into a beaker with Piranha solution for cleaning (Procedure A). For passivation of the coverslips they are transferred into a glass holder and incubated with the Sil-PEG passivation solution in a Schlenk flask at 80 °C in an oil bath. The Schlenk flask can be attached to a vacuum and nitrogen line via its valve to provide an inert reaction container (not shown, Procedure B). After cleaning and drying the coverslips, specific ligands and gold nanoparticles (red solution) are immobilized by incubating a drop of the respective solution in-between two coverslips. To remove the coverslips again from each other, additional water or PBS is injected with a pipette and the upper coverslip is lifted with a pair of tweezers (Procedures C-E). To observe the surfaces at an inverted microscope they are glued with a two-component silicon glue (yellow and blue gel) into a Petri dish with a custom-made hole in the center (2 mm smaller than the coverslip) (Procedure F). The final device is shown in the last figure, where medium and cells can be added before transferring to a microscope. Further information is provided at the steps indicated below the pictures.
- Cleaning of glass surfaces
- Clean and activate coverslips in a Teflon holder in ~100 ml freshly prepared ‘Piranha solution’ [3:1 (v/v) sulfuric acid: hydrogen peroxide] for 1 h. Make sure the coverslips are completely covered by solution (Figure 2, cleaning). Stir the Piranha solution e.g., with an acid-resistant Pasteur pipette.
CAUTION: Piranha solution is very corrosive and reacts exothermic. It must not get in contact with organic solvents. Perform every work step containing Piranha solution under a fume hood with constant ventilation.
Note: From here on transfer and handle the coverslips only with tweezers (Figure 2). - Remove ‘Piranha solution’ from the coverslips by carefully decanting it into an appropriate waste container according to the safety regulations of your institute, e.g., discarding after 10-20x diluted in water.
- Rinse coverslips 3 times with DI water and twice with ultra-pure water.
- Sonicate coverslips in ultra-pure water once for 5 min in ultrasonic cleaner at ~60 watt (standard sonication power).
- Dry surfaces in a nitrogen stream by holding them carefully with tweezers.
Check point: Observe by eye that no stripes or drops of water remain else repeat Steps A3-A4 or discard the coverslip.
- Clean and activate coverslips in a Teflon holder in ~100 ml freshly prepared ‘Piranha solution’ [3:1 (v/v) sulfuric acid: hydrogen peroxide] for 1 h. Make sure the coverslips are completely covered by solution (Figure 2, cleaning). Stir the Piranha solution e.g., with an acid-resistant Pasteur pipette.
- Passivation of glass surfaces
- Transfer dried coverslips to a glass holder with tweezers and avoid touching them in the center.
- Prepare passivation solution as indicated in Table 1 (depending on the desired surface Opt. I-III, see Figure 1A) under continuous protective nitrogen atmosphere in a Schlenk flask (Figure 2, passivation). All steps involving organic solvents (Steps B2-B7) shall be performed following the safety regulations of your institute under a fume hood with constant ventilation. Create a protective atmosphere by connecting the Schlenk flask to a vacuum line and nitrogen supply. First vacuum-empty the flask, then flush with nitrogen and reduce the nitrogen stream in the following to add the powdery PEG, dried toluene and triethylamine.
Table 1. Passivation compositions for the three different type of surfaces (Option I-III). Option I should be chosen when the adhesion of cells to a specific ligand is to be tested, Option II to access the forces at such specific ligands and Option III when ligand-specific adhesion to the surface should be granted independent of the force measurements of specific ligands.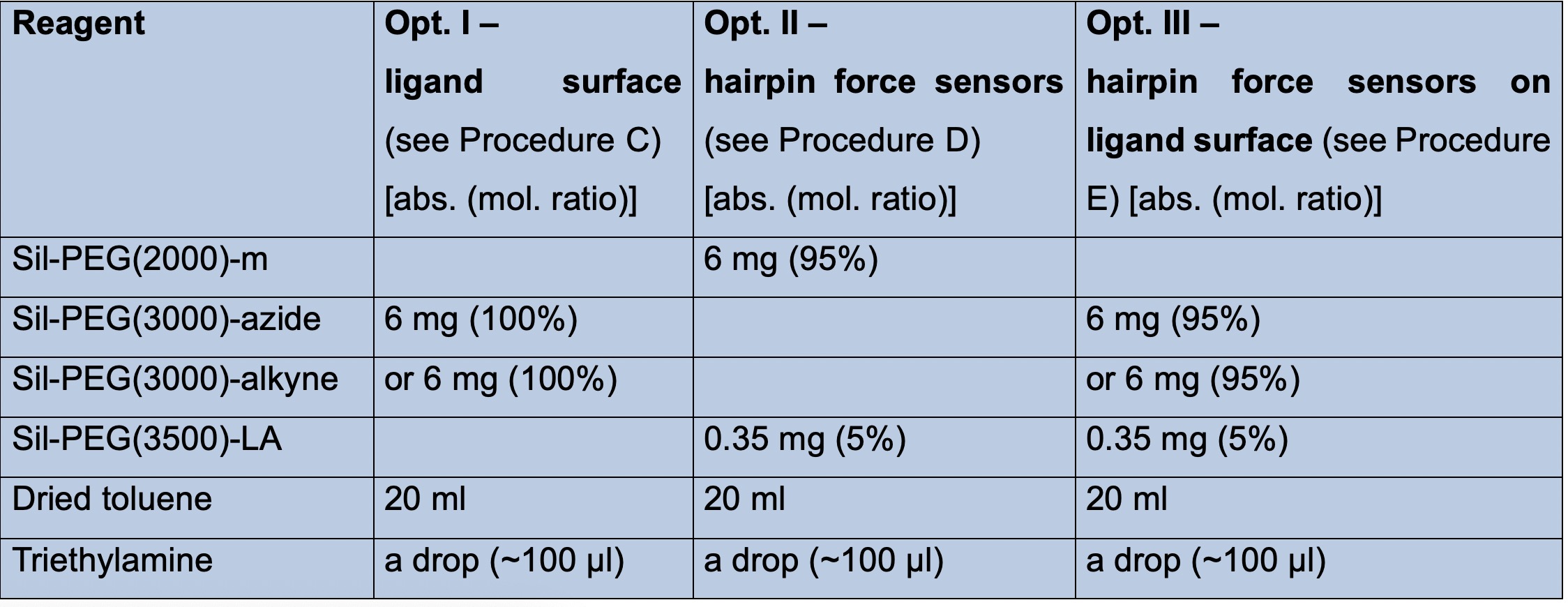
- Add coverslips under protective atmosphere into the Schlenk flask in the solution and close the lid and gas supply. The reaction is now kept under protective atmosphere.
- Heat tube overnight at 78-80 °C in an oil bath. The oil bath can be set up combining the heat-resistant glass tub filled with oil on the magnetic stirrer. The additional thermostat keeps the temperature constant.
- Wash passivated coverslips by sonication in ethyl acetate for 5 min within their glass holder.
- Wash passivated coverslips by sonication in methanol for another 5 min within their glass holder.
Pause point: It is possible to store passivated glass surfaces in methanol for up to 5 days if they are not used immediately. - Dry surfaces in a nitrogen stream by holding them carefully with tweezers. Nitrogen stream can be dosed carefully with a compressed air gun. Make sure not to scratch the passivated surfaces during the entire procedure.
Check point: As control for proper passivation add a dye (e.g., 100 nM StarRed in PBS) or seed adhesive cells on a passivated versus a non-passivated coverslip and observe their attachment to the surface under a microscope. - For reuse, clean the used Schlenk flask and glass sample holder from PEG residue in a potassium hydroxide bath overnight and wash them afterwards with DI water.
- Option I–Immobilization of specific cellular ligands
- Prepare 100 µl of the reaction mixture for copper-catalyzed azide-alkyne cycloaddition (CuAAC, see Recipe 7) containing the desired cellular ligand.
Note: Choose appropriate alkyne or azide functionalized PEG for azide or alkyne-ligands, respectively. - Prepare a humidity chamber with parafilm on the bottom and a wet tissue inside a Petri dish where coverslips are placed onto (Figure 2, immobilization). Parafilm is used to hold the coverslips in their spot and to ease the separation of ‘sandwiched’ glasses later on.
- Place a drop of 100 µl click mixture on top of a passivated and dried coverslip.
- Place a second, identical coverslip on top of the first one.
- Incubate the ‘sandwich’ for 2 h at room temperature (RT).
- Prepare 100 µl of the reaction mixture for copper-catalyzed azide-alkyne cycloaddition (CuAAC, see Recipe 7) containing the desired cellular ligand.
- Option II–Immobilization of DNA hairpin force sensors
- Place a drop of 250-300 µl of the gold nanoparticle solution on top of a passivated and dried coverslip.
- Incubate the coverslips for 30 min at RT.
- In the meantime, hybridize hairpin force sensors using ‘top’ and ‘bottom’ and ‘hairpin’ ssDNA. Use DNA mixture and annealing program as indicated (see Recipe 8). If DNA strands are purchased unlabeled, DNA and fluorophore should be coupled in a previous step (see Recipe 4 for labeling of DNA strands below).
- Separate the surfaces by carefully pipetting 300 µl ultra-pure water in-between the glasses and pick them up with a pair of tweezers (Figure 2, Immobilization). Clean them from supernatant gold nanoparticle solution by thoroughly rinsing with ~10 ml ultra-pure water.
Check point: Confirm successful immobilization of gold nanoparticles by the pink shine of the coverslip. - Remove excess liquid with tissues over the coverslip edges without letting them run completely dry or touching them.
- Place a drop of 100 µl DNA mixture on top of a gold-coated coverslip.
- Place a second, identical coverslip with the gold nanoparticle-coated side facing down on top of the first one.
- Incubate the ‘sandwich’ overnight at 4 °C under exclusion of light.
- Prepare 100 µl of the reaction mixture for CuAAC (see Recipe 7) containing tripeptide/ligand.
- Separate and clean the surfaces carefully from DNA mixture by washing in PBS (analogous to Step D4).
- Remove excess liquid with tissues over the coverslip edges without letting them run completely dry or touching them.
- Place a drop of 100 µl click mixture on top of a DNA-coated coverslip.
- Place a second, identical coverslip with the DNA-coated side facing down on top of the first one.
- Incubate the ‘sandwich’ for 2 h at RT in the humidity chamber under exclusion of light to avoid bleaching of the fluorophores.
- Option III–Combination of specific cellular ligand and DNA hairpin force sensors
- Prepare 100 µl of the 1st reaction mixture for CuAAC (see Recipe 7) containing 1st ligand.
- Prepare a humidity chamber with a wet tissue inside a Petri dish and flat parafilm on the bottom where coverslips are placed onto.
- Place a drop of 100 µl click mixture on top of a passivated and dried coverslip.
- Place a second, identical coverslip on top of the first one.
- Incubate the ‘sandwich’ for 2 h at RT.
- Separate and clean the surfaces carefully from click mixture by washing in PBS (analogous to Step D4).
- Further steps are equal to ‘D) Immobilization of DNA hairpin force sensor immobilization’ Steps D1-D14 using the 2nd ligand at Step D9.
- Sample preparation for microscopy
- Separate and clean the surfaces carefully from click mixture by washing in PBS (analogous to Step D4).
- Remove excess liquid with tissues over the coverslip edges without letting them run completely dry or touching them.
- Glue coverslips with the functionalized surface facing up into Petri dishes with custom made holes (2 mm smaller than the coverslips) in the bottom. Therefore, mix ~200 µl of Twinsil 22 silicon 1:1 e.g., with a pipette tip and apply the glue on the edges of the hole, such as the coverslip completely covers the hole and is surrounded by glue.
- Wait curing time of the glue (~ 5 min, check with the remaining glue ). If necessary, clean surface by washing in PBS to remove glue contamination.
Check for successful immobilization of cellular ligands and force sensors by comparing cellular attachment (Procedure G) and homogenous background fluorescence, respectively, of only passivated coverslips versus those with immobilized ligands (Procedure C) or force sensors (Procedure D).
- Assays with cells (U2OS)
- Bring chamber for cell observation to 37 °C and 5% CO2 atmosphere.
- Fill 2-3 ml cellular medium (0.5% FBS) (see Recipe 5) in the Petri dishes prepared in step F covering the functionalized coverslip. Use medium with low FBS content to prevent unspecific ligand interactions and improve microscopy. Use a cell culture fume hood to prevent medium or cell culture contamination.
- Harvest adherent cells from culture flask by washing with PBS and detach with Accutase following standard protocols (e.g., Basic Cell Culture Protocols (Helgason and Miller, 2004)).
Note: Accutase gently detaches cells for faster spreading results. - Seed cells in diluted concentration to allow an undisturbed spread. In case of U2OS 105 cells reach an adequate density (calculated from T-25 and 35 mm dish flask surface for around 1:10 dilution).
- Wait until cells settle down and start productively spreading on the surfaces (10-30 min).
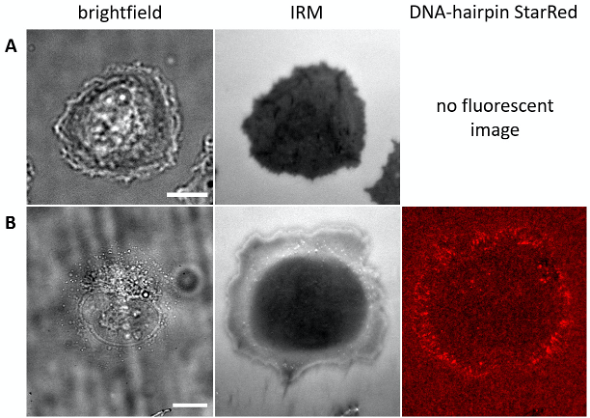
Figure 3. U2OS cells adhering and exerting forces on specific ligand presenting surfaces. Representative bright-field, interference reflection microscopy (IRM) and fluorescence wide-field images of U2OS cells 15-20 min after seeding on the surfaces with (A) option I: cRGD-functionalization and (B) option II: cRGD-linked DNA hairpin sensors (4.2 pN opening force; StarRed-fluorophore). Scale bars = 10 µm. - Bring chamber for cell observation to 37 °C and 5% CO2 atmosphere.
- Assays with sporozoites
- Try to keep the edges of the coverslip as dry as possible, whereas the center of the functionalized surface should be kept humid. This eases the silicon chamber founding.
- Press a silicon chamber with its sticky side on the edges of the functionalized coverslip to minimize the cellular distribution area (Note: silicon sticks best on dry glass). Purchased multi-well chambers can be cut to a fitting size. This step can be skipped, but eases to generate an adequate parasite density.
- Fill 15 µl sporozoite medium (6% BSA) (see Recipe 6) into the silicon chamber.
- Add 15 µl ice-cooled PBS containing freshly isolated sporozoites from 6-8 pairs of well infected mosquitoes’ salivary glands (for a detailed protocol of mosquito infection and sporozoite collection see Prinz et al., 2017) resulting in 3%-BSA-medium.
Notes:- FBS-reduced cellular medium (0.5% FBS) can be use as well.
- Do not keep salivary glands on ice for prolonged times as this could harm the sporozoite motility.
- Usage of GFP-tagged sporozoites eases the selection of well infected salivary glands as well as their isolation. They can be employed on ligand-presenting surfaces (option I, see Figure 4A), but care must be taken to choose proper filter sets when using them together with force sensors (Figure 4B) to avoid fluorescent bleach through.
- FBS-reduced cellular medium (0.5% FBS) can be use as well.
- Spin cells down to surface in a centrifuge at 2,000 x g for 3 min. Well-founded silicon chamber can stand this procedure.
- Motility can be observed directly after attaching to the surfaces for around 30 min (Figure 4).
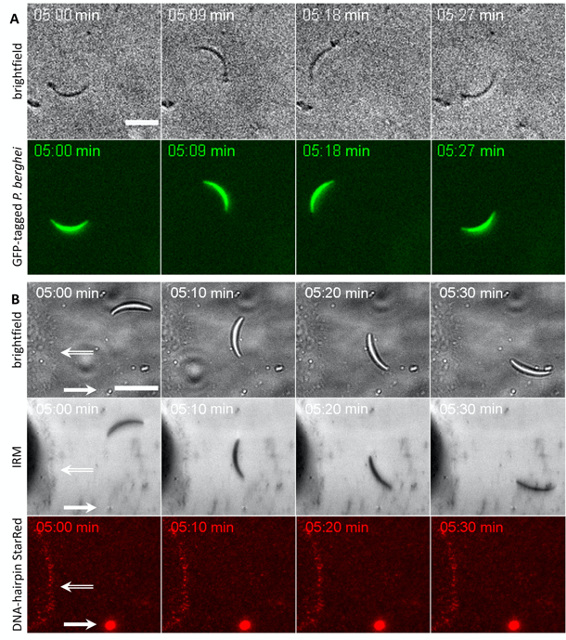
Figure 4. Sporozoites (P. berghei) gliding on specific ligand presenting surfaces. Representative bright-field, interference reflection microscopy (IRM) and fluorescence wide-field time lapse images of (A) GFP-expressing sporozoites 5 min after seeding on the surfaces with option I: cRGD-functionalization and (B) WT sporozoites 5 min after seeding on the surfaces with option II: cRGD-linked DNA hairpin sensors (4.2 pN opening force; StarRed-fluorophore). For Option II Osteosarcoma cells (hollow arrow) were seeded together with the sporozoites as a positive control for the DNA-based force sensors (see Figure 3). Sporozoites adhere and glide on both surfaces, however, no tension signal was detected with the DNA hairpin sensors. This might be due to sub-threshold forces, few surface interactions or too short adhesion times (sporozoites move at ~2 µm/s), which do not allow for detectable unfolding of the tension probes, while osteosarcoma cells adhering on the same substrates gave rise to fluorescent signals (hollow arrow). Bright fluorescent spots (full arrow) are auto fluorescent remnants from the purification of Sporozoites out of mosquitoes’ salivary glands. Scale bars = 10 µm.
- Microscopy settings
- Imaging was performed on a fluorescence wide-field microscope (Olympus) equipped with a custom-made environmental chamber for cellular conditions (37 °C; 5% CO2). Sporozoites were usually imaged at RT without CO2 supply but we also successfully performed gliding motility assays under cellular conditions.
- Brightfield images were taken with a polarized light filter set.
- Fluorescence microscopy images were taken with excitation bandpass filter 632/22 nm and emission filter 679/34 nm.
- Interference reflection microscopy (IRM) images were taken with excitation bandpass filter 542/27 nm in IRM mode (set emission bandpass filter 525/48 nm) with minimal exposure time and intensity. IRM observation can be thus used as a low-bleaching control for cell adherence to the surface during the imaging process.
Data analysis
Receptor surfaces (option I), which do not contain force sensors, can be used for a qualitative testing of the cellular adherence properties to defined ligands on an otherwise non-adherent, PEG-ylated surface. Brightfield imaging or IRM allows to validate if cells adhere tightly to the surface, such as no fluorescence microscopy is required for this approach.
Furthermore, quantitative analysis of the settling and spreading time of cells on the surface (data analysis see Schaufler et al., 2016) or of the gliding motility in case of motile sporozoites (data analysis see Prinz et al., 2017) can be used for comparable data analysis between different cell/parasite lines or different ligands.
DNA force sensor surfaces (options II and III) provide a digital ON-OFF signal at a specific threshold force. Fluorescence microscopy images of the sensor surface provide spatial, temporal, and semi-quantitative information on the exerted forces, indicated by the transient appearance (hairpins) of the fluorescent signal. Therefore, fluorescence microscopy images were first adjusted for brightness and contrast for better visibility of the signals with the Fiji software. For time lapse imaging, ‘Stack contrast adjustment’ plugin is used to correct for the photobleaching. A semi-quantitative force readout can be generated from the local brightness reflecting the relative amount of dsDNA opening and displayed via a color-coded LUT e.g., “spectrum” (for more details see Zhang et al., 2014, Supplementary Figure 7). However, this quantitative analysis is limited by the optical resolution, since typically multiple sensors will be contained per diffraction limited spot.
To narrow down the absolute traction values, the experiment can be repeated with DNA sensors of different rigidity by varying the DNA sequence. Multiple of these sensors coupled to different fluorophores can be combined on one surface for multiplex results (see Zhang et al., 2014).
Notes
- Prepare aliquots of appropriate volumes for frozen medium or solution to avoid repeated freeze thaw cycles and elongate their shelf lifetime.
- Separate and clean the surfaces from reaction mixture by carefully adding 300 µl PBS inside the sandwich with a pipette. Subsequently lift off top coverslip and wash them in PBS. Make sure you never scratch the surfaces during the entire procedure.
- Avoid drying out of the surfaces at any time since retracting liquids can produce artefacts caused by surface tension on surfaces.
- Other fluorescent dyes and quenchers with NHS function can be used for the labeling of DNA depending on the availability of filters at the microscope. Consider dyes with a high quantum yield and an emission wavelength close to the particle resonance peak of gold nanoparticles for stronger quenching (Kang et al., 2011). Note, that cellular autofluorescence, especially from nucleus, can overlap sensor signals from underneath the cell. For smaller cells with a high nuclear to body ratio (e.g., Plasmodium sporozoites), far-red dyes like StarRed can minimize this signal-autofluorescence interference.
Recipes
- PBS buffer
- Prepare PBS as indicated:
NaCl 8.006 g (cfinal: 137 mM)
KH2PO4 1.375 g (cfinal: 10.1 mM)
KCl 201.3 mg (cfinal: 2.7 mM)
Na2HPO4 255.5 mg (cfinal: 1.8 mM) - Add 0.8 L ultra-pure water
- Adjust pH to pH 7.4 by adding a few Milliliter HCl (1 M)
- Add ultra-pure water to a total volume of 1 L
- Store buffer at RT
- Prepare PBS as indicated:
- Sil-PEG(3500)-LA
- Solve 145 µmol NH2-PEG(3400)-LA in 3 ml DMF in a small Schlenk flask under nitrogen atmosphere with a magnetic stir bar
- Add 159.5 µmol 3-(trimethoxysilyl)propylisocyanate (1.1-times the molecular amount of PEG)
- Stir overnight at room temperature
- Cool solution down to 4 °C
- Add cooled diethyl ether for 1 h
- Wash the reactant with cooled diethyl ether over a glass frit (No. 3)
- Dry gained Sil-PEG-LA from frit overnight under vacuum
- Store the powder at -20 °C under inert gas
- Solve 145 µmol NH2-PEG(3400)-LA in 3 ml DMF in a small Schlenk flask under nitrogen atmosphere with a magnetic stir bar
- Dried toluene
- Dry molecular sieve thoroughly in an oven
- Vacuum an empty Erlenmeyer tube and add a nitrogen atmosphere
- Add toluene under protection atmosphere and close flask properly
- Wait several hours to get rid of excessive water in the toluene
- Store at room temperature under nitrogen atmosphere
- Dry molecular sieve thoroughly in an oven
- Labeling of DNA strands
- Dissolve DNA in ultra-pure water to a concentration of 1 mM
- Dissolve dye-NHS respectively quencher-NHS in dried DMSO to 10 mM
- Dissolve 105.99 mg Na2CO3 in 1 ml ultra-pure water
- Mix 60 µl solved DNA top strand (cfinal: 0.2 mM), 60 µl solved dye-NHS (cfinal: 2 mM) and 30 µl Na2CO3 solution (cfinal: 0.1 mM) in 150 µl PBS. Proceed the same way with DNA bottom strand and the quencher-NHS
- Adjust pH to 8.5
- Stir mixture overnight at room temperature under the exclusion of light
- Purify mixture in NAP-5 column as indicated in the user manual of the manufacturer
- Purify mixture in HPLC (gradient high-pressure liquid chromatography). Use Luna 5u C18(2)-RP-HPLC column, mobile phases are trietyhlammonium acetate (TEAA) (0.1 M) and acetonitrile with a gradient from 10% to 50% in 90 min and from 50% to 90% in 20 min
- Collect fraction showing both maximum absorption peaks at the same time for DNA (260 nm) and dye
- Freeze product to -80 °C and lyophilize it after that to get rid of the solvents
- Resuspend pure product in ultra-pure water
- Determine the concentration with a nanodrop spectrophotometer measuring the absorbance ADNA; 1mm of DNA at 260 nm and the absorbance Adye; 1mm at the excitation wavelength of the respective dye/quencher
- Calculate concentration from three different measurements via:
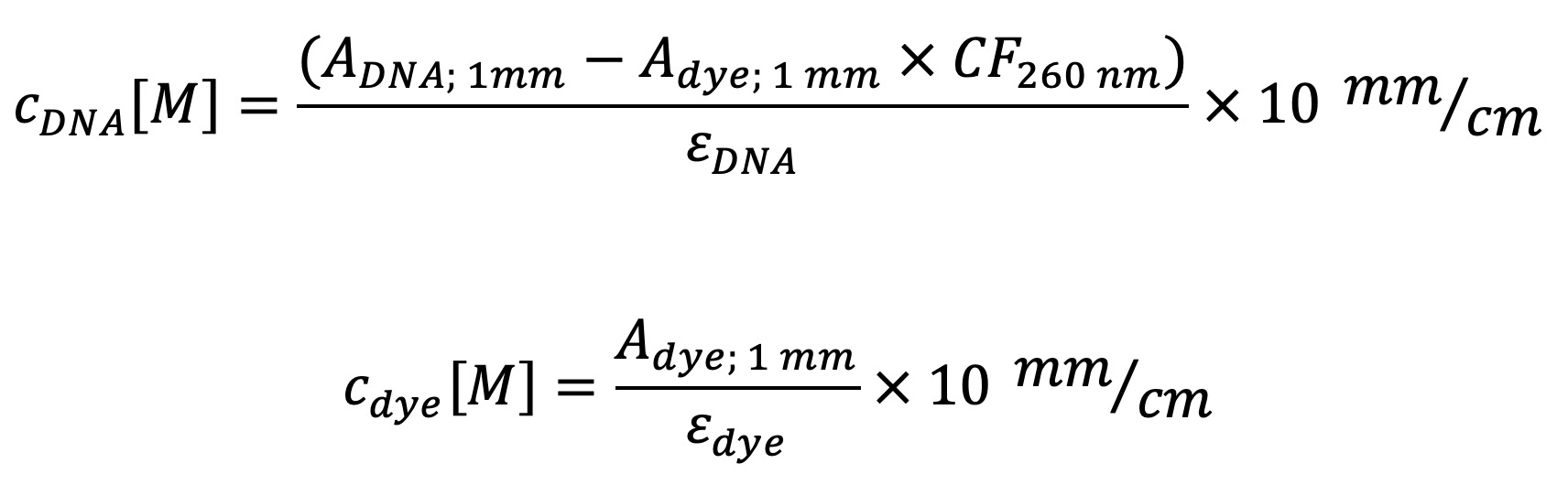
Note: Take the extinction coefficient εdye and correction factor CF260nm for the dye spectrum at 260 nm wavelength into account. - Store resulting strand at 4 °C under the exclusion of light
- Dissolve DNA in ultra-pure water to a concentration of 1 mM
- Microscopy U2OS medium
- Prepare cell medium (0.5% FBS) as indicated:
MEM 48.75 ml
Pyruvate 0.5 ml
Penicillin/Streptavidin 100x 0.5 ml
FBS 0.25 ml - Mix and filter medium under sterile conditions (hood)
- Store medium at 4 °C
- Prepare cell medium (0.5% FBS) as indicated:
- Microscopy sporozoite gliding medium
- Prepare sporozoite medium (RPMI + 6% BSA) always freshly as indicated:
RPMI 44.5 ml Penicillin-streptomycin 2.5 ml Albumin fraction V 3 g - Dissolve albumin thoroughly
- Spin down unsolvable particles at 13k rpm for 5 min in the Heraeus Biofuge
- Prepare sporozoite medium (RPMI + 6% BSA) always freshly as indicated:
- Click reaction mixture (CuAAC) (adapted from Hong et al., 2009)
- Prepare click reaction buffer as indicated:
NaCl 87.7 mg (cfinal: 150 mM) Na2HPO4 14.7 mg (cfinal: 80.8 mM) KH2PO4 25.9 mg (cfinal: 19 mM) Add 8 ml ultra-pure water - Adjust the buffer to pH 7.4 by adding a few Microliter HCl (1 M)
- Add ultra-pure water to a total volume of 10 ml
- Dissolve aminoguanidine in ultra-pure water to a concentration of 100 mM
- Dissolve copper sulfate in ultra-pure water to a concentration of 20 mM
- Dissolve cRGD-alkyne in ultra-pure water to a concentration of 10 mM
Note: Other tripeptides and ligands containing an alkyne function can be used equivalently. For azide-functionalized molecules the alkyne reaction partner is substituted and treated equally in this recipe. - Dissolve sodium ascorbic acid in ultra-pure water to a concentration of 100 mM
- Dissolve THPTA in ultra-pure water to a concentration of 50 mM
- Premix 3 µl THPTA with 1.5 µl copper solutions thoroughly
- Mix 87.5 µl click reaction buffer with 1 µl cRGD-alkyne (cfinal: 0.1 µM), 1.5 µl THPTA/copper premix (cfinal: 0.5 µM/0.1 µM), 5 µl aminoguanidine (cfinal: 5 µM) and 5 µl ascorbate (cfinal: 5 µM)
- Store buffer at RT. Store aminoguanidine and copper solutions at 4 °C, peptide-alkyne and THPTA solutions at -20 °C. Prepare ascorbate solution always directly before use
- Prepare click reaction buffer as indicated:
- DNA annealing
- Dissolve DNA strands in ultra-pure water to concentration of 10 µM. In case of labeled top DNA strand, use the calculated values form the spectrophotometer measurement after resuspending. Store DNA strands at 4 °C, store hairpin strand at -20 °C
- For hairpin sensors, mix 3 µl top DNA (cfinal: 0.3 µM), 3 µl bottom DNA (cfinal: 0.3 µM) and 3.3 µl hairpin DNA (cfinal: 0.33 µM) to 70.3 µl NaCl buffer (1 M)
- Anneal the strands in thermal cycler. Program the thermal cycler: heat 10 min at 95 °C (denaturation), hold 4 s at 95 °C (annealing), repeat last step 399x while cooling down 0.2 °C at each step, final hold at 12 °C
- Add 20.4 µl additional bottom DNA (cfinal: 2.04 µM) to the annealed DNA
- Dissolve DNA strands in ultra-pure water to concentration of 10 µM. In case of labeled top DNA strand, use the calculated values form the spectrophotometer measurement after resuspending. Store DNA strands at 4 °C, store hairpin strand at -20 °C
Acknowledgments
Thanks to all the coworkers from AG Frischknecht and AG Cavalcanti-Adam for enriching debates and suggestions.
This work was funded by fellowships from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)-Projektnummer 240245660-SFB 1129, P1 and P15, and the Max Planck Society.
Competing interests
The authors declare that no competing financial interests exist.
References
- Blakely, B. L., Dumelin, C. E., Trappmann, B., McGregor, L. M., Choi, C. K., Anthony, P. C., Duesterberg, V. K., Baker, B. M., Block, S. M., Liu, D. R. and Chen, C. S. (2014). A DNA-based molecular probe for optically reporting cellular traction forces. Nature Methods 11(12): 1229-1232.
- Bonnet, G., Krichevsky, O. and Libchaber, A. (1998). Kinetics of conformational fluctuations in DNA hairpin-loops. Proc Natl Acad Sci U S A 95(15): 8602-8606.
- Brockman, J. M., Blanchard, A. T., Pui-Yan, V. M., Derricotte, W. D., Zhang, Y., Fay, M. E., Lam, W. A., Evangelista, F. A., Mattheyses, A. L. and Salaita, K. (2018). Mapping the 3D orientation of piconewton integrin traction forces. Nat Methods 15(2): 115-118.
- Frischknecht, F. and Matuschewski, K. (2017). Plasmodium sporozoite biology. Cold Spring Harb Perspect Med 7(5).
- Goktas, M. and Blank, K. (2016). Molecular force sensors: from fundamental concepts toward applications in cell biology. Adv Material Interfaces: 1600441.
- Helgason, C. D. and Miller, C. L. (Eds.) (2013). Basic Cell Culture Protocols. 4th edition. Humana Press, New Jersey.
- Hong, V., Presolski, S. I., Ma, C. and Finn, M. G. (2009). Analysis and optimization of copper-catalyzed azide-alkyne cycloaddition for bioconjugation. Angew Chem Int Ed Engl 48(52): 9879-9883.
- Hynes, R. O. (1992). Integrins: versatility, modulation, and signaling in cell adhesion. Cell 69(1): 11-25.
- Iskratsch, T., Wolfenson, H. and Sheetz, M. P. (2014). Appreciating force and shape-the rise of mechanotransduction in cell biology. Nat Rev Mol Cell Biol 15(12): 82 5-833.
- Janmey, P. A. and Miller, R. T. (2011). Mechanisms of mechanical signaling in development and disease. J Cell Sci 124(Pt 1): 9-18.
- Kang, K. A., Wang, J., Jasinski, J. B. and Achilefu, S. (2011). Fluorescence manipulation by gold nanoparticles: from complete quenching to extensive enhancement. J Nanobiotechnology 9: 16.
- Lang, M. J., Fordyce, P. M., Engh, A. M., Neuman, K. C. and Block, S. M. (2004). Simultaneous, coincident optical trapping and single-molecule fluorescence. Nat Methods 1(2): 133-139.
- Liu, Y., Blanchfield, L., Ma, V. P.-Y., Andargachew, R., Galior, K., Liu, Z., Evavold, B. and Salaita, K. (2016). DNA-based nanoparticle tension sensors reveal that T-cell receptors transmit defined pN forces to their antigens for enhanced fidelity. Proc Natl Acad Sci U S A 113(20): 5610-5615.
- Michálek, J., Čapek, M. and Kubínová, L. (2007). Fast Algorithm for Matching of Image Pairs for Constant Brightness Applied to Stacks of Confocal Microscope Images.
- Münter, S., Sabass, B., Selhuber-Unkel, C., Kudryashev, M., Hegge, S., Engel, U., Spatz, J. P., Matuschewski, K., Schwarz, U. S. and Frischknecht, F. (2009). Plasmodium Sporozoite Motility Is Modulated by the Turnover of Discrete Adhesion Sites. Cell Host Microbe 6: 551-562.
- Muthinja, J. M., Ripp, J., Krüger, T., Imle, A., Haraszti, T., Fackler, O. T., Spatz, J. P., Engstler, M. and Frischknecht, F. (2018). Tailored environments to study motile cells and pathogens. Cell Microbiol 20(3).
- Nan, B. (2017). Bacterial Gliding Motility: Rolling Out a Consensus Model. Curr Biol 27(4): R154-R156.
- Natarajan, R., Thathy, V., Mota, M. M., Hafalla, J. C., Menard, R. and Vernick, K. D. (2001). Fluorescent Plasmodium berghei sporozoites and pre-erythrocytic stages: a new tool to study mosquito and mammalian host interactions with malaria parasites. Cell Microbiol 3(6): 371-379.
- Parsons, J. T., Horwitz, A. R. and Schwartz, M. A. (2010). Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol 11(9): 633-643.
- Polacheck, W. J. and Chen, C. S. (2016). Measuring cell-generated forces: a guide to the available tools. Nat Methods 13(5): 415-423.
- Quadt, K. A., Streichfuss, M., Moreau, C. A., Spatz, J. P. and Frischknecht, F. (2016). Coupling of Retrograde Flow to Force Production During Malaria Parasite Migration. ACS Nano 10(2): 2091-2102.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P. and Cardona, A. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7): 676-6 82 .
- Schoen, I., Pruitt, B. L. and Vogel, V. (2013). The Yin-Yang of rigidity sensing: how forces and mechanical properties regulate the cellular response to materials. Annu Rev Mater Res 43(1): 589-618.
- Totaro, A., Castellan, M., Battilana, G., Zanconato, F., Azzolin, L., Giulitti, S., Cordenonsi, M. and Piccolo, S. (2017). YAP/TAZ link cell mechanics to Notch signalling to control epidermal stem cell fate. Nat Commun 8: 15206.
- Wang, N., Butler, J. P. and Ingber, D. E. (1993). Mechanotransduction across the cell surface and through the cytoskeleton. Science 260(5111): 1124-1127.
- Wang, X. and Ha, T. (2013). Defining single molecular forces required to activate integrin and notch signaling. Science 340(6135): 991-994.
- Wiegand, T., Fratini, M., Frey, F., Yserentant, K., Liu, Y., Weber, E., Galior, K., Ohmes, J., Braun, F., Herten, D. P., Boulant, S., Schwarz, U. S., Salaita, K., Cavalcanti-Adam, E. A. and Spatz, J. P. (2020). Forces during cellular uptake of viruses and nanoparticles at the ventral side. Nat Commun 11(1): 32.
- Woodside, M. T., Behnke-Parks, W. M., Larizadeh, K., Travers, K., Herschlag, D. and Block, S. M. (2006). Nanomechanical measurements of the sequence-dependent folding landscapes of single nucleic acid hairpins. Proc Natl Acad Sci U S A 103(16): 6190-6195.
- Zhang, T. B., Zhang, C. L., Dong, Z. L. and Guan, Y. F. (2015). Determination of base binding strength and base stacking interaction of DNA duplex using atomic force microscope. Sci Rep 5: 9143.
- Zhang, Y., Ge, C., Zhu, C. and Salaita, K. (2014). DNA-based digital tension probes reveal integrin forces during early cell adhesion. Nat Commun 5(1): 5167.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Wack, M., Wiegand, T., Frischknecht, F. and Cavalcanti-Adam, E. A. (2020). An in vitro DNA Sensor-based Assay to Measure Receptor-specific Adhesion Forces of Eukaryotic Cells and Pathogens. Bio-protocol 10(17): e3733. DOI: 10.21769/BioProtoc.3733.
Category
Cell Biology > Cell-based analysis > Cell adhesion
Microbiology > Microbe-host interactions > Ex vivo model
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.