- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Co-culture of Murine Neurons Using a Microfluidic Device for The Study of Tau Misfolding Propagation
.JPG)

Published: Vol 10, Iss 16, Aug 20, 2020 DOI: 10.21769/BioProtoc.3718 Views: 5120
Reviewed by: Max SchelskiSébastien GillotinAnonymous reviewer(s)
Original research article
The authors used this protocol in:
Nov 2019
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
The deposition of misfolded, aggregated tau protein is a hallmark of several neurodegenerative diseases, collectively termed “tauopathies”. Tau pathology spreads throughout the brain along connected pathways in a prion-like manner. The process of tau pathology propagation across circuits is a focus of intense research and has been investigated in vivo in human post-mortem brain and in mouse models of the diseases, in vitro in diverse cellular systems including primary neurons, and in cell free assays using purified recombinant tau protein. Here we describe a protocol that takes advantage of a minimalistic neuronal circuit arrayed within a microfluidic device to follow the propagation of tau misfolding from a presynaptic to a postsynaptic neuron. This assay allows high-resolution imaging as well as individual manipulation of the releasing and receiving neuron, and is therefore beneficial for investigating the propagation of tau and other misfolded proteins in vitro.
Keywords: Protein misfolding propagationBackground
The propagation of misfolded protein throughout the brain underlies the spread of pathology in several neurodegenerative disorders, including Alzheimer’s disease, Parkinson’s and Prion disease (Goedert et al., 2010; Davis et al., 2018; Hallinan et al., 2019a). Understanding the underlying mechanisms may help limit disease progression and is therefore an area of intense research. Several in vivo and in vitro assays exist that monitor this process, including mouse models that develop neurodegeneration in response to overexpressing mutant human tau (Allen et al., 2002; Ramsden et al., 2005; SantaCruz et al., 2005; Gibbons et al., 2017). In vitro models include cell lines expressing biosensors that monitor aggregation (Kfoury et al., 2012; Holmes et al., 2014) and primary neuronal cultures, including those in microfluidic devices (Wu et al., 2013 and 2016, Takeda et al., 2015). In this protocol we describe in detail an in vitro assay to monitor the seeding and propagation of tau misfolding across connected neurons. This protocol builds on previously established assays, as described above, but additionally allows to monitor not just the passage of misfolded tau from one neuron to the next, but also the seeding in the previously unaffected cell, thus fully recapitulating inter-neuronal propagation. The microfluidic setup further allows individual manipulation of the pre- and postsynaptic cell, and high resolution live and fixed cell imaging of both connected neurons. This assay can be adapted for the study of the spread of other proteinopathies, such as prion or α-synuclein pathology. In addition, the basic directionalised and connected setup allows the culture of a range of different neuronal subtypes and thus the study of various other trafficking- and spread-related neurotoxic agents, including that of viruses and environmental insults such as nanoparticle toxicity.
Materials and Reagents
- Microfluidic device preparation
- Microfluidic devices
Note: Microfluidic devices are commercially available from Xona Microfluidics, or can be manufactured as previously described in Taylor et al., 2005 and Holloway et al., 2019. For unidirectional outgrowth we recommend using the “arrow” devices described in Holloway et al., 2019. - 22 x 50 mm rectangle coverslips (0.16-0.19 mm thickness, Smith Scientific, catalog number: NPS16/2250 )
- 94/16MM Petri dishes, non-vented (Greiner Bio-one, catalog number : 632180 )
- 3 M Scotch-tape
- poly-D-Lysine hydrobromide (PDL, Sigma, catalog number: P7886 )
- Sterile ddH2O
- 70% Ethanol (EtOH)
- Neurobasal medium (Gibco, catalog number: 21103-049 )
- Microfluidic devices
- Primary neuron culture and transfection
- 15 ml Falcon Conical Tubes 17/120MM (Cellstar, catalog number: 188271 )
- 50 ml Falcon Conical Tube 30/115 (Cellstar, catalog number: 227261 )
- 40 µm sterile cell strainer (Fisher Scientific, catalog number: 15360801 )
- Timed-pregnant female mouse, E15-E18
Note: We routinely use C57Bl/6 mice. - Glutamax supplement (Gibco, catalog number: 35050-038 )
- Neurobasal Media (NBM) (Gibco, catalog number: 21103-049 )
- Fetal Bovine Serum (FBS) (Gibco, catalog number: 10270-106 )
- Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, catalog number: 41965-039 )
- Opti-MEM reduced serum medium with glutamax supplement (Gibco, catalog number: 51985-026 )
- DNase (Sigma, catalog number: DN25 ; optional)
- B27 Media Supplement, 50x (Gibco, catalog number: 17504-044 )
- Lipofectamine2000 (Gibco, catalog number: 1168027 )
- 0.5% Trypsin-EDTA (Gibco, catalog number: 15400-054 )
- Phosphate Buffered Saline (DPBS), no Calcium (Ca2+) and Magnesium (Mg2+) (Gibco, catalog number: 14190-094 )
- PDL solution (see Recipes)
- Culture medium (complete NBM) (see Recipes)
- DNase solution (see Recipes)
- Fixation and imaging
Software
- FIJI (https://fiji.sc) or ImageJ (NIH, https://imagej.nih.gov/ij/, version 1.51 was used)
- Matlab (MathWorks, version R2017a was used)
- SoftWoRks software v6 (or suitable image acquisition software for the microscope to be used)
- Excel (or any spreadsheet) to collect data prior to analysis.
Equipment
- Dumont forceps #5, 11 cm, straight (Fine Science Tools, catalog number: 11254-20 )
- Dumont #5/45 forceps, 11 cm, 45 Degree Angle (Fine Science Tools, catalog number: 11251-35 )
- Scissors
- Incubator
- Laminar flow hood
- Dissection microscope
- Hemocytometer (Fisher, catalog number: 11704939 )
- Autoclave
- Tissue culture hood
- 4 °C refrigerator
- -20 °C freezer
- 37 °C water bath
- Cell culture incubator set at 37 °C, 5% CO2
- Orbital shaker (Fisher, catalog number: 10759145 )
- DeltaVision Elite microscope
Note: Alternatively, any inverted high-resolution fluorescent microscope can be used.
Procedure
Note: Perform all tissue culture steps in a sterile environment. Carry out dissections in a laminar flow hood on a dissection microscope. Perform microfluidic device preparation, tissue dissociation and cell culture in a vertical flow hood.
- Preparation of coverslips and microfluidic devices
- Glass coverslips
- Place coverslips in a plastic container containing 1 N NaOH and wash for 1 h on an orbital shaker at approximately 120 rpm, careful to avoid spills.
- Remove NaOH from the container and wash coverslips with ddH2O several times until the pH returns to neutral.
- Store coverslips in 70% ethanol.
- Place coverslips in a plastic container containing 1 N NaOH and wash for 1 h on an orbital shaker at approximately 120 rpm, careful to avoid spills.
- Microfluidic devices
- Cover the structured surfaces of the device with 3M tape to remove any particulates and fibres.
- Peel off the tape and store devices in a container containing 70% ethanol.
- Cover the structured surfaces of the device with 3M tape to remove any particulates and fibres.
- Assemble devices (1 day before dissection)
- Remove two glass coverslips from the 70% ethanol-containing container and place into a sterile Petri dish using forceps.
- Allow the glass to dry (~5 min in open air in hood) before applying 1 ml of 0.1 mg/ml PDL onto each the glass for at least 1 h to overnight (Figure 1).
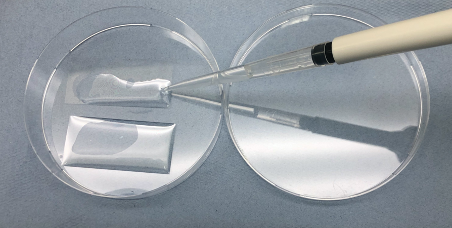
Figure 1. Coating of glass coverslips - Remove PDL solution and wash the glass with autoclaved ddH2O three times.
- Allow the PDL-coated glass to air-dry inside a sterile tissue culture hood for ~1 h prior to the addition of the microfluidic device.
Note: Glass coverslips that have been PDL-coated, washed and dried can be stored in Petri dishes at -20 °C for up to two weeks. - Remove the devices from the 70% ethanol and place the devices at an angle on the edge of a sterile Petri dish, channel-side up (i.e., upside down, see Figure 2A).
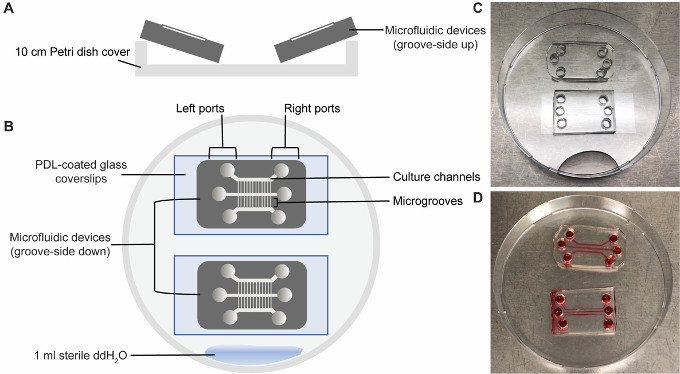
Figure 2. Assembly of microfluidic devices. (A, B) Schematic of the setup. A. The devices are dried groove-side up in a 10 cm Petri dish in a sterile environment. B. They are then assembled with the structured side facing the glass coverslip, gently pushing the device down in the non-structured areas to allow good contact for leak-free sealing between PDMS and the glass substrate. (C, D) Photos of two assembled devices. C. Dry devices before media addition. D. Two filled devices showing one well sealed (top) and a leaky (bottom) device. Devices were filled with food dye for better contrast. - Allow the devices to completely dry (~1 h).
- Using sterile forceps, set down the dry devices (channel-side down) on to the centre of the PDL-coated glass coverslips. Use the blunt end of the forceps to gently press down the devices onto the coverslips to ensure even PDMS-glass contact, removing any air pockets (see Figure 2B).
- Observe that the devices adhere to the surface of the glass, ensuring a tight seal.
- Add 10 µl of 70% ethanol to inlets at one side of the device and observe as the channels fill from one port to the next.
- Place lid on the Petri dish and examine the assembled device on a light microscope making sure that there is no leakage or bubbles trapped inside the channels and/or microgrooves.
- Next, rinse out ethanol by adding 50 µl of supplemented Neurobasal medium to each channel, allowing the device to fill. Remove ethanol from outlet and top up inlets with supplemented Neurobasal medium. Place in the incubator overnight.
- Remove two glass coverslips from the 70% ethanol-containing container and place into a sterile Petri dish using forceps.
- Glass coverslips
- Hippocampal dissection and primary cell preparation
- Sacrifice the pregnant female using CO2 followed by cervical dislocation. Remove the uterus, place it into sterile Petri dish filled with PBS and move to a horizontal flow hood for fine dissection
- Isolate the embryos and collect into fresh PBS. Make sure to rinse off remaining blood.
- Isolate embryonic brains. Using a dissection microscope, remove the meninges and dissect out the hippocampi. On average, one embryo yields ~500,000 hippocampal neurons, with between 4-10 embryos per pregnant female.
Note: A helpful illustration of murine embryonic hippocampal dissection can be found at Seibenhener and Wooten, 2012. - Collect the isolated hippocampi in 1 ml PBS into a 15 ml conical tube and move to a vertical flow hood.
- Add 100 µl Trypsin-EDTA and incubate in a water bath at 37 °C for 8 min to disrupt cell-cell contacts.
- Add 1 ml serum to inactivate the trypsin and spin cells for 3 min at 300 x g. Remove supernatant and resuspend cells in 1 ml culture medium.
- Triturate cells with a 1 ml pipette about 6-8 times. This will dissociate tissue into a single cell suspension.
Note: If the cell suspension begins to clump (often observed with older embryos), add 50 µl DNAse solution for trituration. - Filter the dissociated cells through a 40 µm cell strainer to remove non-dissociated larger chunks and count cells using a hemacytometer.
- Cell Culture
- Transfer 300,000 cells into an Eppendorf tube.
Note: If you plate more than 2 channels per device adjust cell number and media volume accordingly. Use 60,000-70,000 cells per channel. - Spin the tube at 300 x g for 3 min.
- Gently resuspend the cell pellet in the Eppendorf tube in 50 µl complete NBM.
- Remove the primed devices from the incubator and transfer into a sterile culture hood.
- Remove all the media from all of the inlets in the device.
- Starting from the left port (Al) add 10 µl of cell suspension and remove 1 µl from the adjacent connecting right port (Ar) to encourage flow of cells (see Figure 3A).
- Close the Petri dish and use a light microscope to check that cells have entered the main culture channels. Return Petri dish into a sterile hood.
- Continue on to fill the other channels (e.g., Bl) as above.
- Allow the cells to settle close to the microgrooves by propping the Petri dish containing the device at an angle (see Figure 3B). After 5-10 min transfer the Petri dishes to the incubator for an additional 45 min to allow the cells to adhere to the glass.
- Top up medium by adding 50 µl of complete NBM to all the ports.
- Add 1 ml of sterile ddH2O on the side of the Petri dish to maintain humidified atmosphere before replacing the lid and place cells in incubator. This counts as day in vitro (DIV) 0.
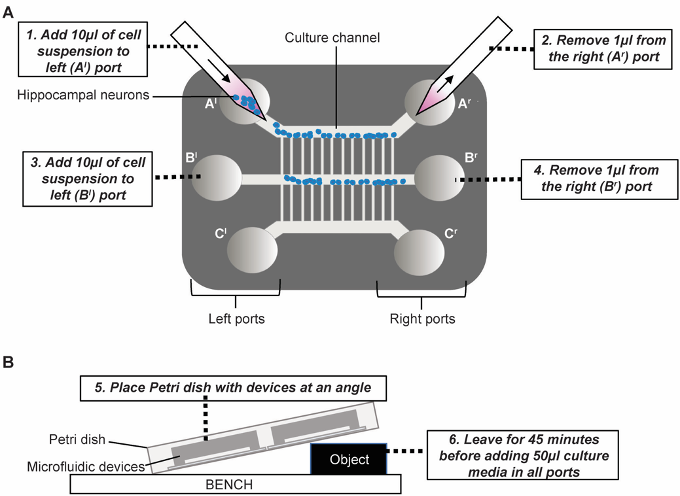
Figure 3. Plating of neurons into the device. A. Add neuronal suspension into port, as close to the channel as possible, and encourage flow into the channel by removing a small volume from the other side. B. Once plated, place devices at a gentle angle so that gravity guides the cells to settle near the microchannels. For this, the culture channels should be horizontal with channel A on top, so that cells in culture channel A settle near the microgrooves connecting to channel B and those in channel B near the microgrooves connecting to channel C.
- Transfer 300,000 cells into an Eppendorf tube.
- Transfection
On the following day (DIV1), transfect the cultures in the devices with the relevant constructs. To assay tau propagation, we transfected human tau 0N4R constructs described in Hoover et al., 2010 as follows: GFP-tauE14 in channel A, and RFP-tauWT in channel B (Figure 4A).- Add 25 µl of OptiMEM each in two separate Eppendorf tubes, labeled 1 and 2.
- Add 0.5 µg of plasmid in tube 1 and 0.5 µl Lipofectamine2000 in tube 2. Mix by pipetting up and down.
- Make the transfection reagent by mixing tube 1 and 2 by gentle bubbling. Do not vortex. Allow to stand inside a sterile hood for 20 min.
- Take the device from the incubator into a sterile TC hood and remove all media in both ports of channel A (the channel to be transfected) (see Figure 4B for workflow).
- Top up ports connected to culture channel B with complete NBM. A high volume of media within the ports of channel B will fluidically isolate this channel and prevent flow of the transfection reagent from channel A to channel B.
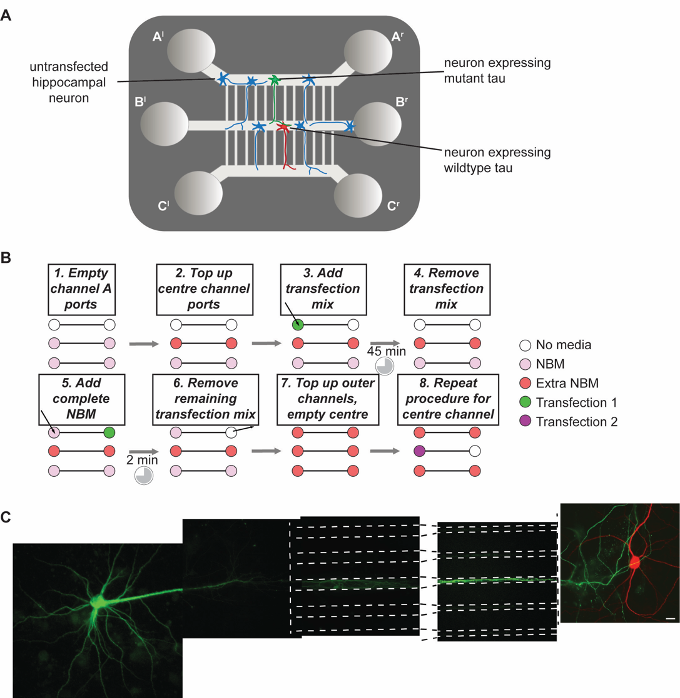
Figure 4. Transfection within the device. A. Diagram of the transfected device. Individual presynaptic neurons in channel A express mutant tau (green), while the majority of cells remain untransfected (blue). Transfected projecting neurons make contact with postsynaptic neurons expressing fluorescent wildtype tau. B. Workflow of the transfection procedure. C. Partial reconstruction of two transfected, connecting neurons within a device. Scale bar = 10 µm. - Add 50 µl transfection reagent on the left feeding port of channel A.
- Close the Petri dish lid and incubate for 40 min in the incubator at 37 °C.
- Remove all media from ports A and add 50 µl of fresh complete NBM in all the left ports to flush remove the remaining transfection reagent.
- Leave for 2 min before removing excess media in the right port.
- Now, add 50 µl of complete NBM to all ports.
- Repeat to transfect channel B.
- Add 1 ml sterile ddH2O on the side of the Petri dish to prevent drying out before placing back into the incubator overnight.
- Feed cultures every 2-3 days by topping up one port. Remove excess media from opposite port as necessary.
- Add 25 µl of OptiMEM each in two separate Eppendorf tubes, labeled 1 and 2.
- Fixation
Note: Devices can be imaged live in phenol red-free media, or fixed for later observation. We typically fix the devices unless observing dynamic processes.- On the desired day in vitro, stop the experiment by fixing cells.
- Remove the media from all ports.
- Fill port Ar with 50 µl PFA. Allow flow into the main channel and through the microgrooves.
- Top up port Ar and fill port Br.
- Leave to fix for 15 min.
- Remove PFA solution and fill device with PBS or TBS.
- Fixed devices can be stored short-term in the fridge (up to 2 weeks), but ensure that they do not dry out by topping ports up with PBS or TBS.
- Microscopy
Note: while we performed the imaging on a DeltaVision microscope, any high resolution inverted fluorescence microscope is suitable. We typically use a 60x oil objective, and 1 pixel = 0.1075 µm.- Clean bottom of the glass coverslip with 70% EtOH.
- Image cells on an inverted fluorescence microscope using a 60x oil objective and adequate filter settings for the GFP and RFP channels.
- For every connecting pair, ensure you image the corresponding cell bodies and distal axonal segments. Ideally capture corresponding differential interfering contrast (DIC) or phase image for quality control.
Notes:- For the analysis you will need an axonal stretch that is in focus for a length of ≥ 75 µm.
- Any axonal stretch that is ≥ 300 µm from the cell body can be used as “distal axon”. Ensure you only use one axonal stretch per neuron for analysis.
- Ensure you do not image blebbed cells as this will interfere with the analysis (see Figure 5). In doubt check axonal integrity on transmitted light channel to ensure that the axon structure is smooth.

Figure 5. Examples of two blebbed axonal stretches expressing GFP-TauE14 that should be excluded from analysis. Scale bar = 10 µm.
- For the analysis you will need an axonal stretch that is in focus for a length of ≥ 75 µm.
- Capture image at highest pixel depth possible (we used 16bit).
Data analysis
- Reading out fluorescence intensities along the axon in ImageJ/FIJI
Note: The data analysis is based on the observation that aggregation of fluorescent tau leads to a fluorescence redistribution within the cell, with brighter spots and stretches appearing that represent accumulation (Figures 5 and 9).- Open original microscopy file of a distal axonal segment in FIJI or ImageJ.
- To create a plot profile on the opened image, right click on the line tool (Figure 6) and select “Segmented Line” in the drop-down menu (Figure 6A).
- Create a segmented line plot along the length of the axon (Figure 6B). Double-click to stop the creating a segmented line.
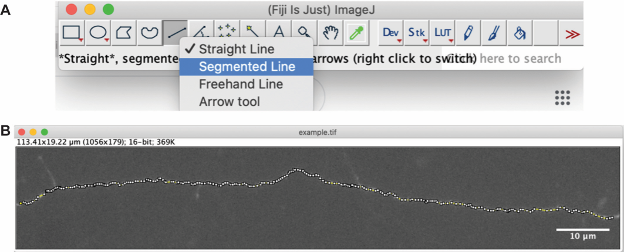
Figure 6. Trace the axon using the segmented line tool - To quantify the fluorescence values along the axon that has been plotted, go to Analyze→Plot Profile (Figure 7). This will generate a list of pixel intensity values corresponding to the length of the axon that was plotted (Figure 8).
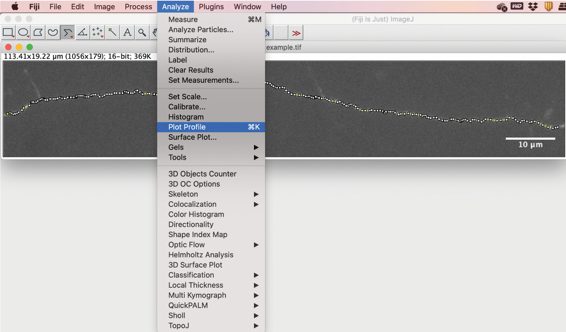
Figure 7. Generate a plot profile to read out fluorescence intensity values along the length of the axon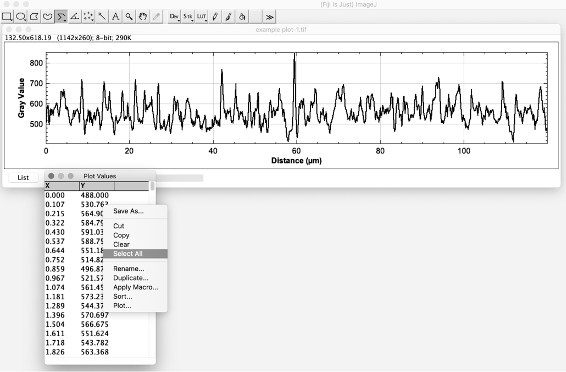
Figure 8. To save the values for further analysis, click on List and Select All values to copy. Collect as columns in a spreadsheet, ensuring you keep track which column of data belongs to which original axonal image. - Click on the “List” (as shown in the blue arrow in Figure 8) to view the individual values. To save the data: Select all→Copy.
- Paste into columns in an excel spreadsheet.
- Figure 9 shows an example of an analysed stretch next to its plot profile.
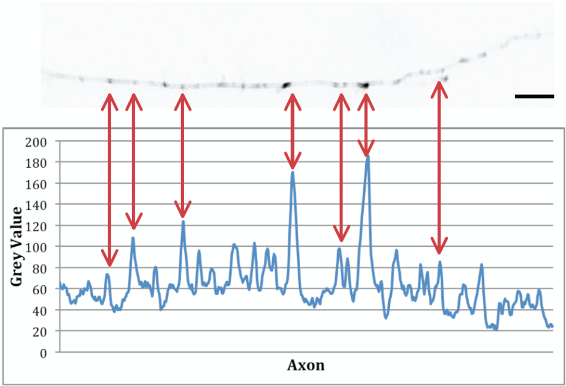
Figure 9. Example of an imaged axon and its corresponding plot profile
- Paste all measured stretches from one experiment into one file. Use one spreadsheet per condition.
- You will initially also need one spreadsheet with a range of control values from different time points.
- Open original microscopy file of a distal axonal segment in FIJI or ImageJ.
- Fluorescence distribution analysis
Note: To begin this part of the analysis you need a range of “control” axons from different experimental sets across the time points analysed. We recommend a minimum of 30 imaged axonal stretches. This will determine the allowed baseline variability within your experimental data. It also allows for individual control axons to score positive in the later analysis.- Establishing the allowed baseline variation across experiments
- Copy a random selection of values from control axons across different experiments into a spreadsheet.
- Open the MATLAB script “baseline” found at https://www.mathworks.com/matlabcentral/fileexchange/74503-baseline-analysis.
- Import data into the MATLAB script and run the script.
- The script will first ensure that different expression levels in individual cells do not skew the analysis by substracting the 10th percentile values from each series (Figure 10).
Note: Explanation of script “baseline” (https://www.mathworks.com/matlabcentral/fileexchange/74503-baseline-analysis). This script is designed to determine the signal variability within the controls across biological repeats and time points. For this, you need sample control axons randomly picked from the different experiments and paste these into individual columns in an excel spreadsheet. When you run the script it imports the columns as “data”. First, the 10th percentile from each column is determined and deducted to eliminate variability due to different expression levels. The result is designated “zero”. These values are then listed in a single column, and the mean and standard deviation are determined. Choose mean plus 5 standard deviations as allowed baseline variation for script “axon_analysis”.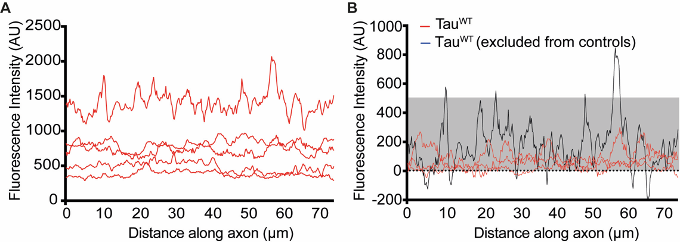
Figure 10. Visual representation of the first part of the data analysis, determining allowed baseline variability. In all analysis, different expression levels (as seen in raw plot profiles displayed in A) are taken into account by substracting the 10th percentile of all values in each trace (the result is displayed in B). The cutoff determined in figure 10 results in the grey shaded box in B and allows for control axons (such as the black trace) to score positive in the later analysis. - It will then determine the average fluorescent intensity and the standard deviation. Choose mean plus 5 standard deviations as allowed baseline variation. This ensures your analysis is robust against minor variations in focus along the axonal stretch.
- Copy a random selection of values from control axons across different experiments into a spreadsheet.
- Scoring aggregation within one experiment
- Use MATLAB script “axon_analysis” found here.
Note: Explanation of the script “axon_analysis” (available here). This script is designed to detect increased variability in fluorescence distribution, which is observed upon formation of fluorescent accumulations/ aggregates. You need paired control and experimental axons from one repeat, the values pasted as columns into two separate spreadsheets. Important: adjust the number in line 25 to the number determined following script “baseline_analysis”. The script imports your control (e.g., wildtype tau) values as “control” and your experimental (e.g., mutant tau) values as “experimental”. As above, all values are zeroed to the 10th percentile to eliminate variability due to different expression levels (lines 29, 32 and 64, 67). The first part of the script determines the variability within this control dataset. To allow for control axons to score positive, only those axons falling within the allowed baseline variability determined in the script “baseline_analysis” are included (line 25). A logical array (line 51) then tests for each individual fluorescent value whether it is larger than the mean plus 5 standard deviations of those axons that were included into the control group in line 41. Those that score positive in each column are assigned a “1” and are added up and divided by the total number of individual values per column. This gives a fraction of axonal values scoring positive. If this fraction is > 0.1 (10% of the values) the axon is counted as positive. Line 60 determines the percentage of control axons meeting this criteria.
The second part of the script tests the experimental dataset. Following deduction of the 10th percentile (lines 64, 67), the script tests all individual values against the maximum allowed baseline value determined from the controls (line 71), adds up the positive scores from each axon (line 74), normalizes these to the size of the dataset (lines 77, 78) to determine the percentage of axons scoring positive in this experiment (line 80). - Adjust in line 25 to the value determined as allowed baseline variation above.
- Import control and experimental data set from one experiment as “control” and “experimental”.
- The script will first determine the observed variation of all axons that are within the allowed variability across experiments in this particular experiment (Figure 10B, grey shaded area), and then use this value to determine the percentage of control axons that have more than 10% of their values outside the allowed variability and thus score positive (such as the black trace in Figure 10B).
- It will then determine the percentage of experimental axons that have more than 10% of their values outside the allowed variability and thus score positive (see Figure 11).
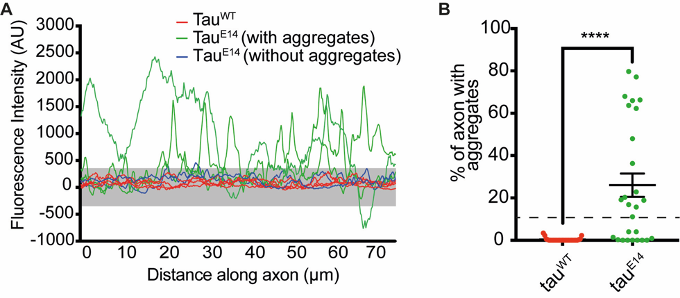
Figure 11. Visual representation of the aggregate analysis. First, intensity values outside the allowed variability are determined (A, values outside the grey box). Then the script calculates what percentage of axonal stretch falls within this category. Results for individual axons are plotted in B. The axons are scored as positive if more than 10% of their values are in this category. This allows to rule out false-positives such as due to untransfected axons crossing. Finally, the percentage of axons that score positive within one experiment is calculated. - You now have the results for one repeat.
- Use MATLAB script “axon_analysis” found here.
- Establishing the allowed baseline variation across experiments
Notes
- Transfection efficiency is generally low, expect ~2-3 transfected neurons that also project through the microgrooves to the adjacent channel per device.
- Each axon that projects through to the next channel typically branches out widely, thus almost every projecting neuron will make contact with a transfected neuron in the next channel.
- The low transfection efficiency allows for full reconstruction of the two connecting neurons. Viral transduction may be used, but its generally high efficiency hampers the ability to identify individually connecting neurons and thus is more useful for biochemical approaches.
- In our hands the aggregation analysis was highly reproducible between different preparations, as well between individuals performing the experiment.
- We typically observed aggregation, measured as formation of dense fluorescent puncta, from DIV8 onwards.
- Neurons containing fluorescent aggregates are positive for misfolded tau (MC1 staining), but due to low transfection efficiency we have not performed biochemical analysis to better define the nature of these aggregates.
Recipes
- PDL solution
0.1 mg/ml poly-D-lysine in sterile H2O
Can be stored frozen as 10 mg/ml stock or 0.1 mg/ml working aliquots - Culture medium (complete NBM)
Fully supplemented Neurobasal medium
To 49 ml Neurobasal medium, add 1 ml B27 and 125 µl glutamax or glutamine - DNase solution
Resuspend lyophilised powder to 1 mg/ml in neurobasal medium and store frozen in aliquots - PFA
4% paraformaldehyde/20% sucrose in PBS
Acknowledgments
This work was supported by Alzheimer's Research UK (Grants ARUK-PhD2014-10 and ARUK-PPG2017B-001), the Gerald Kerkut Charitable Trust and the Biotechnology and Biological Sciences Research Council (BB/L007576/1). This is the detailed protocol underlying the propagation work published in Hallinan et al., 2019b.
Competing interests
The authors declare no competing financial interests.
Ethics
All experiments were performed in accordance with the Animals (Scientific Procedures) Act 1986 set out by the UK Home Office.
References
- Allen, B., Ingram, E., Takao, M., Smith, M. J., Jakes, R., Virdee, K., Yoshida, H., Holzer, M., Craxton, M., Emson, P. C., Atzori, C., Migheli, A., Crowther, R. A., Ghetti, B., Spillantini, M. G. and Goedert, M. (2002). Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci 22(21): 9340-9351.
- Davis, A. A., Leyns, C. E. G. and Holtzman, D. M. (2018). Intercellular spread of protein aggregates in neurodegenerative disease. Annu Rev Cell Dev Biol 34: 545-568.
- Gibbons, G. S., Banks, R. A., Kim, B., Xu, H., Changolkar, L., Leight, S. N., Riddle, D. M., Li, C., Gathagan, R. J., Brown, H. J., Zhang, B., Trojanowski, J. Q. and Lee, V. M. (2017). GFP-mutant human tau transgenic mice develop tauopathy following CNS injections of Alzheimer's brain-derived pathological tau or synthetic mutant human tau fibrils. J Neurosci 37(47): 11485-11494.
- Goedert, M., Clavaguera, F. and Tolnay, M. (2010). The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci 33(7): 317-325.
- Hallinan, G. I., Pitera, A. P., Patel, P., West, J. and Deinhardt, K. (2019a). Minimalistic in vitro systems for investigating tau pathology. J Neurosci Methods 319: 69-76.
- Hallinan, G. I., Vargas-Caballero, M., West, J. and Deinhardt, K. (2019b). Tau misfolding efficiently propagates between individual intact hippocampal neurons. J Neurosci 39(48): 9623-9632.
- Holloway, P. M., Hallinan, G. I., Hegde, M., Lane, S. I. R., Deinhardt, K. and West, J. (2019). Asymmetric confinement for defining outgrowth directionality. Lab Chip 19(8): 1484-1489.
- Holmes, B. B., Furman, J. L., Mahan, T. E., Yamasaki, T. R., Mirbaha, H., Eades, W. C., Belaygorod, L., Cairns, N. J., Holtzman, D. M. and Diamond, M. I. (2014). Proteopathic tau seeding predicts tauopathy in vivo. Proc Natl Acad Sci U S A 111(41): E4376-4385.
- Hoover, B. R., Reed, M. N., Su, J., Penrod, R. D., Kotilinek, L. A., Grant, M. K., Pitstick, R., Carlson, G. A., Lanier, L. M., Yuan, L. L., Ashe, K. H. and Liao, D. (2010). Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68(6): 1067-1081.
- Kfoury, N., Holmes, B. B., Jiang, H., Holtzman, D. M. and Diamond, M. I. (2012). Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem 287(23): 19440-19451.
- Ramsden, M., Kotilinek, L., Forster, C., Paulson, J., McGowan, E., SantaCruz, K., Guimaraes, A., Yue, M., Lewis, J., Carlson, G., Hutton, M. and Ashe, K. H. (2005). Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J Neurosci 25(46): 10637-10647.
- Santacruz, K., Lewis, J., Spires, T., Paulson, J., Kotilinek, L., Ingelsson, M., Guimaraes, A., DeTure, M., Ramsden, M., McGowan, E., Forster, C., Yue, M., Orne, J., Janus, C., Mariash, A., Kuskowski, M., Hyman, B., Hutton, M. and Ashe, K. H. (2005). Tau suppression in a neurodegenerative mouse model improves memory function. Science 309(5733): 476-481.
- Takeda, S., Wegmann, S., Cho, H., DeVos, S. L., Commins, C., Roe, A. D., Nicholls, S. B., Carlson, G. A., Pitstick, R., Nobuhara, C. K., Costantino, I., Frosch, M. P., Muller, D. J., Irimia, D. and Hyman, B. T. (2015). Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer's disease brain. Nat Commun 6: 8490.
- Taylor, A. M., Blurton-Jones, M., Rhee, S. W., Cribbs, D. H., Cotman, C. W. and Jeon, N. L. (2005). A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods 2(8): 599-605.
- Wu, J. W., Herman, M., Liu, L., Simoes, S., Acker, C. M., Figueroa, H., Steinberg, J. I., Margittai, M., Kayed, R., Zurzolo, C., Di Paolo, G. and Duff, K. E. (2013). Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem 288(3): 1856-1870.
- Wu, J. W., Hussaini, S. A., Bastille, I. M., Rodriguez, G. A., Mrejeru, A., Rilett, K., Sanders, D. W., Cook, C., Fu, H., Boonen, R. A., Herman, M., Nahmani, E., Emrani, S., Figueroa, Y. H., Diamond, M. I., Clelland, C. L., Wray, S. and Duff, K. E. (2016). Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci 19(8): 1085-1092.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Hallinan, G. I., Lopez, D. M., Vargas-Caballero, M., West, J. and Deinhardt, K. (2020). Co-culture of Murine Neurons Using a Microfluidic Device for The Study of Tau Misfolding Propagation. Bio-protocol 10(16): e3718. DOI: 10.21769/BioProtoc.3718.
- Hallinan, G. I., Vargas-Caballero, M., West, J. and Deinhardt, K. (2019b). Tau misfolding efficiently propagates between individual intact hippocampal neurons. J Neurosci 39(48): 9623-9632.
Category
Neuroscience > Nervous system disorders > Cellular mechanisms
Neuroscience > Cellular mechanisms > Cell isolation and culture
Cell Biology > Cell isolation and culture > Microfluidic culture
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.