- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Generation of Functional Mouse Hippocampal Neurons

Published: Vol 10, Iss 15, Aug 5, 2020 DOI: 10.21769/BioProtoc.3702 Views: 9778
Reviewed by: Marion HoggGhulam HUSSAINZheng Zachory Wei
Original research article
The authors used this protocol in:

Aug 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Primary culture of mouse hippocampal neurons is a very useful in vitro model for studying neuronal development, axonal and dendritic morphology, synaptic functions, and many other neuronal features. Here we describe a step-by-step process of generating primary neurons from mouse embryonic hippocampi (E17.5/E18.5). Hippocampal neurons generated with this protocol can be plated in different tissue culture dishes according to different experimental aims and can produce a reliable source of pure and differentiated neurons in less than one week. This protocol covers all the steps necessary for the preparation, culture and characterization of the neuronal culture, including the illustration of dissection instruments, surgical procedure for embryos’ isolation, culturing conditions and assessment of culture’s purity and differentiation. Evaluation of neuronal activity was performed by analysis of calcium imaging dynamics at six days in culture.
Background
The hippocampus is a very well-characterized brain structure critical for important cerebral functions such as memory, spatial navigation, emotional memory and learning. Anatomically, the murine hippocampus has a well-defined, C-shaped structure that is easy to locate and isolate. At the cellular level, it is mainly composed of pyramidal cells with fewer interneurons and glial cells compared to other brain regions (Kaech and Banker, 2006). As such, the hippocampus is an ideal region for generating primary neuron cultures of high purity from wild-type or genetically engineered mouse models that can be used for disease modeling or to investigate multiple aspects of neuronal function such as synaptic transmission and electrophysiological properties, sensitivity to neurotoxicity, differentiation and aging (Busche, 2018; Koyama and Ikegaya, 2018; Molnar, 2011; Wu et al., 2019; Rush et al., 2020).
Many protocols have been developed for generating cortical and hippocampal neuron cultures by co-culturing neurons with glial feeders (Kaech and Banker, 2006) describing three-dimensional neuronal culture systems with astrocytes encapsulated in hydrogel microfibers (Kim et al., 2020), supplementing culture medium with growth factors for long-term neuron cultures (Ray et al., 1993), or generating neurons from post-natal mice (Beaudoin et al., 2012). Each of these methodologies presents advantages and disadvantages depending on the goals of the experiment. Here, we extend and describe in detail a protocol for generating hippocampal neurons that we have developed and successfully employed over the years (e.g., Dorsey et al., 2006; Tomassoni-Ardori et al., 2019). We focus primarily on providing an easy, simple and reliable procedure for obtaining primary hippocampal neurons from mice at the late embryonic stage (E17.5-E18.5). We illustrate the whole preparation process including aspects that have often been omitted in other protocols such as a description of dissection instruments and surgical procedures for embryos removal and dissection. We report analysis of neuronal differentiation and function after 6 days in culture by testing for neuronal differentiation markers and neuronal activity by cellular calcium imaging dynamics. Moreover, we show the levels of neuronal culture purity in relation to glial cells with and without treatment with the glial inhibitor cytosine arabinoside (Ara-C).
This step-by-step protocol allows the experimenter to obtain differentiated hippocampal neurons with a high degree of purity within 6 days.
Materials and Reagents
The following list provides examples of the materials and equipment that we routinely use in our laboratory. Reagents and equipment with similar specifications will work as well.
- Poly-D-lysine/laminin cellware 12 mm round coverslips (Corning, catalog number: 354087 )
- Sterile glass Pasteur pipettes (Thomas Scientific, catalog number: 1215D99 )
- 24-well plate (Corning, catalog number: 3524 )
- 35 mm TC-treated culture dish (Corning, catalog number: 353001 )
- 100 mm TC-treated culture dish (Corning, catalog number: 430167 )
- Cellometer disposable counting slides (SD100 slides) (Nexcelom Bioscience, catalog number: CHT4-SD100-002 )
- 50 ml conical tube (Corning, catalog number: 430828 )
- 15 ml conical tube (Corning, catalog number: 430790 )
- InvitrolonTM PVDF/Filter Paper Sandwiches (for mini gels) (Thermo Fisher Scientific, catalog number: LC2005 )
- E17.5/E18.5 mouse embryos
- Neurobasal medium (Thermo Fisher Scientific, catalog number: 21103-049 )
- B27 supplement (Thermo Fisher Scientific, catalog number: 17504-044 )
- Dulbecco's modified high glucose eagle medium (DMEM) (Thermo Fisher Scientific, catalog number: 11965-092 )
Note: This DMEM formulation contains 4500 mg/L glucose, L-glutamine and phenol red. - Distilled water (Thermo Fisher Scientific, catalog number: 15230-162 )
- Poly-D-lysine (Sigma Aldrich, catalog number: P6407 )
- Fetal bovine serum (not heat-inactivated) (Omega Scientific, catalog number: FB-01 ; or other brand FBS)
- 0.25% Trypsin-EDTA (Thermo Fisher Scientific, catalog number: 25200-056 )
- Penicillin/Streptomycin (P/S) 10,000 U/ml (100x concentrated) (Thermo Fisher Scientific, catalog number: 15140-122 )
- Dulbecco’s phosphate buffered saline (DPBS) (Thermo Fisher Scientific, catalog number: 14190-144 )
- Trypan blue (Sigma-Aldrich, catalog number: 93595 )
- Cytosine arabinoside (Ara-C) (Sigma-Aldrich, catalog number: C6645 )
Note: Stock solution is reconstituted in water at a concentration of 50 mg/ml, aliquoted and stored at -20 °C. - Anti-Tubulin-β-III antibody (mouse monoclonal) (Covance, catalog number: MMS-435P )
- Anti-GFAP antibody (rabbit polyclonal) (Agilent (Dako), catalog number: Z0334 )
- Alexa FluorTM 488 donkey anti-rabbit IgG (H+L) secondary antibody (Thermo Fisher Scientific, catalog number: A-21206 )
- Alexa FluorTM 647 donkey anti-mouse IgG (H+L) secondary antibody (Thermo Fisher Scientific, catalog number: A-31571 )
- DAPI (Invitrogen, catalog number: D1306 )
- Paraformaldehyde (Sigma-Aldrich, catalog number: P6148 )
- TritonTM-X100 (Sigma-Aldrich, catalog number: T8532 )
- Donkey serum (Sigma-Aldrich, catalog number: S30-100ML )
- Bovine serum Albumin (BSA) (Sigma-Aldrich, catalog number: A9647 )
- Fluorescence mounting medium (Dako, catalog number: S3023 )
- Ethanol solution 70%
- Laminin Mouse Protein, Natural (Thermo Fisher Scientific, catalog number: 23017-015 )
- Sample buffer, Laemmli 2x concentrate (Sigma-Aldrich, catalog number: S3401 )
- NuPAGE 4 to 12%, Bis-Tris, Mini Protein Gel (Thermo Fisher Scientific, catalog number: NP0321PK2 )
- Tween 20 (Sigma Aldrich, catalog number: P1379 )
- Non-fat dry milk (Santa Cruz Biotechnology, catalog number: sc-2324 )
- Super Signal West Dura Extended Duration Substrate (Thermo Fisher Scientific, catalog number: 34076 )
- Anti-TrkB antibody (Millipore, catalog number: 07-225 )
- Anti-Rbfox1 antibody clone 1D10 (Millipore, catalog number: MABE985 )
- Anti-Rbfox3 (NeuN) antibody (Millipore, catalog number: MAB377 )
- Anti-mouse IgG, HRP-conjugated (Millipore, catalog number: AP192P )
- Anti-rabbit IgG, HRP-conjugated (Millipore, catalog number: AP182P )
- Fluo-4 DMSO solution 1 mM (Thermo Fisher Scientific, catalog number: F14217 )
- PowerLoad Concentrate 100x (Thermo Fisher Scientific, catalog number: P10020 )
- Probenecid water soluble (Thermo Fisher Scientific, catalog number: P36400 )
Equipment
- Dissection hood (NuAire, model: NU-201-430 )
- Laminar flow hood (NuAire, model: NU-425-400 , Class II, Type A/B3)
- Stereoscopic microscope (Olympus, model: SZH-ILLK )
- Inverted tissue culture microscope (Nikon, model: Diaphot )
- CO2 incubator for cell culture (Sanyo, model: MCO-17AIC )
- Surgical scissors, sharp (Fine Science Tools, catalog number: 14002-16 )
- Student standard pattern forceps (Fine Science Tools, catalog number: 91100-12 )
- Fine scissors, sharp (Fine Science Tools, catalog number: 14060-10 )
- Graefe forceps (Fine Science Tools, catalog number: 11052-10 )
- Dumont #7 forceps (Fine Science Tools, catalog number: 11272-30 )
- Cell counter (Nexcelom Bioscience, Cellometer Vision)
- Centrifuge (Thermo Scientific, model: CL2 )
- Tissue culture hood (NuAire, model: NU-425-400 )
- Confocal microscope system (Zeiss, model: LSM780 )
- Bunsen burner (HanauTM Touch-O-Matic)
- Branson 1800 sonicator (Branson)
- XCell SureLock Mini-Cell protein electrophoresis (Thermo Fisher Scientific, catalog number: EI0001 )
- GeneGnome XRQ-Chemiluminescence Imaging system (Syngene)
Procedure
Notes:
- This methodology describes the critical steps necessary for generating primary hippocampal neurons derived from E17.5/E18.5 mouse embryos. Practical tips after each step are included to better guide the readers through the procedure.
- Depending on the final experimental aims, different tissue culture dishes/plates or glass coverslips can be used for culturing primary hippocampal neurons.
- Coating of tissue culture dishes/plates with poly-D-lysine
- Reconstitute poly-D-lysine in a sterile laminar hood by adding 50 ml of sterile tissue culture grade water to a 5 mg γ-irradiated and cell culture tested poly-D-lysine bottle (final working concentration of 0.1 mg/ml).
- Evenly coat the culture dish surface with 0.1 mg/ml poly-D-lysine solution and incubate at room temperature for 20 min in a laminar flow hood.
- Remove the poly-D-lysine solution by aspiration and rinse the surface thoroughly with sterile tissue-culture grade water twice.
- Before plating the cells, allow the surface to dry completely by leaving the dishes/plates uncovered under the laminar flow hood for about 1 h.
Note: If plating cells on glass coverslips use commercially available poly-D-lysine/laminin pre-coated 12 mm coverslips. Transfer poly-D-lysine/laminin coated coverslips directly into wells of a 24-well plate by using sterile forceps in a sterile laminar flow hood before plating neurons. Alternatively, regular 12 mm coverslips can be coated by placing them into 24-well plate wells and covering them with 0.1 mg/ml poly-D-lysine for 20 min in a laminar flow hood. After that wash twice with PBS and cover with a 10 µg/ml laminin solution for at least 4 h at 37 °C by leaving the coverslips in an incubator until cells are ready to be plated. Rinse twice with PBS just before plating the cell to prevent the culture surface from drying. The 1 mg/ml laminin stock solution is made by adding 1 ml of PBS into 1 mg laminin powder and diluting to 10 µg/ml laminin with PBS. Coating of coverslips in plates avoids the potential to scrape the coating off while transferring between dishes.
- Sacrifice pregnant mouse and remove E17.5/E18.5 embryos
- Euthanize pregnant mouse according to Animal Care and Use Committees (ACUC) approved protocols (CO2 euthanasia or isoflurane anesthesia followed by decapitation or cervical dislocation).
- Pin the animal to a clean surgical area and use 70% ethanol to clean the abdomen.
- In order to expose the embryos, perform an incision in the middle of the abdomen using surgical scissors (Figure 1A) starting from the pubic region and continuing up to the end of the abdominal cavity as indicated by the dashed line in Figure 2A.
Note: Use forceps (Figure 1B) to pinch and pull up the skin layer while performing the incision with surgical scissors. This will help to avoid accidental damage to the embryos located immediately underneath.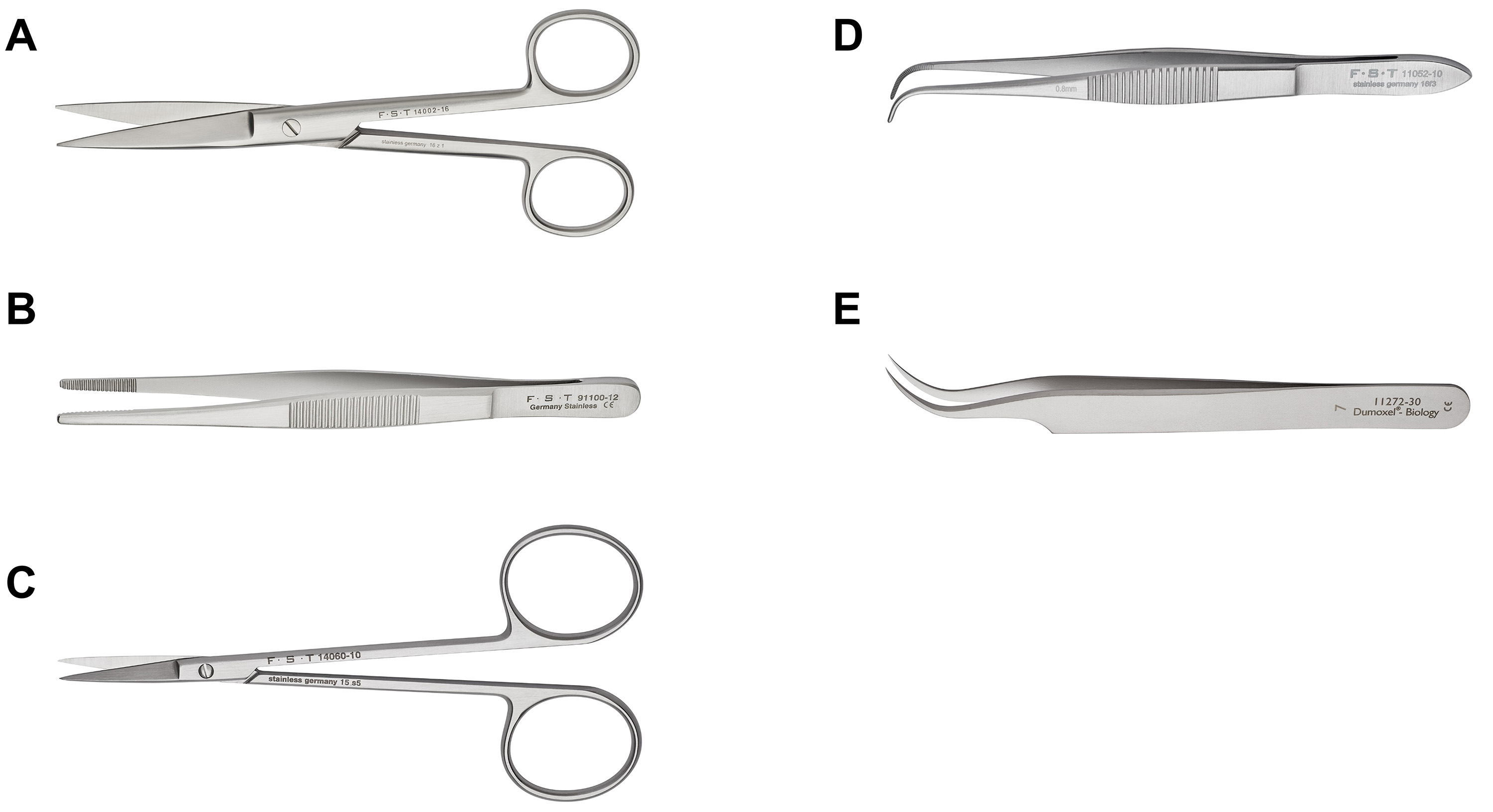
Figure 1. Dissection instruments used for the preparation of hippocampal neurons. A. Surgical scissors used in the protocol Steps B3-B6. B. Standard pattern forceps used in the protocol Steps B3-B6 and C1. C. Fine scissors used in Step C1. D. Curved forceps (Graefe forceps) used in Steps C2-C3. E. Dumont forceps used in Steps C4 and C5.
Figure 2. Preparation of E17.5/18.5 pregnant animal for surgical removal of the embryos. A. Pinned animal showing with a dashed white line the location of the incision (Step B3); B. Exposure of uterus after abdominal incision (Step B3). - Carefully expose the whole uterus (uterine horns) containing the embryos (Figure 2B) by using forceps and surgical scissors (Figures 1A-1B) and immediately transfer the uterus to a 100 mm dish containing ice-cold PBS (Figure 3A).
Note: Use forceps to grab and pull the uterus out of the abdomen while using surgical scissors with the other hand to cut any adherence and the cervix connection (Figure 2B arrows). - Continue to use forceps and surgical scissors to free the single embryos and carefully remove the placenta and yolk sack. Transfer the embryos into a new 100 mm dish containing ice-cold PBS to wash embryos and to remove blood and embryonal fluids (Figure 3B).
- Use forceps (Figure 1B) to hold the embryo and cut the head using surgical scissors (Figure 3C). Transfer the heads into a new 100 mm dish containing ice-cold DMEM complete with 10% fetal bovine serum and 1% antibiotics (P/S).
Note: From this step forward, it is important to keep transferring the biological material to new dishes with clean medium containing antibiotics to lower the possibility of contamination. In the figures shown in this protocol PBS was intentionally used instead of medium containing phenol red only for the purpose of obtaining more clear images (Figure 3A, Figure 4, Figure 5 and Figure 6).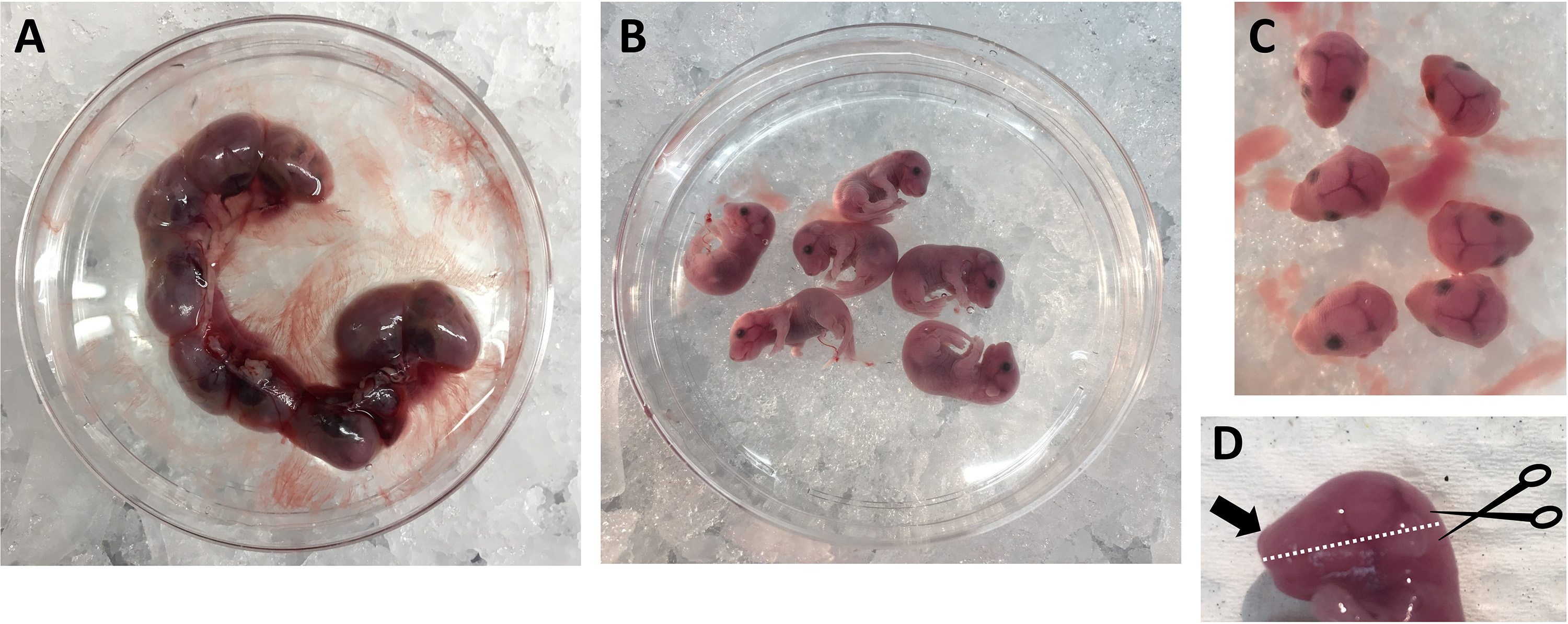
Figure 3. Isolation of the embryos from the uterus. A. Isolated uterus (Step B4); B. Isolated mouse embryos (Step B5); C. Embryo heads (Step B6). D. Side view of embryo’s head. Dashed white line indicates where to execute the transversal cut described in Step C1 starting from the back of the skull (black scissors) while holding the embryo’s head with forceps in the nose area (black arrow).
- Dissection of hippocampi
- Move to a dissection cabinet with sterile laminar air flow and prepare a 100 mm dish containing ice-cold complete DMEM with 10% fetal bovine serum and 1% antibiotics (P/S) for dissection of the brains. Transfer one embryo head at a time into the 100 mm dish containing complete DMEM medium and gently remove the brain from the embryo head (Figure 4A). Specifically, with one hand, use forceps (Figure 1B) to pinch the mouth/nose area of the head (black arrow in Figure 3D). With the other hand, use the fine scissors (Figure 1C) to enter from the back of the skull (black scissors in Figure 3D) and execute a transversal cut (along the transversal plane at the mouth/nose level indicated by the white dashed line in Figure 3D) in order to free the basal brain region. Invert the embryo head to expose the free basal area of the brain. Continue by using two forceps (Figure 1B) to gently pull apart the skull and carefully remove the whole brain (Figure 4A). Transfer and collect the brains into a new 100 mm dish containing clean, ice-cold complete DMEM medium. Replace the medium after each brain dissection to eliminate the blood.
- Transfer one brain at a time to a 35 mm dish lid filled with ice-cold complete DMEM medium and move under a dissection stereoscope.
Note: The lower depth of the lid of a 35 mm dish allows for better movements when using dissection instruments compared to the bottom of the 35 mm dish. - Use two curved forceps (Figure 1D): hold the brain on the side with one forceps (white dots in Figure 4A) and insert the second forceps along the midline (dashed line in Figure 4A) to separate the two hemispheres of the brain. Continue to use curved forceps to gently pull apart and flip the two halves of the brain in order to expose and visualize the hippocampal area (Figures 4B-4C).
Note: Due to the fragility and softness of the embryonal tissues, the brain can be easily damaged while trying to dissect the hippocampal region: always use care when handling and holding embryonal brains. A few practicing sessions at the stereoscope will help to improve dissecting skills quickly.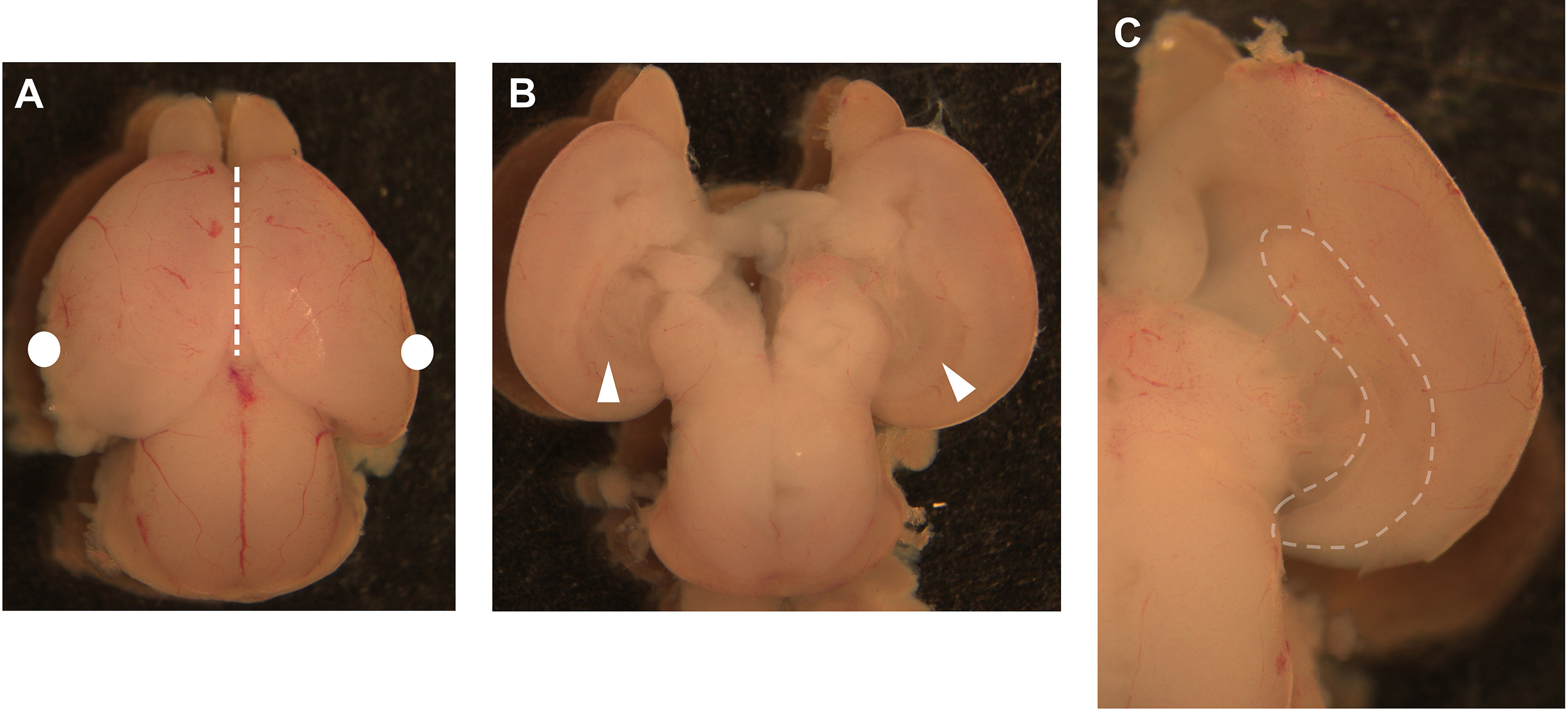
Figure 4. Localization of the hippocampus. A. Dissected embryo brain. Dashed white line indicates where to separate the two brain hemispheres using one curved forceps while holding the brain on the side by using the second curved forceps (white dots) (Step C3). B. Separated brain hemispheres. White arrow heads indicate the location of the hippocampal region on each hemisphere (Step C3). C. Higher power image showing the hippocampal region on one brain hemisphere indicated by a white dashed line. - Carefully remove the meninges from each brain hemisphere (Figure 5).
Notes:- Work by using two Dumont forceps (Figure 1E). Use one forceps to hold the whole brain and use the second forceps to gently peel the meninges off. Start by pinching the olfactory bulb area and gradually pulling back in order to remove meninges completely (Figure 5). Hold the brain gently while performing this operation and it might be necessary to change the holding points many times according to how easily the meninges come off.
- It is important to properly remove the meninges to eliminate as much non-neuronal cell contribution to the neuronal culture as possible.
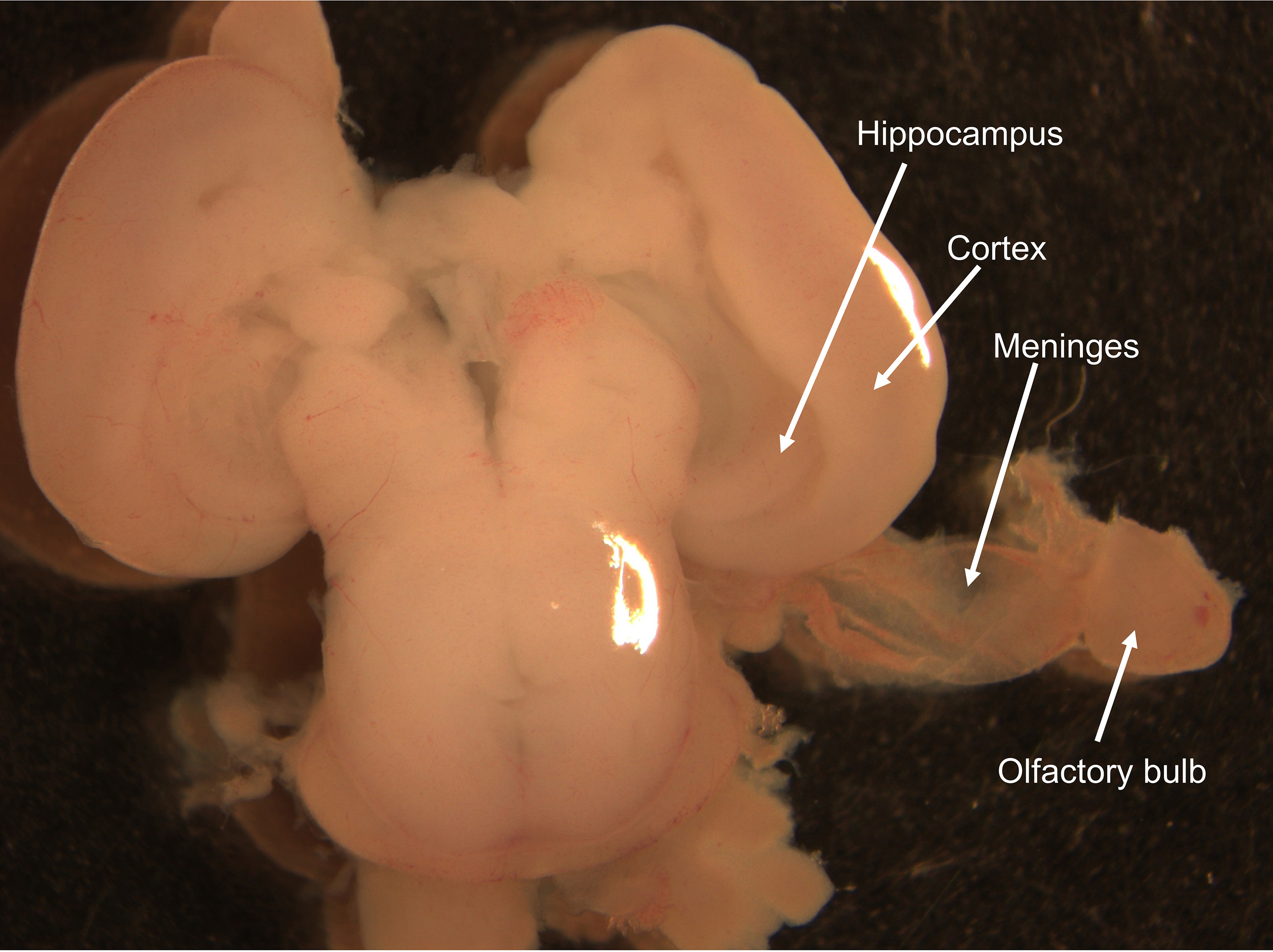
Figure 5. Removal of the meninges. Arrows indicate hippocampal and cortical regions as well as olfactory bulb and meninges removed from one brain hemisphere (Step C4). - Proceed by using Dumont forceps to finely cut along the hippocampus edges to completely free the hippocampal area (Figures 5-6). Use curved forceps to gently pick and transfer each hippocampus to a new 35 mm tissue culture dish containing 2 ml of ice-cold serum-free DMEM medium with 1% antibiotics (P/S) and keep it on ice.
Note: The hippocampus is distinguishable by its typical C-shaped anatomical structure and by a difference in light contrast under a stereoscope compared to the cortical area to which it is connected (Figures 5-6). After removing the hippocampus it is possible to collect cortices from the same embryos for the preparation of cortical neurons if needed (not described in this protocol).
- Trypsinization of the hippocampi
- Mince the hippocampal tissues in the same 35 mm tissue culture dish containing 2 ml of ice-cold serum-free DMEM medium used for the collection by cutting them into small pieces using two Dumont forceps (ideally 5-6 pieces from each hippocampus of approximately 2-3 mm3) (Figure 6C).
Note: It is important to collect and keep the hippocampi in serum-free DMEM medium (Steps C5 and D1) to avoid inhibition of Trypsin-EDTA activity during the next step (Step D2).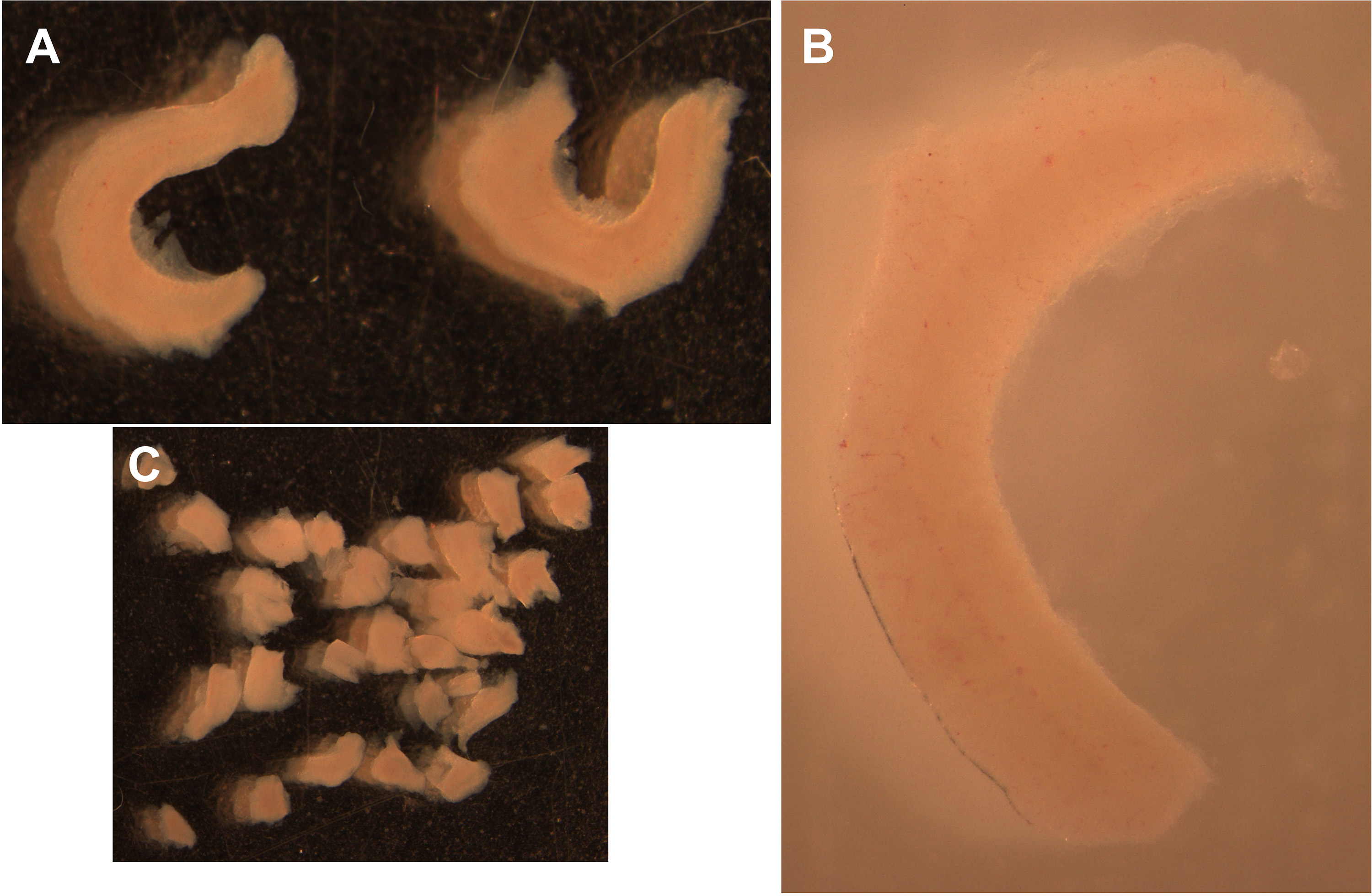
Figure 6. Isolation of the hippocampus (Step C5 and Step D1). A. Isolated hippocampi from one embryonal brain. B. higher power image of the hippocampal region showing the typical C-shaped anatomical structure. C. Minced hippocampi. Hippocampal tissue-chunks generated for trypsin digestion (Step D1). - Move to a tissue culture hood with sterile laminar air flow and use a pipette to transfer the medium containing the minced hippocampi to a 50 ml sterile conical tube. Gently add 2 ml of 0.25% Trypsin-EDTA to the hippocampi and place the tube in a water bath at 37 °C for 25 min (final trypsin concentration of 0.125%).
Note: During the 25 min digestion in trypsin, the minced hippocampal pieces will naturally sediment by gravity to the bottom of the 50 ml conical tube. - Prepare a 15 ml sterile conical tube containing 2 ml of warm (37 °C) DMEM complete with 10% fetal bovine serum and 1% antibiotics (P/S). At the end of the digestion, carefully transfer the 50 ml digestion tube back to a tissue culture hood without disturbing or resuspending the hippocampal sediment and, with a pipette with a 1 ml tip, transfer only the hippocampal sediment to the new 15 ml conical tube containing the 2 ml of complete medium.
Note: Use the 1 ml tip to directly contact the hippocampal sediment at the bottom of the 50 ml tube. Gently aspirate and transfer only the hippocampi at the bottom of the tube without excess liquid from the digestion. The fetal bovine serum contained in the new 2 ml medium will inactivate the trypsin and stop its enzymatic activity.
- Mince the hippocampal tissues in the same 35 mm tissue culture dish containing 2 ml of ice-cold serum-free DMEM medium used for the collection by cutting them into small pieces using two Dumont forceps (ideally 5-6 pieces from each hippocampus of approximately 2-3 mm3) (Figure 6C).
- Trituration of the hippocampal tissue
- Use a Bunsen burner to fire-polish a sterile glass Pasteur pipette and generate a smaller opening and a smoothened tip for dissociating the hippocampal tissue (Figure 7). Use the fire-polished pipette shown in Figure 7B to slowly and gently triturate the hippocampal pieces. Pipet up and down six to eight times.
Note: Trituration of the hippocampi is a critical step to achieve good cell yield and cell viability. After fire-polishing, be sure to let the glass pipette cool down before triturating the hippocampi. Hippocampal chunks should be completely broken up after pipetting up and down six to eight times. - Use the Bunsen burner again to prepare a new fire-pulled glass Pasteur pipette with a longer and tapered tip (Figure 7C). Use this pipette to slowly perform a single up and down pipetting of the hippocampal cell slurry in order to release single cells. After obtaining a single cell suspension proceed immediately to Procedure F. Keep the single-cell suspension in a tissue culture hood with sterile laminar air flow at room temperature while counting the cells.
Note: Performing multiple pipetting with the fire-pulled Pasteur pipette with a longer and tapered tip (Figure 7C) will damage the cells and affect cell viability.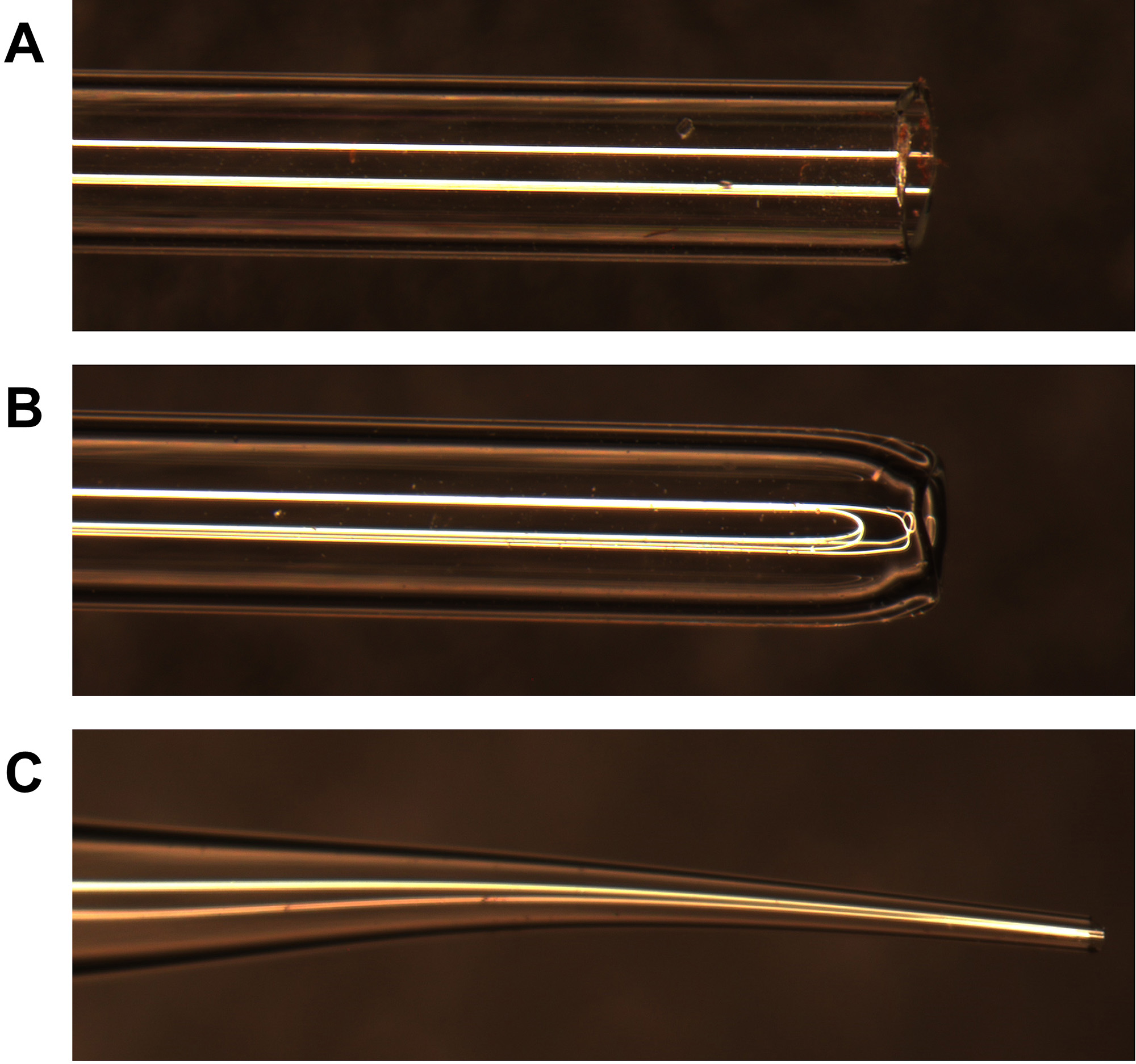
Figure 7. Glass Pasteur pipette tips (Steps E1 and E2). A. Intact glass Pasteur pipette tip. B. Fire-polished glass Pasteur pipette with smoothened and smaller tip. C. Fire-polished glass Pasteur pipette with tapered and extra-small tip.
- Use a Bunsen burner to fire-polish a sterile glass Pasteur pipette and generate a smaller opening and a smoothened tip for dissociating the hippocampal tissue (Figure 7). Use the fire-polished pipette shown in Figure 7B to slowly and gently triturate the hippocampal pieces. Pipet up and down six to eight times.
- Cell counting, cell seeding and cell culture
- Collect 50 μl of the cell suspension and add 5 μl of Trypan blue dye to assess cell viability. Count cells using a cell counter.
Note: Cell viability ranges between 70-90% of live cells. As an example, the cell viability and cell count of a neuronal preparation from 8 embryos is about 4 x 106 live cells from a total of 5 x 106 cells (cell viability = 80%). - Centrifuge the remaining hippocampal cell suspension at 350 x g for 5 min. Discard the supernatant and resuspend the cell pellet by pipetting gently up and down in warm Neurobasal medium with B27 supplement. Adjust the volume of medium according to the tissue culture plates that will be used. Use a seeding density of approximately 260,000 cells/square centimeter. Seed the cells in the tissue culture dish/plate previously coated with poly-D-lysine.
Note: Cell yield is approximately 400,000-600,000 cells/embryo. Prepare complete medium for neuronal culture by adding 10 ml of B27 supplement and 5 ml of P/S antibiotics (100x concentrate) into 500 ml of Neurobasal medium. Some protocols (e.g., Ruhl et al., 2019) suggest to pre-plate neurons for one hour in DMEM medium with 10% FBS to facilitate neuron adhesion to the plate. However, we have found that plating directly in Neurobasal medium with B27 supplement is sufficient for good neuron adhesion. - After 18 to 24 h from the initial plating, add cytosine arabinoside (Ara-C) to the culture to stop the proliferation of dividing cells (non-neuronal cells). Remove half of the volume of the culture medium (Neurobasal medium containing B27 supplement) and replace it with fresh medium containing 2 µM Ara-C (1 µM Ara-C final concentration).
Note: Failing to add Ara-C to a primary neuron culture will considerably affect the purity of the culture (Figures 8B and 8F). Death of some mitotic cells can be observed after adding Ara-C to the neuron culture. - Culture the primary hippocampal neurons by replacing only half of the medium every 48 to 72 h. It is not necessary to continue adding Ara-C when replacing medium after the initial treatment. After 6 days in culture, the primary neurons will be well developed and differentiated as assessed by both morphology but especially expression of typical neuronal markers such as Tubulin-β-III, NeuN, Rbfox1, neurotrophin receptors and several other neuronal proteins (Tomassoni-Ardori et al., 2019) (Figure 8 and Figure 9).
Notes:- Replacing only half of the culture medium maintains the growth factors naturally secreted and present in the neuronal conditioned medium.
- Although absolute prevention of microbial contamination (bacteria, mycoplasma, fungi) during the preparation of primary cell cultures is impossible, these steps can greatly improve the outcome: i) work in a sterile environment; ii) use proper aseptic technique including wearing sterile gloves and using sterile glassware and disposable pipettes and tips; iii) keep all the equipment clean (dissecting microscope, incubators, centrifuges and water baths).
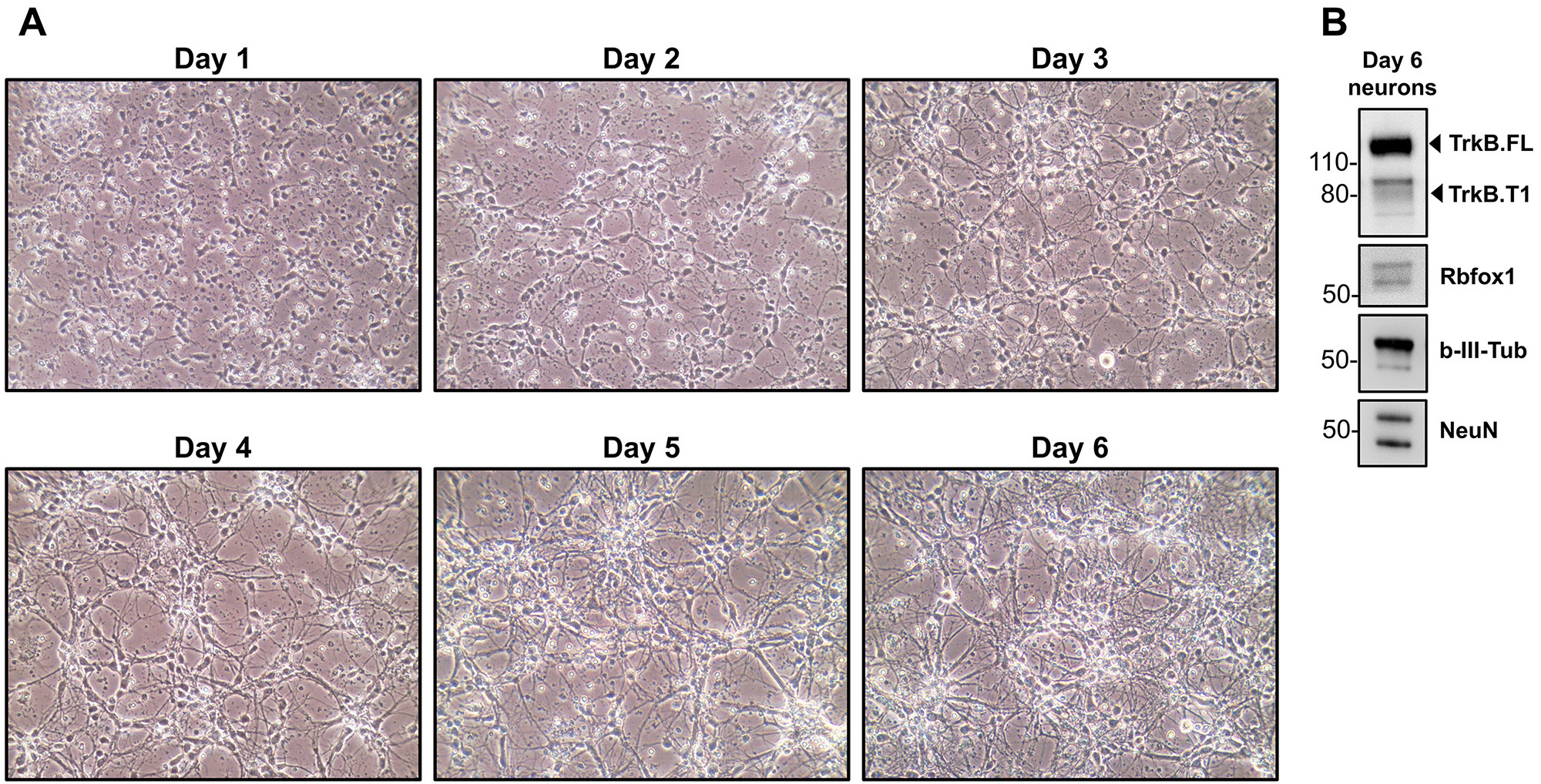
Figure 8. Development and differentiation of neurons in culture. A. Brightfield images of primary hippocampal neuron cultures from day 1 to day 6 in vitro. Note the increasing complexity of the neuronal network over time in vitro (magnification 20x). B. Western blot analysis of hippocampal neurons after 6 days in vitro. Immunoblots were performed with antibodies against markers of differentiated neurons: the BDNF receptor TrkB extracellular domain to detect all TrkB isoforms, Rbfox1, beta-III-tubulin (b-III-Tub) and Rbfox3 (NeuN). Molecular Weight markers are indicated on the left.
- Collect 50 μl of the cell suspension and add 5 μl of Trypan blue dye to assess cell viability. Count cells using a cell counter.
- Western blot analysis for the detection of markers of differentiated neurons (Figure 8B)
- After 6 days of in vitro culture, remove the culture medium from one well of a 24-well plate containing neurons.
- Rinse cells with PBS once.
- Lyse cells by adding 150 µl of 2x Laemmli sample buffer directly into the well.
- Transfer the cell lysate to a 1.5 ml tube and sonicate the sample for 10 min to shred genomic DNA and eliminate viscosity.
- Heat the sample at 95°C for 5 min to promote protein denaturation.
- Load 10 µl of sample per lane into a 4-12% NuPAGE pre-cast gel and run the gel at 120 V.
- Transfer gel to a PVDF membrane.
- Block membrane with 5% non-fat milk in TBS-Tween (0.1%) for 1 h at room temperature.
- Incubate membrane overnight with specific primary antibodies at 4 °C while gently shaking. Dilute primary antibodies 1:1,000 in 5% non-fat milk in TBS-Tween (0.1%) buffer.
- Wash membrane three times for 5 min with TBS-Tween (0.1%) buffer.
- Incubate membrane with HRP-conjugated secondary antibodies at room temperature for 1 h while gently shaking. Dilute secondary antibodies 1:5,000 in 5% non-fat milk in TBS-Tween (0.1%) buffer.
- Wash membrane three times for 5 min with TBS-Tween (0.1%) buffer.
- Incubate membrane with enhanced chemiluminescence (ECL) substrate and acquire images with a chemiluminescence imaging system or by film exposure.
- Phenotypic analysis of hippocampal neurons by immunofluorescence (Figure 9)
- After 6 days in culture, remove the culture medium from the 24-well plate containing the poly-D-lysine/laminin pre-coated 12 mm coverslips with hippocampal neurons. Rinse the cells with PBS and fix the cells with a 4% paraformaldehyde/PBS solution for 30 min at 4 °C.
- Rinse cells with PBS calcium/magnesium free (PBS -/-) 3 times for 5 min.
- Use a blocking solution for 1 h at room temperature.
Note: Blocking solution is prepared using 10% normal donkey serum (v/v), 0.1% TritonTM-X100 (v/v), and 0.1% BSA (w/v) in PBS -/-. - Incubate the cells with specific primary antibodies overnight at 4 °C.
Note: Dilute primary antibody in a solution containing 1% normal donkey serum, 0.1% TritonTM-X100 (v/v), and 0.1% BSA (w/v) in PBS -/-. The dilution of anti-GFAP antibody is 1:1,000 and the dilution of anti-Tubulin-β-III antibody is 1:200 (see Materials and Reagents). - Rinse the cells with PBS -/-, 3 times for 5 min.
- Incubate the cells with a specific secondary antibody and DAPI staining for nucleus visualization (0.5 µg/ml) for 2 h at room temperature. Alexa Fluor 488 Donkey anti-rabbit IgG (H+L): secondary antibody for anti-GFAP antibody; Alexa Fluor 647 Donkey anti-mouse IgG (H+L): secondary antibody for anti-Tubulin-β-III antibody. Dilute secondary antibodies 1:250 using the same solution as in Step G4.
- Rinse the cells with PBS -/-, 3 times for 5 min.
- Invert coverslips and mount them to a glass slide using Dako fluorescence mounting medium. After the mounting medium is dry the samples can be analyzed by confocal imaging.
Note: Although in this protocol we show that differentiated hippocampal neurons can be obtained by 6 days in culture and we recommend the use of Ara-C for improving neuronal culture purity (Figure 8 and Figure 9), in some cases co-culturing neurons with glial cells can further support neuronal maturation and long-term survival (Kaech and Banker, 2006; Kim et al., 2020).
Figure 9. Immunofluorescence staining of primary hippocampal neurons after 6 days in culture (Procedure G). Neurons were cultured with (A-D) or without (E-H) cytosine arabinoside (Ara-C 1 µM) and stained for the glial cell marker Glial Fibrillar Acidic Protein (GFAP in green), the neuronal marker beta-III-tubulin (b-III-TUB in red) and the nuclei dye (DAPI in blue). Note the significant number of GFAP-positive cells in the neuron culture without Ara-C treatment (F) after 6 days. Higher magnification images (I-K) of neurons cultured with Ara-C 1 µM and stained for beta-III-tubulin (b-III-TUB in green) and the nuclei dye (DAPI in blue). Note the neuronal-network complexity after 6 days in vitro.
- Functional phenotypic characterization of hippocampal neurons by calcium imaging (Figure 10 and Video 1)
A further test of functionally differentiated neurons includes imaging of the calcium dynamic occurring in vitro following spontaneous or glutamate-induced depolarization (Grienberger and Konnerth, 2012)- Preparation of Fluo-4 loading buffer: to prepare 1 ml of loading buffer add 1 µl of stock Fluo-4 (Fluo-4 stock: 1 mM in DMSO; final concentration 1 µm) in 10 µl of PowerLoad (100x concentrate). Then add 10 µl of Probenecid (stock solution 250 mM; final concentration 2.5 mM) and 979 µl of ECS buffer (140 mM NaCl, 4.7 mM KCl, 2 mM NaHCO3, 1 mM NaH2PO4, 1.2 mM MgCl2, 1.5 mM CaCl2, 3 mM glucose and 10 mM HEPES, pH 7.4).
- After 6 days in vitro, remove the culture medium from the 24-well plate with the coverslips containing the hippocampal neurons. Incubate each coverslip with 250 µl of Fluo-4 loading buffer at 37 °C for 30 min following by a 30 min wash in ECS buffer at 37 °C in order to allow the de-esterification and activation of Fluo-4.
- Adjust the flow in the recording chamber to 0.5 ml x min and set the temperature to 37 °C.
- Place the coverslip in the recording chamber and select a suitable field.
- Set focus, set laser intensity (480 nm) at a lower possible value and integration time (pixel dwell time) such that the acquisition time is shorter than 500 ms before starting the time series acquisition at the confocal at 2 images x second.
- Record neuronal spontaneous activity and then apply glutamate at 1 mM for 10 s to depolarize and visualize all the cells in the field.
- For image analysis import the file in ImageJ (or Fiji https://imagej.net/Fiji/Downloads); set the lookup table to Rainbow RGB; adjust image brightness and contrast; open the ROI manager (Analyze/Tools/ROI manager); select an appropriate area shape to cover a neuron of interest and then select Images/Stacks/Plot Z-axis Profile to plot the photon density of the selected ROI over time. Add the ROI to the ROI manager and proceed with another ROI. Density plot can be exported to a spreadsheet for further quantification.
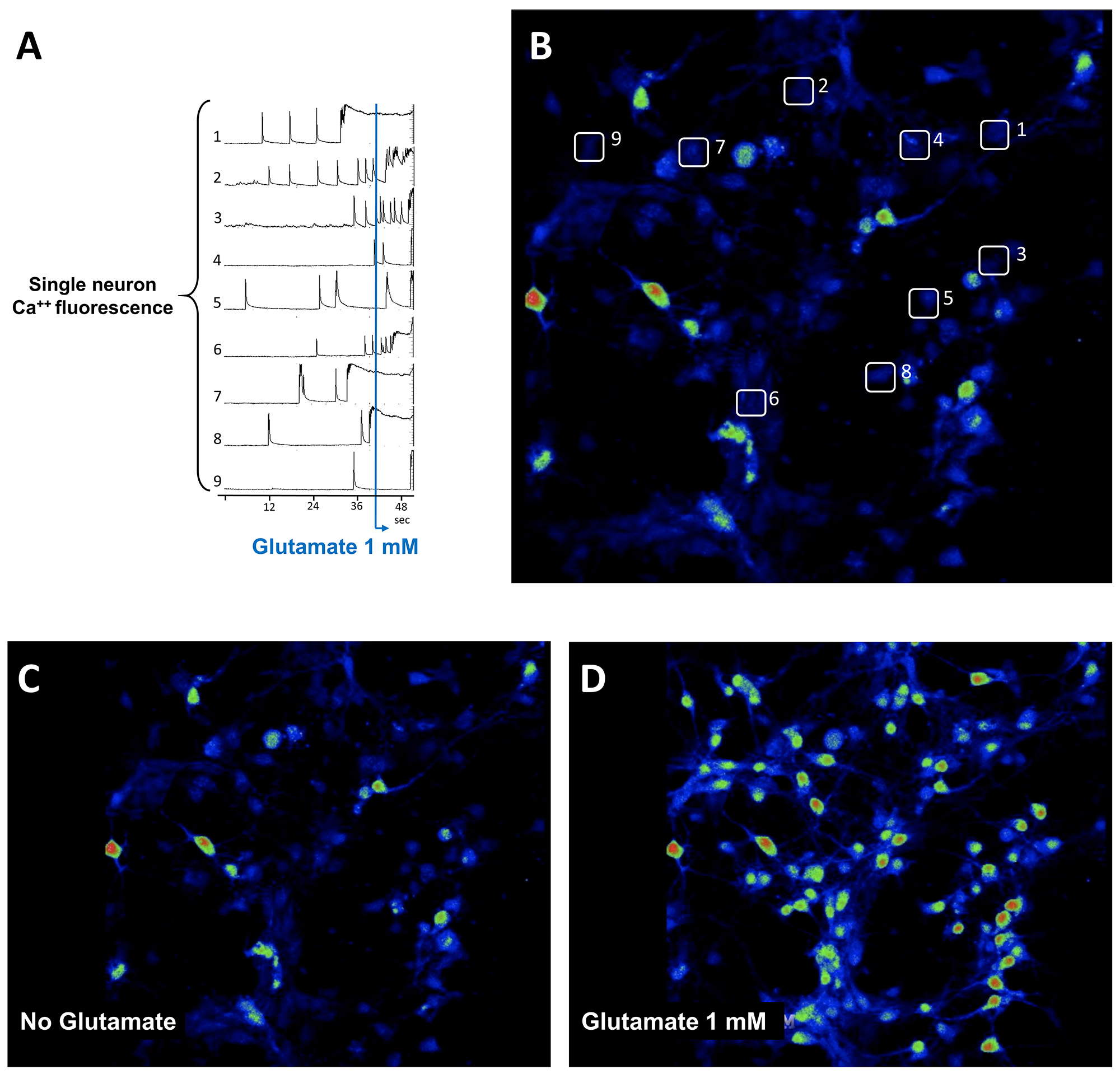
Figure 10. Calcium imaging of primary hippocampal neurons after 6 days in vitro (Procedure I). Single neuron traces of calcium fluorescence intensity over time of primary hippocampal neurons loaded with the calcium indicator Fluo-4 (A). Note the spontaneous activity of neurons before treatment with 1 mM glutamate (blue bar). Single neurons analyzed in (A) are indicated with numbers in (B). Representative images of calcium fluorescence in the neuronal culture before (C) or after (D) stimulation with 1 mM glutamate. Video 1. Calcium live imaging of primary hippocampal neurons after 6 days in vitro (Figure 10; Procedure I)
Video 1. Calcium live imaging of primary hippocampal neurons after 6 days in vitro (Figure 10; Procedure I)
Acknowledgments
We thank Eileen Southon and Andie Kealohi Sato Conching for critical reading of the manuscript. This work was supported by the NIH Intramural Research Program, Center for Cancer Research, National Cancer Institute.
Competing interests
The authors declare no competing interests and mentioning of specific materials, reagents and equipment does not imply endorsement by the National Institutes of Health.
Ethics
The experimental protocols for mouse studies were approved by the Committee of Animal Care and Use of the National Cancer Institute (ACUC) in Frederick, Maryland, USA (Approval number: 17-072; Approval date: 01/23/2018).
References
- Beaudoin, G. M., 3rd, Lee, S. H., Singh, D., Yuan, Y., Ng, Y. G., Reichardt, L. F. and Arikkath, J. (2012). Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat Protoc 7(9): 1741-1754.
- Busche, M. A. (2018). In Vivo Two-Photon Calcium Imaging of Hippocampal Neurons in Alzheimer Mouse Models. Methods Mol Biol 1750: 341-351.
- Dorsey, S. G., Renn, C. L., Carim-Todd, L., Barrick, C. A., Bambrick, L., Krueger, B. K., Ward, C. W. and Tessarollo, L. (2006). In vivo restoration of physiological levels of truncated TrkB.T1 receptor rescues neuronal cell death in a trisomic mouse model. Neuron 51(1): 21-28.
- Grienberger, C. and Konnerth, A. (2012). Imaging calcium in neurons. Neuron 73(5): 862-885.
- Kaech, S. and Banker, G. (2006). Culturing hippocampal neurons. Nat Protoc 1(5): 2406-2415.
- Kim, B. J., Choi, J. Y., Choi, H., Han, S., Seo, J., Kim, J., Joo, S., Kim, H. M., Oh, C., Hong, S., Kim, P. and Choi, I. S. (2020). Astrocyte-Encapsulated Hydrogel Microfibers Enhance Neuronal Circuit Generation. Adv Healthc Mater 9(5): e1901072.
- Koyama, R. and Ikegaya, Y. (2018). The Molecular and Cellular Mechanisms of Axon Guidance in Mossy Fiber Sprouting. Front Neurol 9: 382.
- Molnar, E. (2011). Long-term potentiation in cultured hippocampal neurons. Semin Cell Dev Biol 22(5): 506-513.
- Ray, J., Peterson, D. A., Schinstine, M. and Gage, F. H. (1993). Proliferation, differentiation, and long-term culture of primary hippocampal neurons. Proc Natl Acad Sci U S A 90(8): 3602-3606.
- Ruhl, D. A., Bomba-Warczak, E., Watson, E. T., Bradberry, M. M., Peterson, T. A., Basu, T., Frelka, A., Evans, C. S., Briguglio, J. S., Basta, T., Stowell, M. H. B., Savas, J. N., Roopra, A., Pearce, R. A., Piper, R. C. and Chapman, E. R. (2019). Synaptotagmin 17 controls neurite outgrowth and synaptic physiology via distinct cellular pathways. Nat Commun 10(1): 3532.
- Rush, T., Roth, J. R., Thompson, S. J., Aldaher, A. R., Cochran, J. N. and Roberson, E. D. (2020). A peptide inhibitor of Tau-SH3 interactions ameliorates amyloid-beta toxicity. Neurobiol Dis 134: 104668.
- Tomassoni-Ardori, F., Fulgenzi, G., Becker, J., Barrick, C., Palko, M. E., Kuhn, S., Koparde, V., Cam, M., Yanpallewar, S., Oberdoerffer, S. and Tessarollo, L. (2019). Rbfox1 up-regulation impairs BDNF-dependent hippocampal LTP by dysregulating TrkB isoform expression levels. Elife 8: e48673.
- Wu, Q., Takano, H., Riddle, D. M., Trojanowski, J. Q., Coulter, D. A. and Lee, V. M. (2019). α-Synuclein (αSyn) Preformed Fibrils Induce Endogenous alphaSyn Aggregation, Compromise Synaptic Activity and Enhance Synapse Loss in Cultured Excitatory Hippocampal Neurons. J Neurosci 39(26): 5080-5094.
Article Information
Copyright
![]() Tomassoni-Ardori et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Tomassoni-Ardori et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Tomassoni-Ardori, F., Hong, Z., Fulgenzi, G. and Tessarollo, L. (2020). Generation of Functional Mouse Hippocampal Neurons. Bio-protocol 10(15): e3702. DOI: 10.21769/BioProtoc.3702.
- Tomassoni-Ardori, F., Fulgenzi, G., Becker, J., Barrick, C., Palko, M. E., Kuhn, S., Koparde, V., Cam, M., Yanpallewar, S., Oberdoerffer, S. and Tessarollo, L. (2019). Rbfox1 up-regulation impairs BDNF-dependent hippocampal LTP by dysregulating TrkB isoform expression levels. Elife 8: e48673.
Category
Neuroscience > Cellular mechanisms > Cell isolation and culture
Cell Biology > Cell isolation and culture > Cell differentiation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.