- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A SsrA/NIa-based Strategy for Post-Translational Regulation of Protein Levels in Gram-negative Bacteria


Published: Vol 10, Iss 14, Jul 20, 2020 DOI: 10.21769/BioProtoc.3688 Views: 4380
Reviewed by: Alba BlesaLi ZhangThibaud T. Renault
Original research article
The authors used this protocol in:

Nov 2018

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Strategies to control the levels of key enzymes of bacterial metabolism are commonly based on the manipulation of gene of interest within the target pathway. The development of new protocols towards the manipulation of biochemical processes is still a major challenge in the field of metabolic engineering. On this background, the FENIX (functional engineering of SsrA/NIa-based flux control) system allows for the post-translational regulation of protein levels, providing both independent control of the steady-state protein amounts and inducible accumulation of target proteins. This strategy enables an extra layer of control over metabolic fluxes in bacterial cell factories (see Graphical abstract below). The protocol detailed here describes the steps needed to design FENIX-tagged proteins and to adapt the system to virtually any pathway for fine-tuning of metabolic fluxes.
Graphical abstract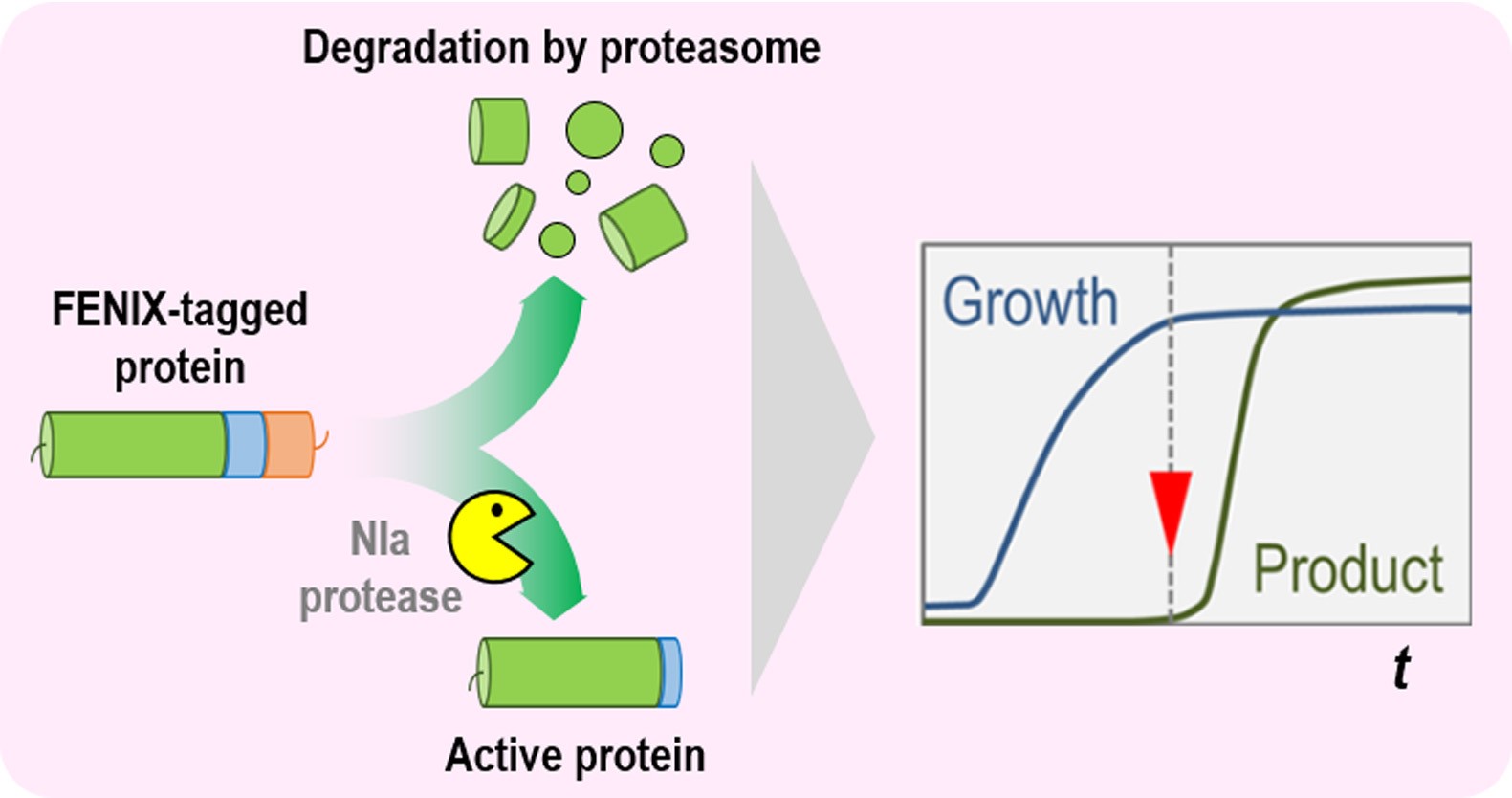
Background
Controlling protein production has become a challenging problem that has been mostly tackled by manipulating the regulation of its production at different levels, such as the DNA level or the amount of protein(s) produced (Wu et al., 2016; Avcilar-Kucukgoze et al., 2017). At the DNA level, both transcription and translation have been widely studied in several microorganisms, enabling the design and development of a large number of synthetic circuits to improve bioproduction processes (Guzmán et al., 1995; Lutz and Bujard, 1997). In contrast, the protein level has been much less studied, with protocols mainly based on RNA interference, riboregulators and specific transcriptional regulators (Isaacs et al., 2004; Lou et al., 2012).
In order to implement a new approach towards the regulation of protein levels, we recently proposed a synthetic biology tool called FENIX (functional engineering of SsrA/NIa-based flux control) (Durante-Rodríguez et al., 2018). FENIX is based on the activity of the protease NIa, capable of recognizing and cleaving a specific sequence of 13 amino acids (the nia target, GESNVVVHQADER) (Verchot et al., 1991; Stevens, 2000; Calles et al., 2019), and the action of the proteasome, which recognizes a sequence of 11 amino acids located in the C-terminal region of a protein (the ssrA target, AANDENYALAA) (Doma and Parker, 2007; Shoemaker et al., 2010). We have designed a synthetic NIa/SsrA tag (GESNVVVHQADER•AANDENYALAA) that can be easily fused to the C-terminal module of any protein of interest via a single cloning step in a standardized vector. The FENIX system relies on the constitutive degradation of the target protein by the proteasome, followed by conditionally restoring protein accumulation by the cleavage of the proteasome tag (ssrA) through the action of the protease NIa. The whole circuit is triggered by the addition of 3-methylbenzoate, which induces the XylS/Pm expression system, thus controlling the individual components of the FENIX system. This novel tool can be also used in Pseudomonas putida for tight control of protein accumulation (Volke et al., 2020). The protocol described below discusses the overall strategy along with specific details on its implementation.
Materials and Reagents
- Kits
- High-Pure plasmid isolation kit (Roche, catalog number: 11754777001 )
- Gene-Clean Turbo kit (Q-BIOgene, catalog number: 1102-400 )
- Enzymes
- Pfu DNA polymerase (Promega, catalog number: M7741 )
- T4 DNA ligase (New England Biolabs, catalog number: M0202L )
- Restriction enzyme NheI (New England Biolabs, catalog number: R3131S )
- Restriction enzyme BsrGI (New England Biolabs, catalog number: R3575S )
- Restriction enzyme SpeI (New England Biolabs, catalog number: R3133L )
- Chemicals and other consumables
- 0.22 μm filter membranes (Merck, catalog number: GSWP02500 )
- Multiwell microtiter plates, 96-well, Flat Bottom (Falcon, catalog number: 353072 )
- 50 ml plastic tubes (Falcon)
- 2-ml microcentrifuge tubes
- 1.5-ml microcentrifuge tubes
- Aluminum foil
- Petri dishes
- Chloramphenicol (Sigma-Aldrich, catalog number: C0378-25G )
- Kanamycin sulfate (Roche, catalog number: 10106801001 )
- Absolute ethanol (Sigma-Aldrich, catalog number: 1009832500 )
- 85% (v/v) glycerol (Sigma-Aldrich, catalog number: 1040942500 )
- NaCl (Merck, catalog number: 1.06404.1000 )
- NaOH (Merck, catalog number: 1.06498.0500 )
- CaCl2 (Merck, catalog number: 1.02382.1000 )
- RbCl (Sigma-Aldrich, catalog number: R2252-100G )
- MnCl2 (Merck, catalog number: 1.05927.0100 )
- Potassium acetate (Merck, catalog number: 1.04820.1000 )
- Acetic acid (glacial) (Merck, catalog number: 1.00063.1000 )
- MOPS (Sigma-Aldrich, catalog number: M1254-250G )
- 3-methylbenzoate (Sigma-Aldrich, catalog number: 89890 )
- Agar (Conda, catalog number: 1806 )
- Agarose D1 (Conda, catalog number: 8019.22 )
- BactoTM tryptone (BD, catalog number: 211705 )
- BactoTM yeast (BD, catalog number: 212750 )
- Competent E. coli DH10B cells (see Recipes)
- Solution for DH10B competent cells TBFI (see Recipes)
- Solution for DH10B competent cells TBFII (see Recipes)
- 30 mg/ml chloramphenicol and 50 mg/ml kanamycin (see Recipes)
- LB broth (see Recipes)
- LB agar with 30 μg/ml chloramphenicol and/or 50 μg/ml kanamycin (see Recipes)
- 0.5 M 3-methylbenzoate (see Recipes)
- 50% (v/v) and 70% (v/v) ethanol (see Recipes)
Equipment
- ABI Prism 377 automated DNA sequencer (Applied Biosystems Inc.)
- Electroporator (Gene Pulser, Bio-Rad)
- Orbital shaker
- Flasks (50 ml, 1 L)
- Pipettes (Gilson, models: PIPETMAN P2, P10, P20, P100, P200, P1000, P5000)
- Centrifuge (Tomy, model: MX-301 )
- Thermal Cycler (Applied Biosystems, model: 2720Thermal Cycler )
- Temperature chamber (Taitec, model: Thermo minder SM-10R )
- Water bath shaker (Taitec, model: Personal-11 )
- Plate reader (Corona, model: MTP-880Lab )
- Autoclave (Tomy Seiko, model: LSX-500 )
Software
- Microsoft Excel (Microsoft)
- BioEdit Sequence Alignment Editor
- ApE-A plasmid Editor (https://jorgensen.biology.utah.edu/wayned/ape)
Procedure
The design and construction of a target gene tagged according to the FENIX system could be divided into the following 4 steps (A to D), summarized in Figure 1.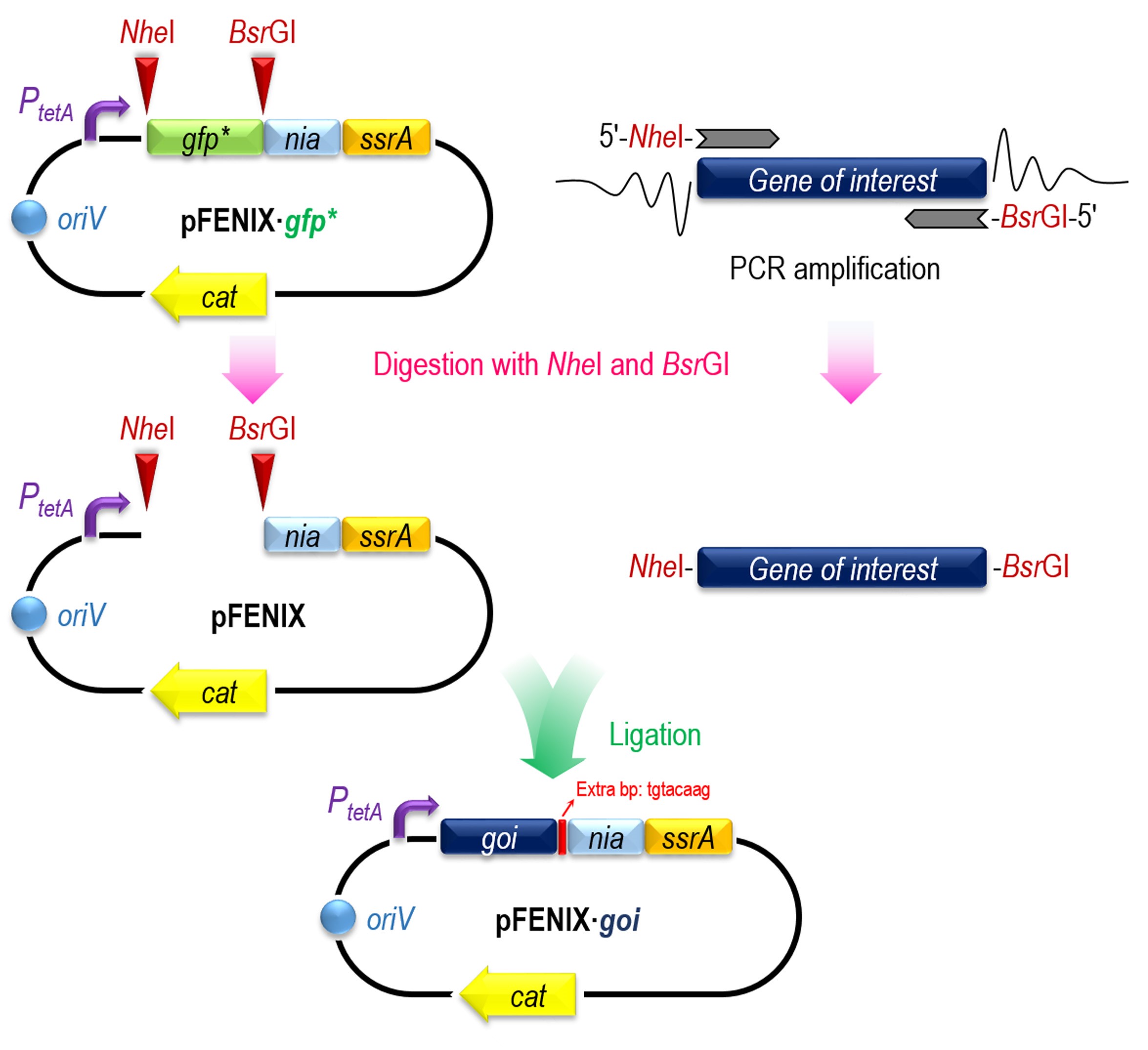
Figure 1. Rationale and construction of the FENIX system for one-step cloning and tagging of individual target proteins. The gene encoding the target polypeptide (gene of interest, goi) is amplified by PCR with specific oligonucleotides that include NheI and BsrGI restriction sites. The resulting amplicon can be directly cloned into plasmid pFENIX·gfp* (which contains a nia/ssrA tagged version of the green fluorescent protein, see Table 1 and Supplementary file) upon digestion with these two restriction enzymes. The NheI restriction site is located between the RBS and the ATG of gfp gene so the N-terminal region of goi will keep without any modification, and the C-terminal region will contain eight extra bp (in red) between goi and nia sequences, translated as XYK amino acids (X could be L, M or V amino acid depending on the last bp of the goi). After ligation, the new plasmid pFENIX·goi is transformed into competent E. coli DH10B cells. In all pFENIX plasmids, the expression of the nia/ssrA-tagged variant of the goi depends on the constitutive PtetA promoter.
Table 1. Bacterial strains and plasmids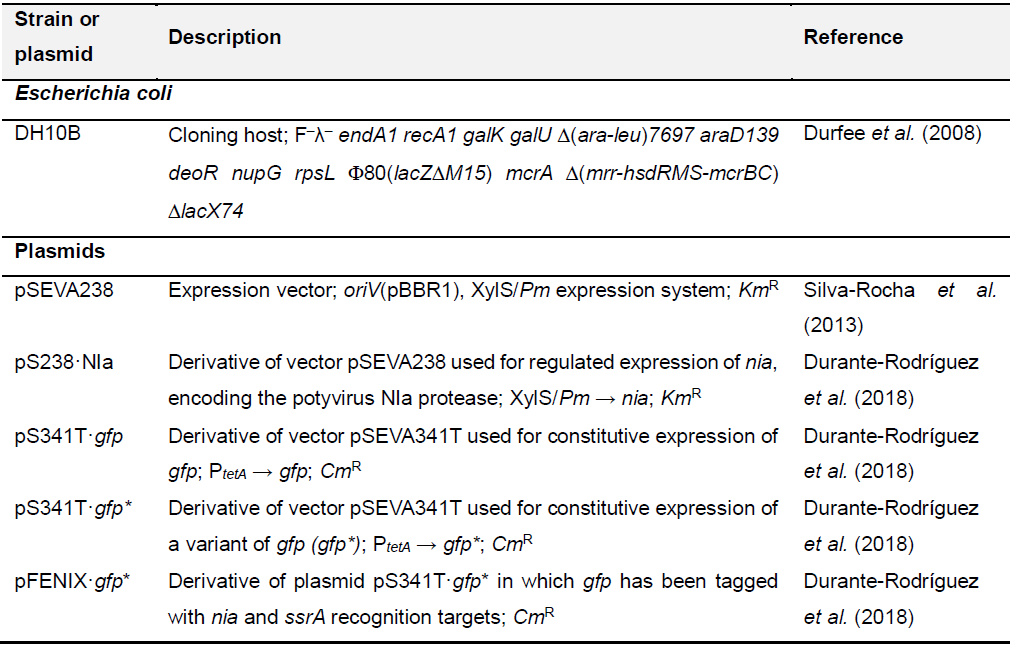
- Construction of pFENIX plasmids for one-step cloning and tagging of individual target proteins
- Select the gene encoding the target polypeptide (gene of interest, goi).
- Design specific oligonucleotides to amplify the goi, including NheI (5'-GCTAGC-3') and BsrGI (5'-TGTACA-3') restriction sites in the 5' and 3' termini, respectively.
- Set up a 100-μl PCR reaction using proof-reading Pfu DNA polymerase (Promega). Standard reaction conditions can be used as described in the provider manual’s for Pfu DNA polymerase. In most cases, thermal cycling conditions are as follows: after heating at 98 °C for 5 min, set 30 cycles of 98 °C for 30 s (denaturation), 57 °C for 30 s (annealing, temperature varies depending on the actual Tm of the oligonucleotides used), and 72 °C for 1 min (elongation, time varies depending on the size of the goi to be amplified, typically 2 min/kb).
- Purify the PCR product by using the Gene-Clean Turbo kit (Q-BIOgene).
Note: When the PCR amplification yields non-specific bands, the target product is cut from an agarose gel after electrophoresis by loading the total volume of PCR and the amplicon is gel-purified by means of the Gene-Clean Turbo kit. - Digest plasmid pFENIX and the PCR product of the goi with the restriction enzymes NheI and BsrGI, and then purify both DNA fragments using the Gene-Clean Turbo kit.
- Clone the goi in the pFENIX backbone by the T4 DNA ligase (New England Biolabs), adding a goi concentration 5-10 times higher than the plasmid pFENIX in a final volume of 20 μl.
- Cell transformation with the pFENIX-goi plasmid
- Keep competent E. coli DH10B cells on ice. This protocol has been carried out with strain DH10B taking into account its high competence skills, but any E. coli strain routinely used for cloning (e.g., DH5α) could be likewise used.
- Take the 20-μl ligation mixture from the step before and add it into 0.1 ml of the cell suspension of competent E. coli DH10B (see Recipes), and then incubate the mixture on ice for 30 min.
- Heat-shock the cells at 42 °C for 45 s, and immediately add 0.9 ml of LB broth to the mixture.
- Incubate 1 ml of the suspension of E. coli DH10B transformants at 37 °C for 60 min.
- Spread 0.1 ml and/or the rest of the suspension of E. coli DH10B transformants onto LB agar plates with 30 μg/ml chloramphenicol, and then incubate the plates at 37 °C overnight.
- Isolate a transformant of E. coli DH10B harboring the cloned candidates on the LB agar with 30 μg/ml chloramphenicol.
- Inoculate a single colony into 10 ml of LB broth containing 30 μg/ml chloramphenicol in a 50-ml conical tube, and incubate the culture in an orbital shaker at 37 °C with shaking (180 rpm) overnight.
- Isolate the plasmid from the transformant cells of 10 ml overnight culture by means of the High-Pure plasmid isolation kit (Roche).
- Check the DNA sequence of the tagged goi cloned into the pFENIX plasmid with an ABI Prism 377 automated DNA sequencer (Applied Biosystems Inc.), using the universal primers R24 (5'-AGCGGATAACAATTTCACACAGGA-3') and F24 (5'-CGCCAGGGTTTTCCCAGTCACGAC-3').
- Compare the sequence of the clones with that of the reference sequence to ensure the absence of mutations. An alignment software such as Bioedit or ApE-A could be used.
- Long-term storage of bacterial strains transformed with these plasmids is routinely done as 15% glycerol stocks according to well-established procedures (Sambrook and Russell, 2001).
- Electroporation of pFENIX recombinants with the plasmid encoding the NIa protease
- Incubate a 20-ml culture of E. coli DH10B harboring plasmid pFENIX·goi in LB medium overnight at 37 °C with shaking (180 rpm) in an orbital shaker.
- Distribute the whole culture in a 50-ml sterile conical tube and centrifuge at 2,300 x g and 4°C for 10 min.
- Remove supernatant, resuspend the pellet gently in 10-ml of cold sterile water (4 °C) and centrifuge at 2,300 x g and 4 °C for 10 min.
- Discard supernatant, add 1 ml of sterile cold water, resuspend the pellet and transfer the suspension to a 2-ml microcentrifuge tube.
- Centrifuge at 3,800 x g and 4 °C for 5 min. Repeat this step 2 times.
Note: The cells are ready to be electroporated when they form a very soft and loose pellet after the centrifugation with cold water. - Dispose of the supernatant and add 300 μl of cold sterile water, resuspend and distribute 100 μl of the resulting suspension into 2-mm gap width electroporation cuvettes.
- Add 200 ng of the plasmid pS238·NIa to the electroporation cuvette containing 100 μl of the suspension of E. coli DH10B cells harboring the plasmid pFENIX·goi, mix gently and proceed to electroporate in an electroporator (Gene Pulser, Bio-Rad).
Note: The parameters of electroporation are resistance, 200 Ω; capacitance, 25 µF; and voltage, 2.5 kV. - Add 0.9 ml of LB medium to the cell suspension and incubate for 1 h at 37 °C with shaking (180 rpm) in an orbital shaker.
- Plate the whole culture onto LB medium plates added with 30 µg/ml chloramphenicol and 50 μg/ml kanamycin.
- Streak a few colonies and check for the presence of both plasmid pFENIX·goi and pS238·NIa in the transformant bacteria by plasmid extraction (High-Pure plasmid isolation kit, Roche).
- Digest the mixture of both plasmids extracted with SpeI (5'-ACTAGT-3') and run an agarose gel electrophoresis to check the relevant sizes. Expect a band of approximately 3.6 kb plus the size of the corresponding goi with pFENIX·goi, and a band of approximately 5.8 kb of the empty pS238·NIa vector.
- Protocol for activation of the protease NIa in the FENIX native system with GFP* (Figure 2)
- Select a single colony of the E. coli transformants harbouring the plasmids listed below (Table 1):
pS238·NIa (negative control)
pS341T·gfp (positive control)
pSEVA238 and pFENIX·gfp* (control strain without the protease NIa)
pS238·NIa and pFENIX·gfp* - Individually inoculate the four recombinant strains in 50-ml conical tubes with 10 ml of LB medium containing 30 μg/ml chloramphenicol (for the positive control), 50 μg/ml kanamycin (for the negative control), or both antibiotics (for strains containing both plasmids).
- Incubate the pre-culture overnight at 37 °C with shaking (180 rpm).
- Inoculate 4 μl of each overnight pre-culture in a 96-well microtiter plate containing 200 μl of fresh LB medium per well, including 30 μg/ml chloramphenicol and/or 50 μg/ml kanamycin, and in presence or absence of 1 mM 3-methylbenzoate as the inducer of nia expression (in the case of strains harbouring the pFENIX·gfp* plasmid). Each experiment should be carried out in three independent wells as technical replicates.
- Incubate the 96-well microtiter plate at 37 °C with shaking in a plate reader during 24 h, till the stationary phase.
- Measure the OD600 and the GFP fluorescence in the plate reader at the endpoint of the growth curve (Figure 2). GFP is excited at 488 nm, the fluorescence signal is recovered at 511 nm, and it was used the bottom reading mode.
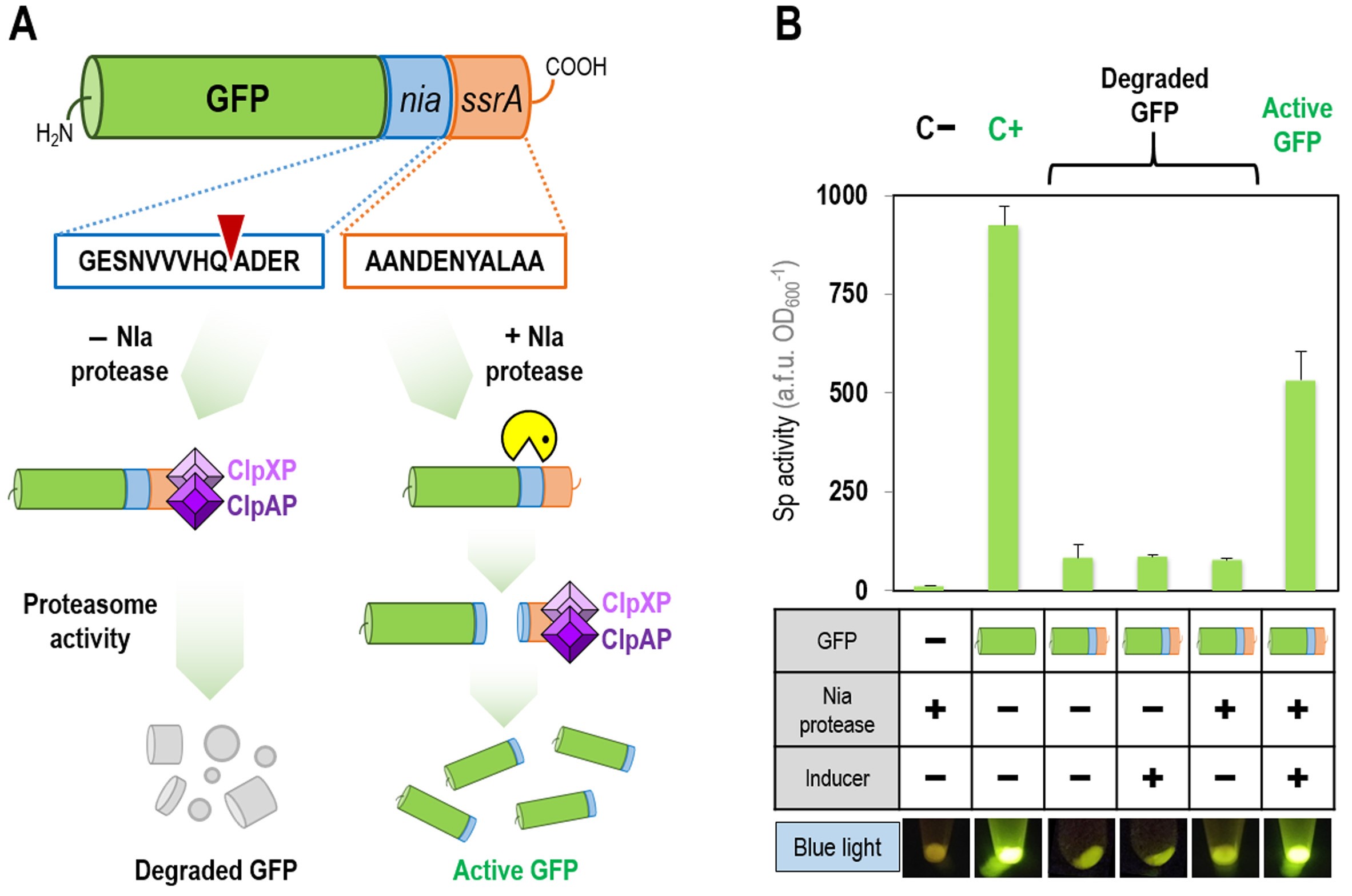
Figure 2. Performance of the FENIX system with GFP fluorescence as the readout. A. Scheme of NIa and SsrA-dependent post-translational control of GFP with the FENIX system. The gene encoding the green fluorescence protein (GFP) is added with a synthetic, hybrid nia/ssrA tag, resulting in a tagged GFP variant in which the C-terminus displays the GESNVVVHQADER·AANDENYALAA amino acid sequence. The SsrA tag is directly recognized by the ClpXP and ClpAP proteases of the bacterial proteasome in vivo, thus degrading the protein. Upon action of the specific potyvirus NIa protease (the recognition site in the synthetic nia/ssrA tag is indicated with an inverted red triangle in the diagram), the SsrA tag is released and GFP can be accumulated, resulting in the appearance of GFP+ cells. B. Evaluation of the FENIX system in recombinant E. coli using GFP protein. Plasmid pFENIX·gfp*, which contain the nia/ssrA-tagged version of GFP (indicated with blue and orange strips in the first row of the table), were transformed into E. coli DH10B carrying either plasmid pS238·NIa or the empty pSEVA238 vector (indicated as + and −, respectively, in the second row of the table). The first two columns represent a negative and positive control, respectively, carried out with E. coli DH10B transformed either with the empty pFENIX vector (i.e., no GFP protein) or with a plasmid constitutively expressing the gene encoding gfp* (pS341T·gfp*, Table 1). Strains indicated in the next four columns contain the nia/ssrA-tagged GFP. These experiments were done in 96-well microtiter plates containing LB medium with the necessary antibiotics and additives (1 mM 3-methylbenzoate as the inducer of nia expression, as indicated in the third row of the table), and were inoculated with a culture of the corresponding strain previously grown in LB medium with the necessary antibiotics overnight. Cells were incubated at 37 °C with rotary agitation, and fluorescence and bacterial growth (expressed as the optical density measured at 600 nm, OD600) were recorded after 24 h. The specific (Sp) activity of the fluorescent proteins under study was calculated as the arbitrary fluorescence units (a.f.u.) normalized to the OD600. Each bar represents the mean value of the Sp activity ± standard deviation calculated from at least three independent experiments. The lower panel shows bacterial pellets harvested from shake-flask cultures after 24 h of incubation under the same growth conditions indicated for the microtiter-plate cultures as observed under blue light.
- Select a single colony of the E. coli transformants harbouring the plasmids listed below (Table 1):
Data analysis
- Extract raw numeric data of both OD600 values and the measurement of GFP fluorescence in absence or presence of the inducer as a text file.
- Open a text file containing the raw numeric data in a software for data management such as Microsoft Excel, R or Sigma Plot.
- Normalize the values of OD600 and the net intensities of the GFP fluorescence.
- Calculate the ratio of fluorescence to OD600 as specific activity of the promoter of each culture by the following formula:Sp = IF/OD600
Sp is the specific activity
IF is the intensity of fluorescence in arbitrary units
OD600 is the cell concentration in units of optical density - Average the ratios from three independent experiments with the technical replicates that are used to calculate the mean specific activity along with the deviations typical corresponding.
- To confirm a significant difference, calculated P-values by Student’s t-test (one-tailed and unpaired) of statistical analysis should be evaluated at less than 0.01 (Durante-Rodríguez et al., 2018).
Recipes
- Competent E. coli DH10B cells
- Inoculate a single colony of E. coli DH10B in 10 ml of LB broth in a 50-ml Erlenmeyer flask
- Incubate the pre-cultures overnight at 37 °C with shaking (180 rpm)
- Dilute the pre-culture 100-fold in 250 ml of fresh LB broth in a 1-L Erlenmeyer flask
- Incubate the cultures at 37 °C with shaking (180 rpm) until mid-exponential phase (optical density measured at 600 nm ~0.5-0.6)
- Keep the cell suspension in an ice batch for 30 min
- Distribute the 250 ml of culture to sterile 50-ml conical tubes
- Collect the cells by centrifugation (2,300 x g, 4 °C, 5 min) and combine the pellets in a single tube
- Suspend the cells in 30 ml of TBFI (see Recipe 2) per 100 ml of original cell suspension
- Incubate the cells in an ice bath for 2 h
- Collect the cells by centrifugation (2,300 x g, 4 °C, 5 min)
- Suspend the cells in 4 ml of TBFII (see Recipe 3) per 100 ml of cells
- Transfer the suspension to pre-chilled 1.5-ml microcentrifuge tubes in aliquots of 100 μl
- Use an aliquot (~100 μl) of the resuspended E. coli DH10B for transformation
- Inoculate a single colony of E. coli DH10B in 10 ml of LB broth in a 50-ml Erlenmeyer flask
- Solution for DH10B competent cells TBFI
- To prepare 250 ml of final volume of TBFI, dissolve 0.74 g of potassium acetate, 0.37 g of CaCl2 and 43.1 ml of 85% (v/v) glycerol in distilled water
- Adjust the pH to 6.0 by dropwise addition of 0.2 M acetic acid
- Add 2.47 g of MnCl2 and 3.02 g of RbCl, checking that the pH value stay at 5.8 (if not, re-adjust by dropwise addition of 0.2 M acetic acid)
- Sterilize the solution by filtration with a 0.22-μm filter membrane (Merck)
- Store the sterilized solution at 4 °C wrapped in aluminum foil
- To prepare 250 ml of final volume of TBFI, dissolve 0.74 g of potassium acetate, 0.37 g of CaCl2 and 43.1 ml of 85% (v/v) glycerol in distilled water
- Solution for DH10B competent cells TBFII
- To prepare 250 ml of final volume of TBFII, dissolve 2.75 g of CaCl2, 0.3 g of RbCl, 0.52 g of MOPS and 43.1 ml of 85% (v/v) glycerol in distilled water
- Adjust the pH to 6.8 by dropwise addition of 10 N NaOH
- Sterilize the solution by filtration with 0.22-μm filter membrane (Merck)
- Store the sterilized solution at 4 °C wrapped in aluminum foil
- To prepare 250 ml of final volume of TBFII, dissolve 2.75 g of CaCl2, 0.3 g of RbCl, 0.52 g of MOPS and 43.1 ml of 85% (v/v) glycerol in distilled water
- 30 mg/ml chloramphenicol and 50 mg/ml kanamycin
- Dissolve 0.3 g of chloramphenicol (Sigma) and 0.5 g of kanamycin sulfate (Sigma) in 10 ml of 70% (v/v) ethanol or distilled water, respectively
- Sterilize the solutions by filtration with 0.22-μm filter membrane (Merck)
- Store the sterilized solutions at -20 °C
- Dissolve 0.3 g of chloramphenicol (Sigma) and 0.5 g of kanamycin sulfate (Sigma) in 10 ml of 70% (v/v) ethanol or distilled water, respectively
- LB broth
- Dissolve 10 g of BactoTM tryptone, 5 g of BactoTM yeast extract, and 10 g of NaCl in 800 ml of distilled water
- Adjust pH to 7.5 with 10 N NaOH
- Adjust volume to 1 L with distilled water
- Autoclave the solution (set at 121 °C and 20 min in an LSX-500 autoclave)
- Store the sterile LB broth at room temperature
- Dissolve 10 g of BactoTM tryptone, 5 g of BactoTM yeast extract, and 10 g of NaCl in 800 ml of distilled water
- LB agar with 30 μg/ml chloramphenicol and/or 50 μg/ml kanamycin
- Dissolve 7.5 g of agar in 1 L of LB broth
- Autoclave (set 121 °C and 20 min in LSX-500 )
- Add chloramphenicol and/or kanamycin after cooling in the final concentration of 30 μg/ml and 50 μg/ml, respectively
- Pour the media into Petri dishes, and then let the LB agar solidify by cooling at room temperature
- Store the LB agar plates upside down at 4 °C
- Dissolve 7.5 g of agar in 1 L of LB broth
- 0.5 M 3-methylbenzoate
- Dissolve 6.75 g 3-methylbenzoate (Sigma) in 100 ml of 50% (v/v) ethanol
- Sterilize the solution by filtration with 0.22-μm filter membrane (Merck)
- Store the solution at 4 °C
- Dissolve 6.75 g 3-methylbenzoate (Sigma) in 100 ml of 50% (v/v) ethanol
- 50% (v/v) and 70% (v/v) ethanol
- Dilute ethanol to 50% (v/v) or 70% (v/v) with sterile water
- Store at room temperature
- Dilute ethanol to 50% (v/v) or 70% (v/v) with sterile water
Acknowledgments
Financial support from The Novo Nordisk Foundation (NNF10CC1016517 and NNF 18CC0033664), the Danish Council for Independent Research (SWEET, DFF-Research Project 8021-00039B), and the European Union’s Horizon2020 Research and Innovation Program under grant agreement No. 814418 (SinFonia) to P.I.N. is gratefully recognized. This work was funded also by the MADONNA (H2020-FET-OPEN-RIA-2017-1-766975), BioRoboost (H2020-NMBP-BIO-CSA-2018), SYNBIO4FLAV (H2020-NMBP/0500) and MIX-UP (H2020-Grant 870294) Contracts of the European Union and the S2017/BMD-3691 InGEMICS-CM Project of the Comunidad de Madrid (European Structural and Investment Funds) to VdL. This protocol was adapted from Durante-Rodríguez et al. (2018) for the method to design and construct a post-translational metabolic switch driven to decoupling the bacterial growth from biopolymer production in E. coli.
Competing interests
The authors declare no conflicts of interest.
References
- Avcilar-Kucukgoze, I., and Ignatova, Z. (2017). Rewiring host activities for synthetic circuit production: a translation view. Biotechnol Lett 39(1): 25-31.
- Calles, B., Goñi-Moreno, A. and de Lorenzo, V. (2019). Digitalizing heterologous gene expression in Gram-negative bacteria with a portable ON/OFF module. Mol Syst Biol 15(12): e8777.
- Doma, M. K. and Parker, R. (2007). RNA quality control in eukaryotes. Cell 131(4): 660-668.
- Durante-Rodríguez, G., de Lorenzo, V. and Nikel, P. I. (2018). A post-translational metabolic switch enables complete decoupling of bacterial growth from biopolymer production in engineered Escherichia coli. ACS Synth Biol 7(11): 2686-2697.
- Durfee, T., Nelson, R., Baldwin, S., Plunkett, G., 3rd, Burland, V., Mau, B., Petrosino, J. F., Qin, X., Muzny, D. M., Ayele, M., Gibbs, R. A., Csörgő, B., Pósfai, G., Weinstock, G. M. and Blattner, F. R. (2008). The complete genome sequence of Escherichia coli DH10B: insights into the biology of a laboratory workhorse. J Bacteriol 190(7): 2597-2606.
- Guzmán, L. M., Belin, D., Carson, M. J. and Beckwith, J. (1995). Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177(14): 4121-4130.
- Isaacs, F. J., Dwyer, D. J., Ding, C., Pervouchine, D. D., Cantor, C. R. and Collins, J. J. (2004). Engineered riboregulators enable post-transcriptional control of gene expression. Nat Biotechnol 22(7): 841-847.
- Lou, C., Stanton, B., Chen, Y. J., Munsky, B. and Voigt, C. A. (2012). Ribozyme-based insulator parts buffer synthetic circuits from genetic context. Nat Biotechnol 30(11): 1137-1142.
- Lutz, R. and Bujard, H. (1997). Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res 25(6): 1203-1210.
- Sambrook, J. and Russell, D. W. (2001). Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, USA.
- Shoemaker, C. J., Eyler, D. E. and Green, R. (2010). Dom34:Hbs1 promotes subunit dissociation and peptidyl-tRNA drop-off to initiate no-go decay. Science 330(6002): 369-372.
- Silva-Rocha, R., Martinez-Garcia, E., Calles, B., Chavarría, M., Arce-Rodríguez, A., de Las Heras, A., Páez-Espino, A. D., Durante-Rodríguez, G., Kim, J., Nikel, P. I., Platero, R. and de Lorenzo, V. (2013). Nucleic Acids Research. 41(Database issue): D666-675.
- Stevens, R. C. (2000). Design of high-throughput methods of protein production for structural biology. Structure 8(9): R177-185.
- Verchot, J., Koonin, E. V. and Carrington, J. C. (1991). The 35-kDa protein from the N-terminus of the potyviral polyprotein functions as a third virus-encoded proteinase. Virology 185(2): 527-535.
- Volke, D. C., Turlin, J., Mol, V. and Nikel, P. I. (2020). Physical decoupling of XylS/Pm regulatory elements and conditional proteolysis enable precise control of gene expression in Pseudomonas putida. Microb Biotechnol 13(1): 222-232.
- Wu, G., Yan, Q., Jones, J. A., Tang, Y. J., Fong, S. S., and Koffas, M. A. G. (2016). Metabolic burden: cornerstones in synthetic biology and metabolic engineering applications. Trends Biotechnol 34(8): 652-664.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Durante-Rodríguez, G., Calles, B., Lorenzo, V. D. and Nikel, P. I. (2020). A SsrA/NIa-based Strategy for Post-Translational Regulation of Protein Levels in Gram-negative Bacteria. Bio-protocol 10(14): e3688. DOI: 10.21769/BioProtoc.3688.
Category
Microbiology > Microbial metabolism > Other compound
Microbiology > Microbial biochemistry > Protein
Molecular Biology > Protein > Targeted degradation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.