- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Mapping mRNA-18S rRNA Contacts Within Translation Initation Complex by Means of Reverse Transcriptase Termination Sites and RNAseq
Published: Vol 10, Iss 16, Aug 20, 2020 DOI: 10.21769/BioProtoc.3713 Views: 5233
Reviewed by: Alexandros AlexandratosSanjeeva S MetikalaJens Kretschmer
Original research article
The authors used this protocol in:

Dec 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The nucleotides involved in RNA-RNA interaction can be tagged by chemical- or UV-induced crosslinking, and further identified by classical or modern high throughput techniques. The contacts of mRNA with 18S rRNA that occur along the mRNA channel of 40S subunit have been mapped by site-specific UV crosslinking followed by reverse transcriptase termination sites (RTTS) using radioactive or fluorescent oligonucleotides. However, the sensitivity of this technique is restricted to the detection of those fragments that resulted from the most frequent crosslinkings. Here, we combined RTTS with RNAseq to map the mRNA-18S rRNA contacts with a much deeper resolution. Although aimed to detect the interaction of mRNA with the ES6S region of 18S rRNA, this technique can also be applied to map the interaction of mRNA with other non-coding RNA molecules (e.g., snRNAs, microRNAs and lncRNAs) during transcription, splicing or RNA-mediated postranscriptional regulation.
Background
Interaction of mRNA with non-coding RNAs is involved in every step of mRNA life cycle, from its biogenesis and processing into the nucleus to translation and ultimate degradation in the cytoplasm. These interactions can occur either within large molecular machines as ribosome and spliceosome, and in smaller complexes such as RISC (RNA-induced silencing complex) or during lncRNA-mediated gene expression regulation (Pisarev et al., 2008; Engreitz et al., 2014; Sharma et al., 2016). For translation, the use of in vitro reconstructed initiation complexes or drugs that freeze ribosomes during the initiation or elongation steps have allowed to snapshot those residues in mRNA and ribosomal RNAs involving in RNA-RNA contact. The incorporation of photoactivatable nucleotides (e.g., 4-thio-UTP) surrounding the AUGi in the mRNA allows site-specific crosslinking upon UV irradiation at 360 nm without affecting non-labeled residues. The resulting crosslinking adducts block primer extension by reverse transcriptase, thus allowing the identification of the crosslinking sites by sequencing the 3′ end of the resulting cDNA fragment (Kielpinski et al., 2013). Recently, a combination of reverse transcriptase termination sites (RTTS) with RNAseq has been described, allowing the identification of every premature stop during reverse transcriptase (Díaz-López et al., 2019). Apart from UV-induced crosslinking, there are other sources of RT stops, including accidental fragmentation of RNA template during extraction or preexisting chemical modification in mRNA residues (methylation and pseudourylation) that must be controlled during the experiment. Here, we describe in more detail this protocol that could be potentially applied to map any RNA-RNA interaction with nucleotide resolution using a labeled RNA bait. Our data working on mRNA-rRNA interaction during translation initiation indicates that the sensitivity of this protocol could be enough to detect even transient RNA-RNA interactions that regulate gene expression.
Materials and Reagents
Note: Make sure all reagents and materials are RNase-free. Materials are stored at room temperature unless otherwise indicated.
- 1.5 ml Eppendorf tubes (Eppendorf, catalog number: 00 30120086 )
- 1 ml thickwall polycarbonate ultracentrifuge tubes (Beckman, catalog number: 343778 )
- Parafilm
- Ultrapure nuclease-free water (Invitrogen, catalog number: 10977-035 )
- Sucrose (Sigma, catalog number: 84097 )
- Agarose (Pronadisa, catalog number: 8010 .00)
- Ethanol (Merck, absolute for analysis, catalog number: 51976 )
- Isopropanol (Merck, absolute for analysis, catalog number: I9516 )
- Phenol:clorophorm:isoamyl alcohol (25:24:1) (Sigma, catalog number: P2069 , store at 4 °C)
- Chroma-Spin STE-10 purification columns (Takara, catalog number: 636055 )
- Magnesium chloride 1 M solution (Sigma, catalog number: M1028 )
- Magnesium acetate (Sigma, catalog number: M5661 )
- Potasium acetate (Sigma, catalog number: P9541 )
- Sodium acetate (Sigma, catalog number: S2889 )
- Trizma base (Sigma, catalog number: 93362 )
- EDTA (Sigma, catalog number: 0 3677 )
- LiDS (Sigma, catalog number: L9781 )
- Nuclease-treated rabbit reticulocyte lysates (including amino acid mix 1mM) (Promega, catalog number: 4960 , store at -70 °C)
- GMP-PNP (Calbiochem, catalog number: sc-215113 , store at -20 °C)
- RNase inhibitor (New England Biolabs, catalog number: M0307S , store at -20 °C)
- HighScribe T7 polymerase kit (New England Biolabs, catalog number: E2040S , store at -20 °C)
- 4-thio-UTP (Jena Biosciences, catalog number: NU-1156S , sotre at -20 °C)
- Oligo d(T) 25 magnetic beads (New England Biolabs, catalog number: S1419S , store at 4 °C)
- Glycogen (Illumina, catalog number: 15073019 , store at -20 °C)
- QIAquick Gel extraction kit (QIAGEN, catalog number: 28704 )
- SuperScript III First-strand synthesis system for RT-PCR (Invitrogen, catalog number: 18080-051 , store at -20 °C)
- RNase H (Invitrogen, catalog number: 18021-014 , store at -20 °C)
- Supreme 2x green master mix (NZYtech, catalog number: MB05403 , store at -20 °C)
- Phusion DNA polymerase (New England Biolabs, catalog number: M0530S , store at -20 °C)
- CHROMA SPIN-30+TE-30 (Takara, catalog number: 636069 )
- DNA clean & Concentrator-25 (Epigenetics, catalog number: D4005 )
- Trizma base (Sigma, catalog number: 93362 )
- Agencourt AMPURE XP (Beckman coulter, catalog number: A63880 )
- DNA Kb ladder (New England Biolabs, catalog number: N3232L )
- Circligase (Illumina, catalog number: ASLPA1212 , store at -20 °C)
- Primers (all from Sigma, store at -20 °C):
18S_rev: CACCTCTAGCGGCGCAATACG
adapter_ligation: [phos] AGATCGGAAGCGTCGGACTGTAGAACTCTGAACGTGT
18S adapter: AGACGTGTGCTCTTCCGATCTCACCTCTAGCGGCGCAATACG
PR_fwd: ATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGAG
INDEX: CAAGCAGAAGAACGGCATACGAGANNNNNNNGTGACTGGAGTTCAGACGT GTGCTCTTCCGATCT
Flat_fw: GGATCCTAATACGACTCACTATAGGGACCCACCAACACAGCACCATGAACAACGAGCCACCGACAGGTGATGAGTGATGACGGAGGCACACACGACAGACAACCGAGAGAGCAGAACGAGACCACAC
U_rev polyA: (T)25 GTGTGGTCTCGTTCTGCTC
T7_U_fw: GGATCCTAATACGACTCACTATAGGG - Polysome buffer (see Recipes)
- TE buffer (see Recipes)
- 1x TAE buffer (see Recipes)
- SuperScript III cDNA synthesis mix (per reaction) (see Recipes)
- Circligase enzyme mix (per reaction) (see Recipes)
- Lysis/binding buffer (see Recipes)
- Wash Buffer 1 (see Recipes)
- Wash Buffer 2 (see Recipes)
- Low Salt Buffer (see Recipes)
Equipment
- P20, P200 and P1000 Pipetman automatic pipettes (Gilson)
- TL100 ultracentrifuge (Beckman)
- TLA 100.2 rotor
- Lamp holder with 2 x 6W 360 nm lamps (Philips, model: TL6W ) (home made)
- Vortex (Biosan, vortex V-1 plus)
- -20 °C freezer (Liehberr)
- Fume hood
- Thermomixer (Eppendorf, comfort)
- Thermoblock equiped with metalblocks (LabNet, Accublock)
- NanoDrop spectrophotometer (Thermo Scientific, Model 1000)
- Magnetic separator rack (New England Biolabs, catalog number: S1506S )
- Benchtop centrifuge (Thermo, model: Megafuge 16R )
- Minifuge (Hettich, Mikroliter)
- Gel electrophoretic system (Bio-Rad, model: Mini-Sub Cell GT System )
- Bioanalyzer (Agilent, model: 2100 )
- NextSeq 550 system (Illumina)
Software
- FASTX toolkit version 0.0.13 (http://hannonlab.cshl.edu/fastx_toolkit)
- Bowtie 2 v2.3.5.1 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml) (Langmead and Salzberg, 2012)
- Featurecounts. Available in http://www.bioconductor.org (Liao et al., 2014)
Procedure
- Preparation of mRNA containing 4-thio-UTP (4sU-mRNA) by in vitro transcription
We synthesized a 128-nt mRNA containing minimal secondary structure bearing a short (19 nt) 5′ UTR an 3' polyadenylated. Photoactivatable 4-thio-UTP can be incorporated at positions +2 (the A of AUG is denoted as +1), +24, +27, +31 or +34 of mRNA. We use 4-thio-UTP at a four-fold higher concentration than UTP to ensure that every mRNA molecule is labeled with a least one 4-thio-UTP (Figure 1A).
Figure 1. Overview of the protocol for the identification of mRNA:18S rRNA contacts within 48S complex assembled in vitro. A. Schematic diagram of the mRNA used to assemble translation initiation complex (48S-PIC) in RRL. The positions in the mRNA labeled with 4-thio-U are numbered respect to the AUGi. B. Overview of the experimental method to identify mRNA-18S rRNA contacts by specific crosslinking of 4-thio-U, followed by reverse transcriptase termination site (RTTS) assay and next-generation sequencing (NGS).- DNA template preparation
- Mix primers Flat_fw and U_rev polyA at 1 μM final concentration in 50 μl of 1x Supreme green master mix and incubate at 95 °C for 5 min, followed by 1 min at 45 °C and 10 min at 72 °C.
- Then, use 2 μl of this mixture to program a PCR amplification with T7_U_fw and U_rev polyA primers (both at 0.5 μM final concentration) in 50 μl 1x Supreme green master mix (30 cycles, 95 °C 2 min, 45 °C 2 min, 72 °C 45 s, final extension of 72 °C 10 min).
- Use a 2 μl sample to check the size of the PCR product (about 200 nt) by electrophoresis in 1.5% agarose gel in TAE 1x buffer.
- Then, purify the PCR fragment with DNA clean & Concentrator-25 columns, that typically yields 25 μl at 0.3-0.5 μg/μl.
- mRNA synthesis and purification
- Program an in vitro transcription using the HighScribe T7 polymerase kit in the presence of 4-thio-UTP.
- Mix the following components in a final volume of 20 μl:
2 μl of 10x Reaction buffer
1 μl of ATP/CTP/GTP 20mM each (1 mM final concentration)
1 μl UTP 5 mM (0.25 mM final concentration)
1 μl 4-thio-UTP 20 mM (1 mM final concentration)
0.5 μl RNasin® Plus RNase Inhibitor
1 μg DNA template
2 μl T7 RNApol - Bring to 20 μl with H2O and incubate in the dark for 2-3 h at 37 °C.
- Digest the DNA template with 1 μl of DNase I (1U/ μl) for 15 min at 37 °C. Then, bring the final volume of the mixture to 40 μl with H2O.
- Then load the mixture in a prepacked chromaspin-30 column and centrifuge at 700 x g for 5 min in a benchtop centrifuge at room temperature.
- Recover the elute and use 1 μl to measure the concentration of the RNA in a NanoDrop spectrophotometer (typically 0.2-0.5 μg/μl). Store the mRNA at -70 °C.
- DNA template preparation
- Assembly of translation initiation complex (48S-PIC) in rabbit reticulocyte lysate (RRL) and UV crosslinking
Program two 100 μl translation mixtures with 4sU-mRNA in the presence of non hydrolyzable GMP-PNP to promote the accumulation of 48S preinitiation complex (PIC) (Figure 1). One of the samples will not receive irradiation (control).- 48S-PIC assembly
- Mix the following components per duplicate:
70 μl of RRL (70% of final volume)
16 μl of H2O
2 μl aminoacid mix (1 mM)
8 μl of magnesium acetate 20 mM (2.5 mM final concentration)
2 μl of GMP-PNP 100 mM (2 mM final concentration)
2 μl of RNase inhibitor - Pre-incubate 5 min at 30 °C and then add 0.5 μg of 4sU-mRNA.
- Mix gently and incubate another 25 min at 30 °C.
- Place the tube on ice to stop the reaction.
- Mix the following components per duplicate:
- UV crosslinking.
- Cut a piece of parafilm and place it between two metal blocks with the plastic cover facing up.
- Fix the assembly carefully with elastic rubber bands and precool for at least 2 h at -20 °C.
- Carefully remove the upper block and the plastic cover, and place the bottom block with the parafilm on an ice box as indicated below (Figure 2):

Figure 2. UV lamp assembly on sample for crosslinking. Sample (red dot) is placed on parafilm for UV irradiation as indicated. Up to six samples can be irradiated simultaneously. - Using a P200 pipet, place one of the samples on the parafilm and extend it with the tip to form a 2 cm diameter smear.
- Place the lamp holder on the box so that UV lamps are 3 cm away from the sample and irradiate 20 min.
- Let the other sample on ice (not irradiated control).
- 48S-PIC assembly
- Poly (A) mRNA isolation from whole ribosomal pellet
- Transfer both the irradiated and not irradiated samples (100 μl) to new Eppendorf tubes on ice and slowly add 800 μl of polysome buffer (Recipe 1)
- Mix well and carefully place the mix (850-900 μl) on sucrose cushion made in a 1 ml ultracentrifuge tube (100 μl 20% sucrose solution in polysome buffer).
- Place the tubes in a TLA 100.2 rotor and centrifuge at 250,000 x g for 3 h at 4 °C. At this step, the ribosomal pellet resembles a translucent lentil.
- Remove the supernatant with a P1000 pipette and gently wash the ribosomal pellet with 200 μl of TE buffer for 3-5 s avoiding touching the pellet with the tip.
- Repeat washing twice more. Add 500 μl of Lysis-Binding Buffer (Recipe 6) and let pellet dissolve for 15 min at room temperature.
- Save a 30 μl aliquot from the not irradiated sample to isolate total RNA (see below).
- To the rest, add 200 μl of oligo d(T)25 magnetic beads (0.25 ml of 5 mg/ml suspension) previously equilibrated in 500 μl Lysis/Binding Buffer for 5 min.
- Incubate at room temperature for 15 min in a rotating carousel at 30 rpm.
- Place the tube into magnetic rack and pull magnetic beads to the side of the tube.
- Then remove and discard the supernatant and add 0.5 ml of Wash Buffer 1 (Recipe 7) to the beads.
- Mix with rotation for 1 min (as describe in Step C8) and capture the beads as described above.
- Repeat washing once more with Wash Buffer 1, twice with Wash Buffer 2 (Recipe 8) and once with Low Salt Buffer (Recipe 9).
- Finally, add 100 μl of H2O to the beads and heat at 50 °C for 2 min in a thermomixer to elute the RNA.
- Measure the concentration in a NanoDrop (typically 50-100 ng/μl) and store at -70 °C.
- Total RNA extraction
- To extract total RNA, bring the 30 μl aliquot saved before (Step C6) to 200 μl with H2O.
- Then, add 200 μl of phenol:chloroform:isoamyl alcohol and vortex vigorously for 1 min.
- Let the tube stand for 5 min and repeat the vortexing three more times.
- Centrifuge at maximum speed for 5 min in a minifuge at room temperature and transfer the upper phase to a new tube avoiding touching the interphase.
- Finally, precipitate the RNA with 2 volumes of 100 % ethanol and 0.2 M of sodium acetate pH 5.2 (final concentration) at -20 °C O/N.
- Centrifuge at maximum speed for 15 min in a minifuge at 4 °C and wash the pellet with 0.5 ml 70% ethanol.
- Repeat the centrifugation at room temperature, remove the ethanol and let the pellet dry 5 min on the bench.
- Then, dissolve the pellet in 20 μl of H2O and store at -70 °C.
- Retrotranscription of 18S rRNA
Use irradiated, not irradiated and total RNA samples for retrotranscription with primer 18S_rev
(targeting 18S rRNA) following the manufacturer specifications:- Mix 0.5 μg of RNA samples (typically 5-10 μl) with 0.5 μM of 18S_rev and 1 μl of 10 mM dNTPs in a final volume of 10 μl.
- Denature the RNA by heating at 65 °C for 5 min and then place the tubes on ice for 1 min.
- Prepare the SuperScript III cDNA synthesis mix (Recipe 4) and add 10 μl to the RNA samples.
- Incubate 50 min at 50 °C and then terminate the reaction by heating at 85 °C for 10 min. Spin down the tubes briefly and keep them on ice.
- Then add 1 μl of RNase H to the tubes and incubate 20 min at 37 °C to degrade the RNA.
- Purify the cDNA using the AMPURE XP magnetic beads following the manufacturer´s recommendation, but using two-fold more beads to keep smaller fragments https://www.beckmancoulter.com/wsrportal/techdocs?docname=B37419.
- Elute the samples in 15 μl of H2O.
- Adapter ligation and first PCR amplification
- Adapter ligation to the 3′ OH of cDNA and circularization is carried out with Circligase in a final volume of 20 μl, containing 10 μl of purified cDNA, 2.5 μM of adapter_ligation primer and 10 μl of Circligase enzyme mix (Recipe 5).
- Incubate the samples 2 h at 60 °C, then 1 h at 68 °C and 10 min at 80 °C.
- Let the samples on ice for at least 2 min.
- Perform the first PCR amplification using Phusion DNA polymerase in a final volume of 10 μl using the PR_fwd and 18S adapter primers (0.5 μM) and 5x Phusion HF buffer.
To optimize the amplification, try three different volumes of the ligated cDNA mix described above (0.5, 1 and 2 μl).- Program the PCR using the following parameters:
3 min initial denaturation at 95 °C, followed by 30 cycles of 15 s at 95 °C, 15 s at 50 °C and 20 s at 72 °C with a final extension of 10 min at 72 °C. - Run a 2 μl aliquot of each sample in an 1% agarose-TAE gel to check the amplification sizes (bands between 100-950 nt are expected).
- Program the PCR using the following parameters:
- Adapter ligation to the 3′ OH of cDNA and circularization is carried out with Circligase in a final volume of 20 μl, containing 10 μl of purified cDNA, 2.5 μM of adapter_ligation primer and 10 μl of Circligase enzyme mix (Recipe 5).
- Second PCR amplification with index primers
Use 3 μl of the first PCR amplification to program a second PCR amplification with PR_fwd and INDEX primers (0.5 μM final concentration). Use a different index primer for each sample (three in this case).- Perform a PCR amplification using the same parameters as described above but reduce the number of cycles to 10.
- Then, bring the samples to 40 μl with H2O and purify the PCR fragments with AMPURE XP beads following the manufacturer specifications.
- Elute the samples in 20 μl H2O and check them in a Bioanalyzer.
- A distribution of peaks in the range of 100-950 nt is expected (see Figure 3A). In some samples a strong peak of around 150 nt is observed, that presumably corresponds to adapter dimers. In this case, perform a size fractionation of the sample in a 1% agarose-TAE gel by cutting the piece of gel above the 150 nt marker. Extract the DNA from agarose piece by using QIAquick gel extraction kit.
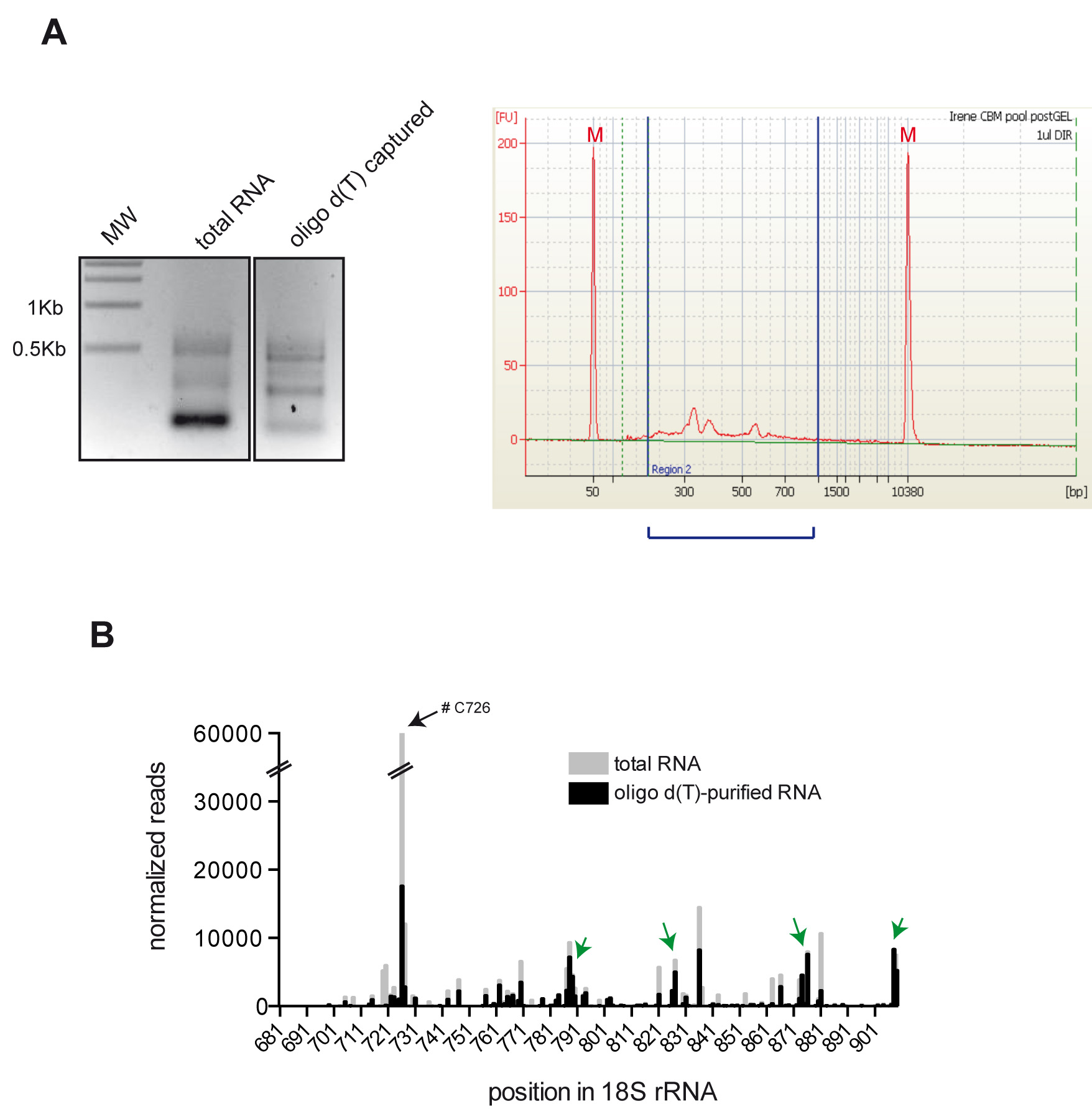
Figure 3. An example of the results obtained. A. A typical picture of agarose gel electrophoresis showing final PCR libraries obtained from RNA total and oligo d(T) captured (right panel). Left panel shows the electropherogram corresponding to oligo d(T) captured sample from above. The sample was analyzed in a bioanalyzer and the range of DNA peaks obtained is indicated by a blue bracket. Size markers are also shown (M). B. A typical histogram of 5′ reverse transcription (RT) arrests on 18S rRNA after crosslinking of mRNA assembled into the 48S-PIC. Total or oligo(dT)-purified RNA was subjected to reverse transcriptase termination site (RTTS) assay of 18S rRNA. Some arrests enriched in oligo(dT)-purified RNA are indicated by green arrows. A strong RT arrest at C726 was frequently observed in total RNA, presumably due to fragmentation of 18S rRNA during the purification process.
- Sequencing
Mix the resulting indexed libraries (three in this case) by adjusting to the sample of lower concentration to get a similar representation of each library in the mix. Send samples for sequencing in a NextSeq550 platform, 1x75 reads. The typical output was 0.5Mreads/sample.
Data analysis
FASTQ files from sequencing are processed under the FASTX software pipeline (http://hannonlab.cshl.edu/fastx_toolkit/galaxy.html):
- Demultiplex the sequencing reads using FASTQ Barcode splitter and index sequences:
Command line: cat sequencing_complete.fastq | fastx_barcode_splitter –bcfile barcodes_sequences.txt –bol –prefix “/sequencing/RTTS_” --suffix “.fastq. - Remove index sequences using FASTQC Clipper option. Remove the sequences lacking the adapter:
Command line: fastx_clipper –a ADAPTER_SEQUENCE –c –i INFILE –o OUTFILE. - Perform a quality-based filtering of sequences using FASTQ Quality Filter option deleting all the sequences below 20 quality score in at least 80% of the molecule:
Command line: fastq_quality_filter –q 20 –p 80 –I INFILE –o OUTFILE). - Align the sequences to rabbit 18S rRNA (accession X06778.1, https://www.ncbi.nlm.nih.gov/nuccore/X06778) using Bowtie 2 with default parameters:
Command line: bowtie2 -q –x rabbit_18S_indexes –U sequences.fq –S aligment.output.sam. - Once aligned, use FeatureCounts software to extract the 5' end position of each read using read2pos5 parameter (command line: featureCounts input_files.sam –o output_file.txt –read2pos5). Then, we used the command cut -f 3 feature_counts_input.txt | sort -g | uniq -c > final_output.txt to generate a table with the frequency representation of every nucleotide of 18S rRNA at the 5' end of reads. The exact crosslinking site is considered the -1 position respect to the mapped 5' end of every read.
- Once mapped the crosslinking sites on 18S rRNA sequence for every sample, identify those arrests which are enriched in oligo d(T)-captured RNA sample. To do this, subtract the data with that obtained using total RNA and not crosslinked samples. Usually, we consider only the crosslinking sites representing > 0.5 % of total reads.
Recipes
- Polysome buffer
Tris-HCl 20 mM pH 7.5
KCl 100 mM
MgCl2 2 mM
DTT 2mM - TE buffer
10 mM Tris pH 8.0
1 mM EDTA - 1x TAE buffer
40 mM Tris-acetate
1 mM EDTA - SuperScript III cDNA synthesis mix (per reaction)
2 μl 10x RT Buffer
4 μl 25 mM MgCl2
2 μl 0.1 M DTT
1 μl RNaseOUT
1 μl SuperScript III RT - Circligase enzyme mix (per reaction)
2 μl CL 10x reaction buffer
1 μl ATP
1 μl MnCl2
1 μl Circligase
5 μl H2O - Lysis/binding buffer
20 mM Tris-HCl (pH 7.5)
500 mM LiCl
0.5% LiDS
1 mM EDTA
5 mM DTT - Wash Buffer 1
20 mM Tris-HCl (pH 7.5)
500 mM LiCl
0.1% LiDS
1 mM EDTA
5 mM DTT - Wash Buffer 2
20 mM Tris-HCl (pH 7.5)
500 mM LiCl
1 mM EDTA - Low Salt Buffer
20 mM Tris-HCl (pH 7.5)
200 mM LiCl
1 mM EDTA
Acknowledgments
The technical support of Laura Barbado is acknowledged. This work was funded by Ministerio de Economía y Competitividad (BFU2017-84955-R) grant.
Competing interests
The authors declare neither conflicts of interest nor competing interests.
References
- Díaz-López, I., Toribio, R., Berlanga, J. J. and Ventoso, I. (2019). An mRNA-binding channel in the ES6S region of the translation 48S-PIC promotes RNA unwinding and scanning. Elife 8: 48246.
- Engreitz, J. M., Sirokman, K., McDonel, P., Shishkin, A. A., Surka, C., Russell, P., Grossman, S. R., Chow, A. Y., Guttman, M. and Lander, E. S. (2014). RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent Pre-mRNAs and chromatin sites. Cell 159(1): 188-199.
- Kielpinski, L. J., Boyd, M., Sandelin, A. and Vinther, J. (2013). Detection of reverse transcriptase termination sites using cDNA ligation and massive parallel sequencing. Methods Mol Biol 1038: 213-231.
- Langmead, B and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4): 357-359.
- Liao, Y., Smyth, G. K. and Shi, W. (2014). featureCounts: and efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30(7): 923-930.
- Pisarev, A. V., Kolupaeva, V. G., Yusupov, M. M., Hellen, C. U. and Pestova, T. V. (2008). Ribosomal position and contacts of mRNA in eukaryotic translation initiation complexes. EMBO J 27(11): 1609-1621.
- Sharma, E., Sterne-Weiler, T., O'Hanlon, D. and Blencowe, B. J. (2016). Global mapping of human RNA-RNA interactions. Mol Cell 62(4): 618-626.
Article Information
Copyright
![]() Díaz-López et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Díaz-López et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Díaz-López, I., Toribio, R. and Ventoso, I. (2020). Mapping mRNA-18S rRNA Contacts Within Translation Initation Complex by Means of Reverse Transcriptase Termination Sites and RNAseq. Bio-protocol 10(16): e3713. DOI: 10.21769/BioProtoc.3713.
- Díaz-López, I., Toribio, R., Berlanga, J. J. and Ventoso, I. (2019). An mRNA-binding channel in the ES6S region of the translation 48S-PIC promotes RNA unwinding and scanning. Elife 8: 48246.
Category
Molecular Biology > RNA > mRNA translation
Molecular Biology > RNA > RNA labeling
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.