- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Method for User-defined Mutagenesis by Integrating Oligo Pool Synthesis Technology with Nicking Mutagenesis

Published: Vol 10, Iss 15, Aug 5, 2020 DOI: 10.21769/BioProtoc.3697 Views: 7042
Reviewed by: Renuka KudvaNicole R. BuanMichael T Veling
Original research article
The authors used this protocol in:

Sep 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Saturation mutagenesis is a fundamental enabling technology for protein engineering and epitope mapping. Nicking mutagenesis (NM) allows the user to rapidly construct libraries of all possible single mutations in a target protein sequence from plasmid DNA in a one-pot procedure. Briefly, one strand of the plasmid DNA is degraded using a nicking restriction endonuclease and exonuclease treatment. Mutagenic primers encoding the desired mutations are annealed to the resulting circular single-stranded DNA, extended with high-fidelity polymerase, and ligated into covalently closed circular DNA by Taq DNA ligase. The heteroduplex DNA is resolved by selective degradation of the template strand. The complementary strand is synthesized and ligated, resulting in a library of mutated covalently closed circular plasmids. It was later shown that because very little primer is used in the procedure, resuspended oligo pools, which normally require amplification before use, can be used directly in the mutagenesis procedure. Because oligo pools can contain tens of thousands of unique oligos, this enables the construction of libraries of tens of thousands of user-defined mutations in a single-pot mutagenesis reaction, which significantly improves the utility of NM as described below.
Use of oligo pools afford an economically advantageous approach to mutagenic experiments. First, oligo pool synthesis is much less expensive per nucleotide synthesized than conventional synthesis. Second, a mixed pool may be generated and used for mutagenesis of multiple different genes. To use the same oligo-pool for mutagenesis of a variety of genes, the user must only quantify the fraction of the oligo-pool specific to her mutagenic experiment and adjust the volume and effective concentration of the oligo-pool for use in nicking mutagenesis.
Background
Evaluation of the sequence dependence of protein function is of tremendous importance for applied and fundamental protein science. In recent years, deep mutational scanning (DMS) has risen to the forefront of protein-based research (Fowler and Fields, 2014). DMS experiments allow for the elucidation of genotype-phenotype relationships and the generation of biomolecular fitness landscapes using large numbers of protein variants assessed using deep sequencing. DMS has been employed for protein engineering (Romero et al., 2015), epitope mapping (Van Blarcom et al., 2015; Kowalsky et al., 2015a), and evolutionary biology (Doud et al., 2017; Faber et al., 2019). The key to DMS is correlating the abundance of cells expressing a particular protein variant to a particular property of that variant. If this can be done then quantitative deep sequencing can measure the property for tens of thousands of protein variants in an in vivo library. The generation of libraries containing large numbers of programmed variants is thus essential to obtaining high quality data from a DMS experiment. Often, a saturation mutagenesis library containing all possible single mutations is the desired starting point.
Methods for saturation mutagenesis have continued to improve over time, starting with uracil-dependent Kunkel and Pfunkel Mutagenesis (Kunkel, 1985; Firnberg and Ostermeier, 2012). More recently, the development of nicking mutagenesis by Wrenbeck et al. (2016), which employs complementary nicking restriction endonucleases has improved on Kunkel and Pfunkel in time and convenience. At the same time, oligonucleotide pool technology, which yields tens of thousands of specifically designed oligos in one pot, has rapidly advanced. Oligo-pools afford practical and economical benefits to the original NM protocol, permitting many more mutations to be designed at lower cost (Medina-Cucurella et al., 2019). The combination of nicking mutagenesis with oligo pool-derived primers may benefit any research where many amino acid substitutions are desired at one or many sites in a target protein, and specifically in directed evolution studies applicable to protein design and evolutionary biology. As an example, this protocol was recently used to program nearly all single point mutants in critical genes in viruses (Faber et al., 2020).
Materials and Reagents
- 245 mm Square BioAssay Dishes (Corning®, catalog number: 431111 )
- PCR Tubes (e.g., VWR, catalog number: 52509-304 )
- Glass Pasteur pipette
- Scraper
- 50 ml conical tube (e.g., Thermo Fisher, catalog number: 14-432-22 )
- High-efficiency electrocompetent E. coli cells (essential that these are > 109 CFU/μg plasmid DNA, e.g., Agilent XL-1-Blue Electroporation Competent Cells) (Agilent, catalog number: 200228 )
- Oligo pool containing mutagenic oligos (or manually pooled mutagenic oligos), e.g., Agilent SurePrint Oligonucleotide Libraries
- Single primer that anneals to the template strand at a non-mutagenized location with an opposite orientation compared with mutagenic oligos (synthesized by e.g., Integrated DNA Technologies)
- Plasmid to mutagenize containing a single BbvCI restriction site (multiple BbvCI restriction sites are acceptable as long as all are in the same orientation)
We have successfully mutagenized plasmids up to 8 kb in length. The plasmid pEDA5-GFPmut3-Y66H described in Wrenbeck et al. (2016) (Addgene ID 80085) is a useful control (see Note 3). - Monarch® PCR & DNA Cleanup Kit (5 μg) (New England BioLabs, catalog number: T1030S/L )
- Miniprep kit (e.g., Monarch® Plasmid Miniprep Kit, New England BioLabs, catalog number: T1010S )
- T4 Polynucleotide Kinase (New England BioLabs, catalog number: M0201S/L )
- Nt.BbvCI (New England BioLabs, catalog number: R0632S/L )
- Nb.BbvCI (New England BioLabs, catalog number: R0631S/L )
- Exonuclease I (E. coli) (New England BioLabs, catalog number: M0293S/L )
- Exonuclease III (E. coli) (New England BioLabs, catalog number: M0206S/L )
- Taq DNA Ligase (New England BioLabs, catalog number: M0208S/L )
- Phusion® High-Fidelity DNA Polymerase (New England BioLabs, catalog number: M0530S/L )
- DpnI (New England BioLabs, catalog number: R0176S/L )
- Nuclease free water (e.g., New England BioLabs, catalog number: B1500S/L )
- Molecular biology grade Dithiothreitol (DTT) (e.g., GoldBio, catalog number: DTT10 )
- 10x CutSmart® Buffer (New England Biolabs, catalog number: B7204S )
- SYBR Safe DNA gel stain (Thermo Fisher, catalog number: S33102 )
- 5x HF Buffer (included with Phusion® polymerase) (New England Biolabs, catalog number: B0518S )
- 10 mM Adenosine 5’-Triphosphate (ATP) (e.g., New England BioLabs, catalog number: P0756S/L ) prepared in single-use aliquots of 10 μl
- 10 mM Deoxynucleotide (dNTP) Solution Mix (e.g., New England BioLabs, catalog number: N0447S )
- β-Nicotinamide adenine dinucleotide (NAD+) (50 mM) (e.g., New England BioLabs, catalog number: B9007S ) prepared in single-use aliquots of 10 μl
- Single-use aliquots (see Recipes)
10 mM ATP
50 mM NAD+
10 mM dNTPs - 50 mM DTT (see Recipes)
Equipment
- -20 °C freezer
- Pipettes
- Thermal Cycler (e.g., Eppendorf, model: MastercyclerTM pro, catalog number: 950040025 )
- Microcentrifuge (e.g., Fisher, model: accuSpin microcentrifuge 17 , catalog number 13-100-675 ; does not need to be refrigerated)
- Electroporator (e.g., Eppendorf Eporator®, catalog number: 4309000027 )
Software
- Python
For making large numbers of specific mutations at many different amino acid positions, automated primer design software is indispensable. We provide a flexible python script for automated primer design in the supplementary information. This script can design mutagenic primers either using a user-specified degenerate codon (e.g., NNK), or by generating one or more mutation specific primers using common codons for a specified organism. The outputs from the script can be specified as tab-delimited or comma-separated values (CSV) files with programmable primer naming. Finally, the script facilitates precise specifications of which residues to mutate using the resfile format from the Rosetta macromolecular modeling suite (Leaver-Fay et al., 2011). Below, we provide some basic examples of inputs and command lines for the script.
The software is written such that the 5′ and 3′ arm lengths are constant, either at a user defined length or a default length of 30 bases. Constant oligonucleotide lengths result in differential melting temperatures for individual members of the oligo pool and is a compromise with length and cost restrictions for oligo pool synthesis. Within a fixed-length pool, the primers with the highest predicted melting temperatures appear to be incorporated up to tenfold more efficiently than those with the lowest predicted melting temperatures (Medina-Cucurella et al., 2019), but the bias is tolerable for all end-uses in our laboratory.
The only required input to the script is a plain text file containing three lines. The first line should contain sequence of the coding sequence of the gene to mutagenize (at least the length of the 5′ homology arm length), the second line should contain the coding sequence (CDS) to mutagenize without a stop codon, and the third line should contain the stop codon and sequence downstream of the coding sequence to mutagenize (at least the length of the 3′ homology arm length). The additional sequence is required in order to design primers that mutagenize the first and last few amino acids, the amount of additional sequence is dependent on the desired 5′ and 3′ arm lengths for the mutagenic primers. The full list of command-line flags is shown in Table S1.
Suppose we want to mutagenize the amino acid sequence MVTAGENSIS, which is encoded (CDS; bold) within the following DNA sequence:
ATAGACAGTAATGGTGACCGCGGGCGAAAACGAAAGCATTAGCTAGACAGTTG
The input file (input.seq) we would use with the script would be:
ATAGACAGTA
ATGGTGACCGCGGGCGAAAACGAAAGCATTAGC
TAGACAGTTG
(The CDS is shown in boldface for clarity.) Note that the third line includes the TAG stop codon.
Example 1:
Make every possible amino acid mutation (including wild-type) at every position in the coding sequence. Generate two primers for each mutation, using the two most common codons in yeast for the mutant amino acid. Design each primer with 28 basepairs of homology to the template before and after the mismatched codon. Output a CSV to the file primers.csv.
python nm_primers.zip --five-prime-arm-length 28 --three-prime-arm-length 28 --organism yeast --separator comma --output primers.csv input.seq
Example 2:
Mutate each residue using the degenerate codon NNK. Output a tab-separated file to primers.tsv. Design each primer with 20 base pairs homology upstream of the mutation and 30 basepairs homology downstream of the mutation.
python nm_primers.zip --five-prime-arm-length 20 --three-prime-arm-length 30 --custom-codon NNK --separator tab –output primers.tsv input.seq
Procedure
A visual depiction of the protocol steps is pictured in Figure 1 followed by detailed written descriptions of each step. Briefly, steps [A] (preparation of plasmid DNA) and [B]-[D] (design, pooling, and phosphorylation of oligonucleotides) are performed ahead of time and can be done in parallel. Steps [E]-[L] are all performed on a single day. ssDNA is prepared from plasmid dsDNA [E] and the mutagenic strand is synthesized from the ssDNA template [F]. The heteroduplex is column purified [G] and the template strand is specifically degraded [H]. The mutagenic strand is regenerated [I], and the mutagenic DNA is enriched [J], column purified [K], and transformed into E. coli [L]. The next day plasmids containing user-defined mutations are prepped from E. coli transformants [M].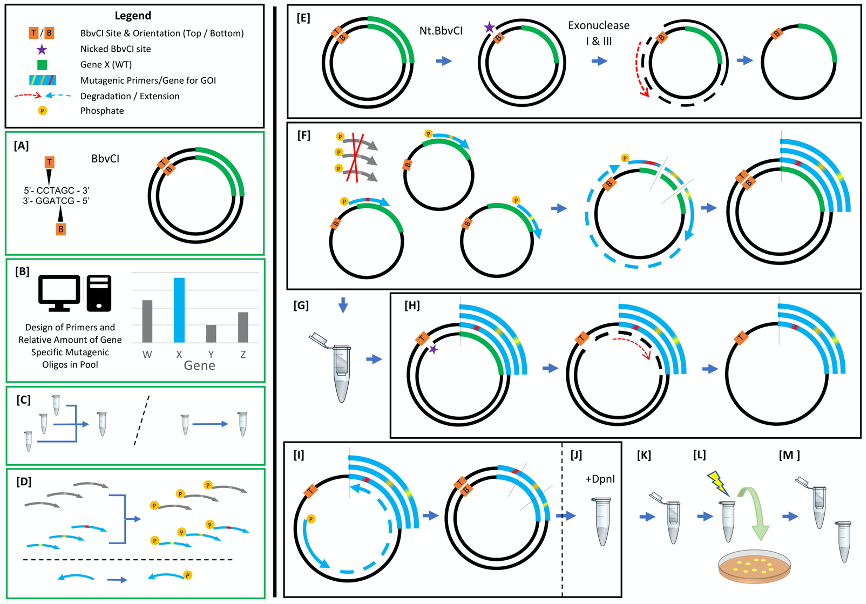
Figure 1. Nicking Mutagenesis Procedure. Steps [A]-[D] shown in green boxes are prepared independently of the rest of the method. [A] Preparation of mutagenesis template plasmid with unique BbvCI restriction site; [B] Automated user-defined design of mutagenic primers; [C] (as needed) Pooling of mutagenic primers; [D] Phosphorylation of oligonucleotides; [E] ssDNA template prep; [F] Mutagenic strand synthesis; [G] Column purification; [H] Bottom template strand degradation; [I] Mutagenic strand regeneration; [J] Removal of input plasmid with DpnI; [K] Column purification; [L] Transformation; [M] Plasmid library preparation.
- Prepare mutagenesis template plasmid
The template plasmid must contain a BbvCI site (CCTCAGC). It is acceptable for the plasmid to contain multiple BbvCI sites only if all are in the same orientation. If not already present, the site can easily be added using any site-directed mutagenesis procedure (e.g., Q5® Site-Directed Mutagenesis). If using the nicking enzymes in the order presented here (Nt.BbvCI first, Nb.BbvCI second), the orientation of the nicking site will determine if the mutagenic primers should match the sense or antisense strand of the plasmid. BbvCI is needed because nickase versions that nick only the top or only the bottom strand are available. In principle other nickases could be used but the buffers may need to be reformulated to allow compatibility with the nucleases and other DNA-modifying enzymes.
Nicked plasmid DNA will be degraded during nicking mutagenesis, so plasmid preparations should be freshly prepared, either immediately prior to use, or not more than one month in advance. The plasmid should not be subjected to more than one freeze-thaw cycle to maximize the fraction of covalently closed molecules. Plasmid prepared and frozen for up to one month in a non-defrosting -20 °C freezer can be used (see troubleshooting tips in Table 1). - Design mutagenic oligonucleotides
Design primers encoding the desired mutations. Primers can encode multiple mutations as long as sufficient homology to the template is included upstream and downstream of the first and last mutations respectively. We typically have these homologous regions be at least 24 bp. Using software makes it possible to design thousands of primers quickly and minimizes errors in design even for small numbers of primers. A primer design script written in Python is provided in the supplementary information (see Software above).
Small numbers of primers can be ordered individually in tubes or 96-well plates, but when making thousands of programmed mutations (e.g., a large number of proximal double mutants), oligo pools are significantly more economical. Because nicking mutagenesis uses femtomole amounts of primers (< 0.05 pmol per reaction), commercial oligo pools can be used directly without PCR amplification. Furthermore, non-homologous primers present in an oligo pool will not anneal to templates or interfere with the procedure, making it possible to synthesize mutagenic primer sets for multiple genes in a single pool. - Pool mutagenic oligonucleotides
If using primers from an oligo pool, resuspend the pool according to the manufacturer’s instructions and compute the fraction of the oligo pool containing relevant primers for this mutagenesis. If pooling individual primers, resuspend each primer and pool all primers so that the final concentration of all primers is 10 μM. For example, if pooling 100 mutagenic primers, the final concentration of each individual primer should be 100 nM, giving a final concentration of 10 μM total primer. - Phosphorylate oligonucleotides
- Assemble the following two reactions in PCR tubes.
Primer pool phosphorylation:
Secondary primer phosphorylation:Component Volume (μl) Final oncentration or Amount Pooled primers (10 μM) 20 8.2 μM 10x T4 Polynucleotide Kinase Buffer 2.44 1x 10 mM ATP 1 0.41 mM T4 Polynucleotide Kinase (10 U/μl) 1 10 U Component Volume (μl) Final Concentration or Amount Nuclease-free H2O 18 N/A 10x T4 Polynucleotide Kinase Buffer 3 1x Secondary primer (100 μM) 7 23.3 μM 10 mM ATP 1 0.33 mM T4 Polynucleotide Kinase (10 U/μl) 1 10 U - Incubate the two reactions at 37 °C for one hour in a thermocycler. As an optional step, inactivate T4 kinase by incubating at 65 °C for twenty min.
- Assemble the following two reactions in PCR tubes.
- Top strand degradation (Template Prep)
- Assemble the following reaction in a PCR tube on ice:
*0.76 pmol of… 3 kb plasmid = 1.41 μg; 4 kb plasmid = 1.88 μg; 5 kb plasmid = 2.35 μg; etc. In order to constrain the volume of plasmid to 1-15 μl, the plasmid must be at a concentration of 51-760 nM. (e.g., a 3 kb plasmid will be between 95 and 1410 ng/μl; a 5 kb plasmid, between 157 and 2350 ng/μl, etc.).Component Volume (μl) Final Concentration or Amount Nuclease-free H2O To 20 μl final volume N/A Fresh plasmid DNA containing BbvCI site 0.76 pmol* 0.76 pmol 10x CutSmart® Buffer 2 1x Exonuclease III (diluted tenfold to 10 U/μl) 1 10 U Exonuclease I (20 U/μl) 1 20 U Nt.BbvCI (10 U/μl) 1 20 U - Run the following program on the thermocycler:
60 min at 37 °C
20 min at 65 °C
Hold at 4 °C
- Assemble the following reaction in a PCR tube on ice:
- Top strand synthesis
- Transfer the 20 μl strand preparation reaction on ice and add the following:
Component Volume (μl) Final Concentration or Amount Nuclease-free H2O 26.7 N/A 5x Phusion HF Buffer 20 1x 1:1,000 diluted phosphorylated oligo pool (final concentration 8.2 nM total primer) 4.3 0.035 pmol 50 mM DTT 20 10 mM 50 mM NAD+ 1 0.5 mM 10 mM dNTPs 2 0.2 mM Phusion polymerase (2 U/μl) 1 2 U Taq DNA Ligase (40 U/μl) 5 200 U - Then run the following protocol on the thermocycler:
- 2 min at 98 °C
- 15 cycles of:
30 s at 98 °C
30 s at 55 °C
30 s/kb at 72 °C
At the end of cycles 5 and 10, add an additional 4.3 μl of 1:1,000 diluted phosphorylated oligo pool. To facilitate this, add pauses after the fifth and tenth cycles of step b above. Adding primers in boluses yields a larger number of transformants while keeping the plasmid-to-primer molar ratio low. - 20 min at 45 °C
- Hold at 4 °C
- 2 min at 98 °C
- Transfer the 20 μl strand preparation reaction on ice and add the following:
- Column purification
- Clean up the reaction using a Monarch® PCR & DNA Cleanup Kit (New England BioLabs) according to the manufacturer’s instructions, with the exception of using five volumes of binding buffer instead of two volumes.
- Elute in 15 μl nuclease-free H2O at room temperature, waiting five min after applying the eluant to the column before centrifugation.
- Bottom strand degradation
- Transfer 14 μl of the column purified reaction eluate to a fresh PCR tube and place on ice. Add the following to the tube (final volume 20 μl):
Component Volume (μl) Final Concentration or Amount 10x CutSmart® Buffer 2 1x 2 U/μl diluted Exonuclease III
(a 50-fold dilution of the stock concentration of 100 U/ μl into 1x CutSmart®)2 4 U Exonuclease I (20 U/μl) 1 20 U 1 U/μl BbvCI.Nb
(a 10-fold dilution into 1x CutSmart® from the stock)1 1 U - Run the following program on the thermocycler:
60 min at 37 °C
20 min at 65 °C
Hold at 4 °C
- Transfer 14 μl of the column purified reaction eluate to a fresh PCR tube and place on ice. Add the following to the tube (final volume 20 μl):
- Bottom strand synthesis
- Place the 20 μl bottom strand degradation reaction on ice and add the following:
Component Volume (μl) Final Concentration or Amount Nuclease-free H2O 27.7 5x Phusion HF Buffer 20 1x 1:20 diluted phosphorylated secondary primer 4.3 5 pmol 50 mM DTT 20 10 mM 50 mM NAD+ 1 0.5 mM 10 mM dNTPs 2 0.2 mM Phusion polymerase (2 U/μl) 1 2 U Taq DNA Ligase (40 U/μl) 5 200 U - Run the following program on the thermocycler:
- 30 s at 98 °C
- 30 s at 55 °C
- 10 min at 72 °C
- 20 min at 45 °C
- Hold at 4 °C
- 30 s at 98 °C
- Place the 20 μl bottom strand degradation reaction on ice and add the following:
- Removal of input plasmid
Add 2 μl DpnI (20 U/μl) to the 100 μl bottom strand synthesis PCR and incubate at 37 °C for one hour in a thermocycler. - Column purification
- Clean up the reaction using a Monarch® PCR & DNA Cleanup Kit (New England BioLabs) according to the manufacturer’s instructions.
- Use five volumes of binding buffer (not two).
- Elute in 6 μl nuclease-free H2O at room temperature, waiting five min after applying the eluant to the column before centrifugation.
- Transformation
- Transform the entire purified product into high efficiency electrocompetent E. coli according to the manufacturer’s instructions.
- Using high efficiency cells (> 109 CFU/μg plasmid DNA) is crucial to the success of the protocol. Plate serial dilutions (e.g., 10−4, 10−5, and 10−6 of the full transformation) on appropriate selective medium in order to determine efficiency.
- Plate the remainder of the transformation on a large square bioassay plate of appropriate selective medium.
- Incubate the plates overnight at 37 °C.
- Plasmid library preparation
A successful transformation should yield a lawn of colonies on the large bioassay plate. A large scraper, as can be prepared by bending a heated glass Pasteur pipette, is useful for resuspending colonies for plasmid preparation.- Apply 5-7 ml of liquid medium (such as LB) to the bioassay plate and scrape the colonies off of the agar surface using the scraper.
- Slightly incline the plate so that the cell suspension pools in one corner and transfer the suspension to a sterile 50 ml conical tube with either a 1 ml pipette or a 10 ml serological pipette. Typically, 1-2 ml of the first liquid media dispensed to the plate is absorbed by the agar.
- Continue to add medium, scrape colonies, and transfer the suspension until the agar is clear of all bacterial growth.
- Mix the cell suspension by aggressive vortexing and/or trituration with a serological pipette to completely disperse aggregated biomass. If the biomass is suspended in roughly 10 ml, the culture will be completely opaque and it will be significantly denser than a saturated E. coli liquid culture in LB.
- Finally, recover plasmid DNA from a small amount of the suspension using of the cell suspension using a miniprep kit (e.g., New England BioLabs Monarch® Plasmid Miniprep Kit T1010S ). For a lawn on a bioassay plate resuspended in 10 ml of LB, 300 μl is usually sufficient to obtain 15 μg of high-copy number plasmid DNA.
Data analysis
After mutagenesis, approximate the number of transformants by counting colonies from the plated dilution series. Specifically, identify the dilution factor where the number of colonies is between 10-100 and multiply the number of colonies on each plate by the corresponding dilution according to the equation below:
To approach complete coverage of the desired set of mutations, a general guideline is to recover at minimum ten times more transformants than there are library variants. Recovering many more transformants is preferable. If the distribution of variant frequencies in the library were uniform, roughly 4.6-fold coverage would be required for 99% coverage (Bosley et al., 2005). However, variant frequencies with nicking mutagenesis are typically distributed log-normally, which necessitates higher fold coverage (Wrenbeck et al., 2016; Medina-Cucurella et al., 2019). Figure 2 shows simulated coverage results demonstrating that 100-fold coverage is a better target. Actual coverage can be measured by next generation sequencing, which is described elsewhere (Kowalsky et al., 2015b).
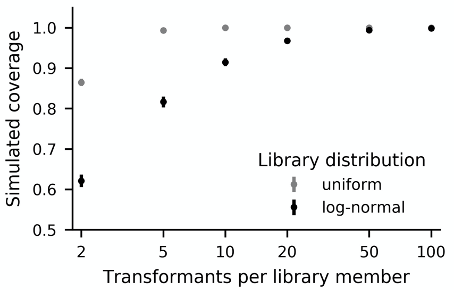
Figure 2. Simulated distribution of coverage for 1000-member libraries with uniform or log-normal variant frequency distributions. Each data point shows mean and standard deviation of library coverage from 100,000 simulated experiments. For most points, the standard deviation is too small to be seen on this scale. The log-normally distributed frequencies were generated by normalizing a lognormal (μ = 4.6, σ = 1.15) distribution seen in experimental datasets (Wrenbeck et al., 2016). For log-normally distributed libraries, 100 transformants per library member is a good target.
Notes
- Plasmid preparation for mutagenesis
Preparation of high quality circular closed dsDNA is crucial to the success of the protocol. Plasmid DNA nicked on the strand opposite to that targeted by the nicking enzyme will be degraded completely rather than to closed ssDNA. It is therefore important that plasmid DNA used as input is not nicked, which is best accomplished by using freshly prepared plasmid that has not been subjected to freeze-thaw cycles. Plasmid quality can be assessed by gel electrophoresis. Supercoiled plasmid DNA should be the dominant species in a high-quality sample. - Efficiency
Using the comprehensive mutagenesis procedure described here, we typically find that 20-50% of transformants are wild type plasmid. Anecdotally, smaller plasmid templates lead to much lower fractions of wild-type transformants (and, as is typical, to much larger numbers of transformants in general). If a large number of transformants is required, it can be helpful to mutagenize only the relevant fragment of a gene in a minimal vector and later subclone the resulting library. If the library will eventually be transformed into yeast, it is particularly useful to transform the yeast with linear vector backbone linear mutagenized fragment library and rely on in vivo homologous recombination. - Troubleshooting
Frequently observed issues with performing the protocol are given in Table 1. Typically, the first step in troubleshooting nicking mutagenesis should be to attempt the procedure using the GFP control plasmid pEDA5-GFPmut3-Y66H described in Wrenbeck et al. (2016) (Addgene ID 80085). The Y66H mutation encoded in the plasmid eliminates the encoded GFP fluorescence and mutagenesis recovers this fluorescence, allowing easy screening for success. - The next step should be to confirm ssDNA template preparation (Procedure E) and regeneration (Procedure F) using your unique plasmid and primer sets with both Nt.BbvCI and Nb.BbvCI. Perform a scaled-up digestion with the nicking enzyme, Exonuclease I, and Exonuclease III. Use 20 μl of this reaction for the 100 μl polymerization/ligation reaction. Finally, analyze uncut plasmid, nicked/degraded plasmid, and regenerated template by agarose gel electrophoresis. It is helpful to use SYBR Safe DNA gel stain (Thermo Fisher S33102 ), because it causes ssDNA to appear orange and dsDNA to appear green when illuminated by a blue-light transilluminator.
Table 1. Common list of issues with troubleshooting suggestions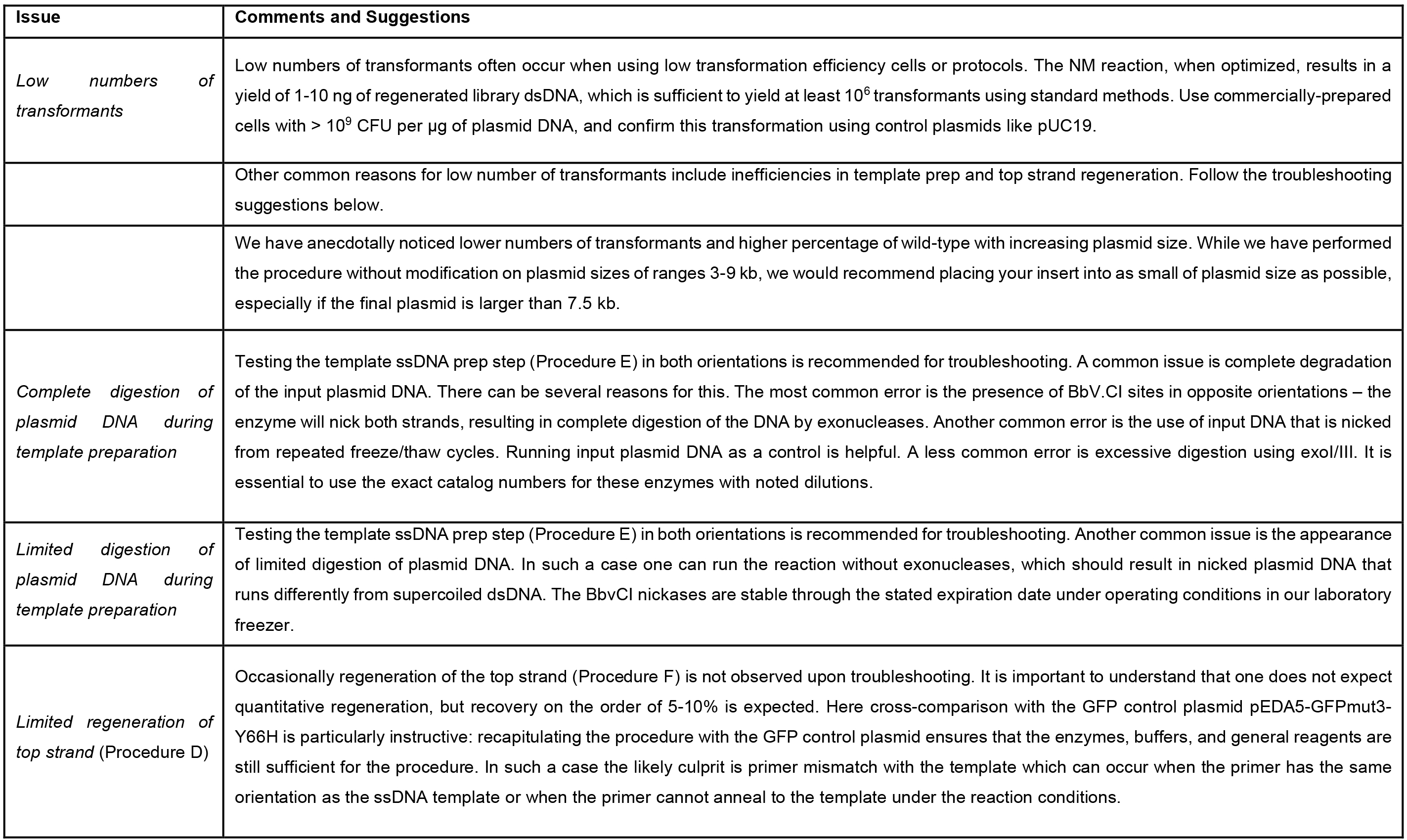
Recipes
- Single-use aliquots
10 mM ATP
50 mM NAD+
10 mM dNTPs
Store at -20 °C
ATP and NAD+ should not be freeze-thawed - 50 mM DTT
50 mM DTT in nuclease-free water and store aliquots at -20 °C
Acknowledgments
Research reported in this publication was supported by the National Institute Of Allergy And Infectious Diseases of the National Institutes of Health under Award Number R01AI141452 to T.A.W. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors wish to acknowledge Emily Wrenbeck, Justin Klesmith, Jim Stapleton, and Angelica Medina-Cucurella for original research upon which this protocol was derived.
Competing interests
T.A.W. is on a U.S. patent application 16/115.029 covering the nicking mutagenesis method. P.J.S and Z.T.B. do not have competing interests.
References
- Bosley, A. D. and Ostermeier, M. (2005). Mathematical expressions useful in the construction, description and evaluation of protein libraries. Biomol Eng 22(1-3): 57-61.
- Doud, M. B., Hensley, S. E. and Bloom, J. D. (2017). Complete mapping of viral escape from neutralizing antibodies. PLoS Pathog 13(3): e1006271.
- Faber, M. S., Van Leuven, J. T., Ederer, M. M., Sapozhnikov, Y., Wilson, Z. L., Wichman, H. A., Whitehead, T. A. and Miller, C. R. (2020). Saturation mutagenesis genome engineering of infective PhiX174 bacteriophage via unamplified oligo pools and golden gate assembly. ACS Synth Biol 9(1): 125-131.
- Faber, M. S., Wrenbeck, E. E., Azouz, L. R., Steiner, P. J. and Whitehead, T. A. (2019). Impact of in vivo protein folding probability on local fitness landscapes. Mol Biol Evol 36(12): 2764-2777.
- Fowler, D. M. and Fields, S. (2014). Deep mutational scanning: a new style of protein science. Nat Methods 11(8): 801-807.
- Firnberg, E. and Ostermeier, M. (2012). PFunkel: efficient, expansive, user-defined mutagenesis. PLoS One 7(12): e52031.
- Kowalsky, C. A., Faber, M. S., Nath, A., Dann, H. E., Kelly, V. W., Liu, L., Shanker, P., Wagner, E. K., Maynard, J. A., Chan, C. and Whitehead, T. A. (2015a). Rapid fine conformational epitope mapping using comprehensive mutagenesis and deep sequencing. J Biol Chem 290(44): 26457-26470.
- Kowalsky, C. A., Klesmith, J. R., Stapleton, J. A., Kelly, V., Reichkitzer, N. and Whitehead, T. A. (2015b). High-resolution sequence-function mapping of full-length proteins. PLoS One 10(3): e0118193.
- Kunkel, T. A. (1985). Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci U S A 82(2): 488-492.
- Leaver-Fay, A., Tyka, M., Lewis, S. M., Lange, O. F., Thompson, J., Jacak, R., Kaufman, K., Renfrew, P. D., Smith, C. A., Sheffler, W., Davis, I. W., Cooper, S., Treuille, A., Mandell, D. J., Richter, F., Ban, Y. E., Fleishman, S. J., Corn, J. E., Kim, D. E., Lyskov, S., Berrondo, M., Mentzer, S., Popovic, Z., Havranek, J. J., Karanicolas, J., Das, R., Meiler, J., Kortemme, T., Gray, J. J., Kuhlman, B., Baker, D. and Bradley, P. (2011). ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol 487: 545-574.
- Medina-Cucurella, A. V., Steiner, P. J., Faber, M. S., Beltran, J., Borelli, A. N., Kirby, M. B., Cutler, S. R. and Whitehead, T. A. (2019). User-defined single pot mutagenesis using unamplified oligo pools. Protein Eng Des Sel 32(1): 41-45.
- Romero, P. A., Tran, T. M. and Abate, A. R. (2015). Dissecting enzyme function with microfluidic-based deep mutational scanning. Proc Natl Acad Sci U S A 112(23): 7159-7164.
- Van Blarcom, T., Rossi, A., Foletti, D., Sundar, P., Pitts, S., Bee, C., Melton Witt, J., Melton, Z., Hasa-Moreno, A., Shaughnessy, L., Telman, D., Zhao, L., Cheung, W. L., Berka, J., Zhai, W., Strop, P., Chaparro-Riggers, J., Shelton, D. L., Pons, J. and Rajpal, A. (2015). Precise and efficient antibody epitope determination through library design, yeast display and next-generation sequencing. J Mol Biol 427(6 Pt B): 1513-1534.
- Wrenbeck, E. E., Klesmith, J. R., Stapleton, J. A., Adeniran, A., Tyo, K. E. and Whitehead, T. A. (2016). Plasmid-based one-pot saturation mutagenesis. Nat Methods 13(11): 928-930.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Steiner, P. J., Baumer, Z. T. and Whitehead, T. A. (2020). A Method for User-defined Mutagenesis by Integrating Oligo Pool Synthesis Technology with Nicking Mutagenesis. Bio-protocol 10(15): e3697. DOI: 10.21769/BioProtoc.3697.
Category
Molecular Biology > Protein > Expression
Biochemistry > Protein > Synthesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.