- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Isolation, Culture, and Differentiation of Primary Myoblasts Derived from Muscle Satellite Cells
Published: Vol 10, Iss 14, Jul 20, 2020 DOI: 10.21769/BioProtoc.3686 Views: 12406
Reviewed by: Chiara AmbrogioEnrico PatruccoShibali Das
Original research article
The authors used this protocol in:

Aug 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
The skeletal muscle is key for body mobility and motor performance, but aging and diseases often lead to progressive loss of muscle mass due to wasting or degeneration of muscle cells. Muscle satellite cells (MuSCs) represent a population of tissue stem cells residing in the skeletal muscles and are responsible for homeostatic maintenance and regeneration of skeletal muscles. Growth, injury, and degenerative signals activate MuSCs, which then proliferate (proliferating MuSCs are called myoblasts), differentiate and fuse with existing multinuclear muscle cells (myofibers) to mediate muscle growth and repair. Here, we describe a protocol for isolating MuSCs from skeletal muscles of mice for in vitro analysis. In addition, we provide a detailed protocol on how to culture and differentiate primary myoblasts into myotubes and an immunofluorescent staining procedure to characterize the cells. These methods are essential for modeling regenerative myogenesis in vitro to understand the dynamics, function and molecular regulation of MuSCs.
Background
Muscle homeostasis, maintained by diverse cellular functions, is vital for preserving muscle integrity. Tissue specific adult stem cells are capable of regenerating local tissues continuously throughout life. In adult skeletal muscles, a population of stem cells called muscle satellite cells (MuSCs) possess a robust regenerative capacity that is key for muscle homeostasis (Yin et al., 2013; Dumont et al., 2016). Quiescent MuSCs reside in a niche beneath the basal lamina juxtaposed to the muscle fiber and are responsible for growth and regeneration of muscle (Yin et al., 2013; Dumont et al., 2016).
MuSCs are a heterogeneous population with diverse cellular and molecular profiles. Quiescent MuSCs, identified by the expression of Pax7, are activated and enter a proliferating stage in response to external stimuli such as growth factors, injury, or pathological conditions (Kuang et al., 2008). The proliferating myoblasts undergo several rounds of cell division and terminally differentiate into mononuclear myocytes, which fuse into myotubes to mediate muscle repair (Rudnicki et al., 2008). A dynamic balance between asymmetric and symmetric division is also important for determining the fate of myogenic cells in terms of self-renewal and expansion (Kuang et al., 2007).
Primary myoblasts (referring to MuSCs that are isolated from muscle) and the derived myotubes are important for modeling MuSC function and muscle physiology in vitro. Several methods for the isolation of primary myoblasts have been reported to investigate their cell biology (i.e., proliferation, self-renewal and differentiation) and molecular regulation. One method involves enzymatic digestion of muscle tissues to liberate single myofibers that carry MuSCs to model in vivo behavior of MuSCs while they are still in their native niche (Shefer et al., 2005). Another method involves fluorescence-activated cell sorting (FACS), an efficient and widely used technique for selecting rare cell populations from various tissues. FACS-mediated purification of MuSCs from other muscle resident cells such as fibroblasts and immune cells involves the use of positive selection markers (e.g., CD34, Integrin α7 and Vcam1) and negative selection markers (e.g., CD45, CD31, CD11b, and Sca1) (Liu et al., 2015). FACS requires a flow cytometer machine that is not available to many labs, and a prolonged sorting process that often reduces the viability of cells. Alternatively, MuSCs and primary myoblasts can be enriched using other more affordable methods, such as magnetic activated cell sorting (MACS) (Motohashi et al., 2014). However, MACS also requires antibodies and beads to eliminate non-muscle cells, and the number of cells that can be isolated is limited by the column and the speed of MACS isolation is slow as the mixture of cells flow through MS column by gravity. Finally, primary myoblasts can be enriched simply through a procedure of serial pre-plating based on differential affinity of different cell types to culture substrate.
Here, we describe a protocol for convenient and efficient purification of primary myoblasts from hind limb muscles of mice (Yue et al., 2017; Jia et al., 2019). This method consists of mechanical mincing followed by enzymatic digestion to release mononuclear cells, and selection of MuSCs by FACS. The process of primary myoblast isolation includes muscle dissection and digestion by collagenase type II and dispase, followed by myoblast purification through multiple rounds of pre-plating. Pre-plating is essential to purify a population of MuSCs on the basis of adhesion characteristics. We will also describe how to culture, differentiate, and validate pre-differentiation myoblasts and differentiated multinucleated myotubes using immunofluorescent staining for the expression of myogenic regulatory markers.
Materials and Reagents
- Dissection boards (Styrofoam board)
- 6-well tissue culture (TC) plates (Falcon, catalog number: 353046 )
- Mounting medium (Diagnostic BioSystems, catalog number: K024 )
- 11 mm x 11 mm cover slides (IMEB, catalog number: CG1-2450 )
- 60 mm TC plates (Falcon, catalog number: 353002 )
- 100 mm TC plates (Falcon, catalog number: 353003 )
- 1.5 ml Eppendorf tubes (DOT Scientific, catalog number: RN1700-GMT )
- 15 ml polystyrene centrifuge tubes (Falcon, catalog number: 352095 )
- 50 ml high Clarity PP centrifuge tubes (Falcon, catalog number: 352098 )
- 70 µm cell strainers (Falcon, catalog number: 352350 )
- 10 ml serological pipettes (Falcon, catalog number: 357551 )
- Mouse aged 4-6 weeks
- Phosphate-buffered saline (PBS, pH 7.4) (Sigma-Aldrich, catalog number: P3813 )
- Tween-20 (VWR, catalog number: 97062332 )
- Dispase II (Roche Applied Science, catalog number: 0 4942078001 )
- Collagenase type II (Worthington, catalog number: LS004177 )
- 0.25% Trypsin (Gibco, catalog number: 25200056 )
- Fetal bovine serum (FBS) (Hyclone, catalog number: SH3007103 )
- Horse serum (HS) (Hyclone, catalog number: SH3007403 )
- Dulbecco’s modified Eagle’s medium (DMEM) (Sigma-Aldrich, D5796 )
- Ham's F-10 Nutrient Mixture (Gibco, catalog number: 11550043 )
- Collagen type I (Sigma-Aldrich, catalog number: C7661 )
- Matrigel (Corning, catalog number: 354234 )
- Paraformaldehyde powder (Polysciences, catalog number: 00 380 )
- 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Sigma-Aldrich, catalog number: D8417 )
- Penicillin-streptomycin solution (Hyclone, catalog number: SV30010 )
- Glycine (Sigma-Aldrich, catalog number: G8898 )
- Ethanol (Decon Labs, catalog number: V1001 )
- Goat serum (Jackson ImmunoResearch, catalog number: 005-000-121 )
- Bovine serum albumin (BSA) (Gemini Bio-Products, catalog number: 700-105P )
- Triton X-100 (Sigma-Aldrich, catalog number: X100 )
- Sodium azide (Fisher Scientific, catalog number: S2271 )
- Basic fibroblast growth factor (bFGF) (Promega, catalog number: G5071 )
- Primary antibody:
Pax7 (Developmental Studies Hybridoma Bank)
MyoD (Santa Cruz Biotechnology, catalog number: sc-760 )
MyoG (F5D) (Developmental Studies Hybridoma Bank)
MyHC (MF20) (Developmental Studies Hybridoma Bank) - Secondary antibody:
Goat anti-Mouse IgG1, Alexa Fluor 568 (Thermo Fisher Scientific, Invitrogen, catalog number: A-21124 )
Goat anti-Mouse IgG2b, Alexa Fluor 488 (Thermo Fisher Scientific, Invitrogen, catalog number: A-21141 )
Goat anti-Rabbit IgG, Alexa Fluor 488 (Thermo Fisher Scientific, Invitrogen, catalog number: A-11034 ) - 70% ethanol (see Recipes)
- Collagen type I coated TC plates (see Recipes)
- Matrigel coated TC plates (see Recipes)
- Digestion Medium (see Recipes)
- Neutralization medium (see Recipes)
- Growth medium (see Recipes)
- Differentiation medium (see Recipes)
- 0.05% Trypsin (see Recipes)
- 4% Paraformaldehyde (PFA) (see Recipes)
- 100 mM glycine solution (see Recipes)
- Blocking buffer (see Recipes)
- bFGF stock (see Recipes)
- PBS-T (see Recipes)
Equipment
- Dissecting scissors, 10-cm, straight (WPI, catalog number: 14393-G )
- Tweezers, 4.75”, straight (Excelta, catalog number: 3-S-SE )
- T-pins (Business source, catalog number: 32351 )
- Swing-bucket centrifuge (Thermo Scientific, model: CL2 )
- Autoclave (Beta Star life sciences, model: small sterilizer series 26” x 26” model )
- Class II, type A2 biosafety cabinet (Labconco, catalog number: 3440009 )
- Thermostat water bath (Thermo Fisher Scientific, catalog number: TSGP02 )
- Cell incubator (Thermo Fisher Scientific, model: Heracell 240 )
- 14.0 MP digital USB microscope camera (OMAX Microscope, catalog number: A35140U3 )
- Leica DMI 6000B fluorescent microscope (Leica Microsystems, model: DMI 6000 B )
- Lumen 200 fluorescence illumination system (Prior Scientific, model: Lumen 200 )
- Coolsnap HQ CCD camera (Photometrics, model: Coolsnap HQ )
- Ultra pipet controller (Corning, catalog number: 4099 )
- 10 μl, 200 μl, 1,000 μl pipettes (Gilson, model: Pipetman )
Software
- Photoshop software (e.g., Photoshop CC)
Procedure
- Preparation prior to isolation
- 70% ethanol (see Recipe 1).
- Autoclave 1.5 ml Eppendorf tubes.
- Sterilize tools with 70% ethanol.
- Collagen type I to coat cell culture plate (see Recipe 2).
- Matrigel to coat cell culture plate (see Recipe 3).
- Plate coating
- Coat 100 mm plates with 5 ml of collagen type I to cover the plate completely for 30 min at room temperature. After 30 min, transfer collagen type I to the original tube, and air-dry the plate. The plate can be stored in A2 biosafety cabinet until ready to use. This plate will be used for pre-plating.
Note: Collagen type I can be reused for future use. - Coat 60 mm plates with 3 ml of Matrigel to cover the plate completely for 20 min at room temperature. After 20 min, transfer Matrigel to the original tube, and air-dry the plate. It is recommended to use the plate right after Matrigel is dried out. This plate will be used for differentiation of myoblasts.
Note: Matrigel can be reused for future use.
- Coat 100 mm plates with 5 ml of collagen type I to cover the plate completely for 30 min at room temperature. After 30 min, transfer collagen type I to the original tube, and air-dry the plate. The plate can be stored in A2 biosafety cabinet until ready to use. This plate will be used for pre-plating.
- Digestion medium (see Recipe 4).
- Neutralization medium (see Recipe 5).
- Growth medium (see Recipe 6).
- Differentiation medium (see Recipe 7).
- 0.05% trypsin (see Recipe 8).
- 4% paraformaldehyde (PFA) fixation solution (see Recipe 9).
- 100 mM glycine solution (see Recipe 10).
- Blocking buffer (see Recipe 11).
- bFGF (see Recipe 12).
- PBS-T (see Recipe 13).
- Muscle dissection
- Sacrifice a mouse aged 4-6 weeks following the approved protocol in your laboratory. In this case, cervical dislocation is used.
Note: Dissection of a mouse does not need to be performed inside an A2 biosafety cabinet. - Spray the mouse with 70% ethanol and pin the mouse face up on a dissection board.
- Cut the skin at the ankle using sterilized scissors. With the mouse in a transverse abdominal position, use tweezers to expose the quadricep muscle. Collect muscles in cold PBS. Flip the mouse and pin it with its back facing up. Remove the skin on lower body, dissect two entire hind limb skeletal muscles and place them in cold PBS.
- Transfer the dissection tools and the plate containing muscle to a biosafety cabinet. Wipe all dissection tools with 70% ethanol to avoid potential contamination from outside of the biosafety cabinet.
 Video 1. Muscle dissection of mouse. This video demonstrates a detailed procedure of how to isolate hind limb muscles, as well as how to remove the epimysium and fat around the muscle and the neural bundles within the muscle. After transferring the muscle to a 60 mm plate containing PBS, it was washed several times in PBS, minced with a pair of scissors or a razor blade, and digested by collagenase type II to liberate MuSCs from myofibers.
Video 1. Muscle dissection of mouse. This video demonstrates a detailed procedure of how to isolate hind limb muscles, as well as how to remove the epimysium and fat around the muscle and the neural bundles within the muscle. After transferring the muscle to a 60 mm plate containing PBS, it was washed several times in PBS, minced with a pair of scissors or a razor blade, and digested by collagenase type II to liberate MuSCs from myofibers.
- Sacrifice a mouse aged 4-6 weeks following the approved protocol in your laboratory. In this case, cervical dislocation is used.
- Muscle digestion
- Wash hind limb muscles with PBS until muscles are free of hair.
Note: It is recommended to wash several times until hair is completely removed from muscle tissue. - Blot dry muscles on tissue paper.
- Transfer muscle to 1.5 ml Eppendorf tube.
- Mince the muscle using dissection scissors inside a 1.5 ml Eppendorf tube.
Note: Finely minced muscle will yield more myoblasts. - Transfer the minced muscle into a 15 ml Falcon tube.
- Digest the muscle with digestion medium (5 ml per mouse) in a 37 °C water bath for 12 min, shaking the tube every 2 min.
- Mix the digested muscle with a 10 ml serological pipette until the mixture can be smoothly pipetted.
- Digest for another 12 min in the 37 °C water bath, shaking the tube every 2 min.
- Stop digestion by adding 5 ml neutralization medium.
- Place a 70 µm sterilized cell strainer on top of a 50 ml tube, and pre-wet the strainer with 3 ml neutralization medium.
Note: Pre-wetting is important for cells to pass smoothly through the cell strainer. - Collect the media containing cell mixture using a 10 ml serological pipette and filter through the 70 µm sterilized cell strainer.
Note: Collect cell filtrate underneath cell strainer using 1 ml pipette. - Spin the cell mixture at 2,000 x g for 5 min at room temperature.
- Discard the supernatant using a vacuum pipette.
Note: Do not aspirate the cell pellet. - Resuspend the cell pellet with 5 ml growth medium.
- Seed the cells from one mouse in a non-coated 100 mm culture plate and incubate at 37 °C for 4 days, and supplement 5 ml of growth medium on top of pre-existing medium each day for 3 days.
Note: A heterogeneous population of cell mixture and broken fibers will be visualized under a microscope on Day 4.
- Wash hind limb muscles with PBS until muscles are free of hair.
- Primary myoblast isolation and pre-plating
- On Day 4, small fibers pieces with rounded primary myoblasts can be seen, and cells are now ready for pre-plating.
- Mechanically detach cells attached to the bottom of plate by pipetting using a 10 ml serological pipette and transfer to a 50 ml tube. Floating cells are also transferred to the 50 ml tube.
Notes:- This method only utilizes mechanical detachment, therefore trypsin is not necessary.
- Confirm if most cells were detached from the plate by checking under a microscope.
- Centrifuge the 50 ml tube at 2,000 x g for 5 min.
Note: Let centrifuge stop without applying brake. This usually takes a couple of minutes. - Aspirate the media from the 50 ml tube.
- Add 3 ml trypsin into the 50 ml tube and resuspend cells carefully and incubate in a 37 °C water bath for 5 min.
Note: Cells are still heterogeneous population. - Centrifuge the 50 ml tube at 2,000 x g for 5 min.
- Discard the supernatant without disturbing the pellet and resuspend the cells with 5 ml of growth medium
- Transfer to a non-coated culture plate and incubate at 37 °C incubator for 45 min.
- After 45 min, transfer supernatant from the non-coated culture plate to collagen-coated plate.
Note: Myoblasts do not adhere to the culture plate in a short time, therefore, transferring supernatant media helps to purify myoblast from other cells such as fibroblasts in the bottom of the plate. - Steps D2-D9 (Pre-plating) can be repeated multiple times to obtain ≥ 95% purity of myoblasts.
Note: Pre-plating can be repeated twice to increase myoblast purity. Myoblasts should appear small and rounded, while fibroblasts will appear elongated and at times have bipolar processes.
- Culturing and differentiation
- Culture the purified primary myoblasts in the growth medium, changing the media every 2 days. Maintain the cell density under 80% confluency to prevent the fusion of primary myoblasts.
Note: Collagen type I coated plate is not required after pre-plating step. - Prior to differentiation, coat a 60 mm culture plate with 3 ml of Matrigel for 20 min in the hood at room temperature.
- After removing Matrigel, allow for complete evaporation in the hood for 30 min.
Note: It is not recommended to reuse Matrigel more than 5 times and should be stored at 4 °C or on ice when not in use. - Incubate cells with 1 ml trypsin for 1 min at room temperature to detach primary myoblasts and seed cells onto Matrigel coated plate.
Note: Primary myoblasts are cultured on Matrigel coated plate with 4 ml of growth medium until the desired cell density is reached. - Once the confluency reaches 80-90%, change myoblast medium to differentiation medium.
- Maintain primary myoblasts with differentiation medium for the following three days.
Note: Daily monitoring morphological change of primary myoblasts under a microscope is necessary. Clear morphological change from rounded primary myoblasts to elongated myotubes can be observed on Day 3.
- Culture the purified primary myoblasts in the growth medium, changing the media every 2 days. Maintain the cell density under 80% confluency to prevent the fusion of primary myoblasts.
- Immunostaining of Primary Myoblasts
- Place an autoclaved cover slide in a well of a 6-well plate.
- Seed myoblasts on the autoclaved cover slide
Notes:- Adherent myoblasts should be visible on the cover slide the following day.
- Myoblasts can be cultured several days to the desired confluence.
- Aspirate growth medium from the culture plate.
- Fix adherent cells by adding 1 ml of 4% PFA and incubate for 15 min at room temperature.
- Aspirate 4% PFA from the culture plate.
- Gently wash 3 times with 1 ml of PBS for 5 min each at room temperature.
- Aspirate PBS from the culture plate.
- Add 1 ml of 100 mM glycine and incubate for 10 min at room temperature.
- Remove 100 mM glycine and wash with 1 ml PBS for 5 min at room temperature.
- Repeat Step 9 for a total of 3 washes.
- Add 1 ml of blocking buffer and incubate for 1-2 h at room temperature.
- Add appropriate amount of primary antibody (Pax7-1:10, MyoD-1:300 in blocking buffer) and incubate overnight at 4 °C.
- Gently wash 3 times each with 1ml of PBS-T for 5 min at room temperature.
- Add appropriate amount of secondary antibody (Goat anti-mouse IgG1 for Pax7, MyoG, Goat anti-mouse IgG2b for MyoD, Goat anti-Rabbit IgG for MF20, DAPI-1:1,000 in PBS-T) at room temperature for 1 h.
Note: Secondary antibodies are sensitive to light, so the steps below should be performed in a dark room. - Gently wash 5 times each with 1 ml of PBS-T for 5 min at room temperature.
Note: More washing is recommended to minimize background staining. - Mount the coverslip with a drop of mounting medium and allow to dry.
- Evaluate staining under a fluorescent microscope.
Data analysis
This protocol includes visualization and evaluation of myoblasts in vitro before and after differentiation, after immunostaining with specific myogenic markers. Primary myoblasts cultured in growth medium were fixed by the addition of 4% PFA and were stained with Pax7 and MyoD, markers for undifferentiated myoblasts (Figure 1). After primary myoblasts were seeded on a Matrigel-coated plate, growth medium was changed to differentiation medium once the confluency of myoblasts reached 80%. After 3 days of differentiation, myoblasts were fixed and stained with MyoG and MF20, markers of early and late stages of myogenic differentiation, respectively (Figure 2). For research purposes, myoblasts can be stained with other antibodies, but it is recommended to co-stain with one of the myogenic markers to confirm the differentiation status of the myoblasts.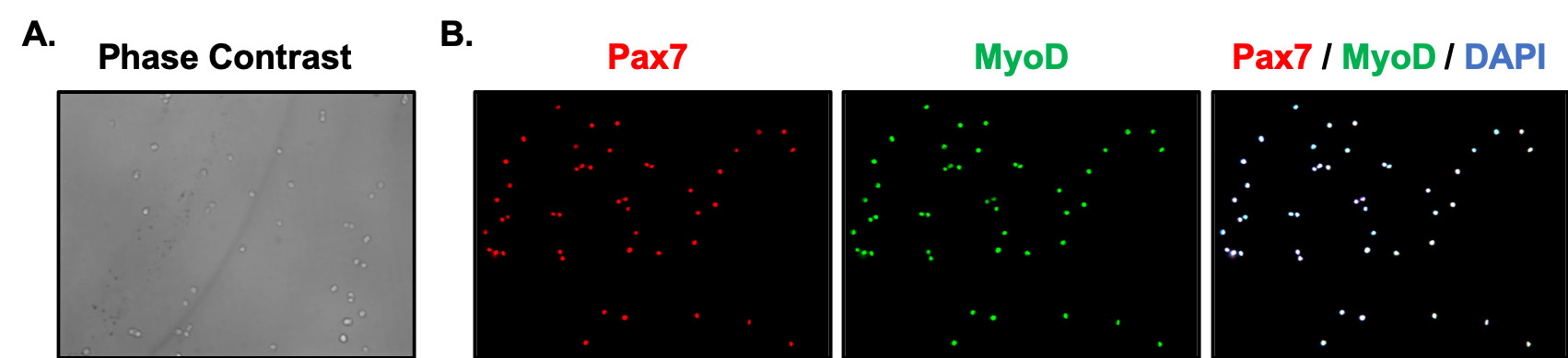
Figure 1. Phase contrast imaging and immunofluorescence staining of undifferentiated primary myoblasts. Representative phase contrast image (20x) demonstrates the morphology of undifferentiated primary myoblasts (A). Primary myoblasts were fixed for immunostaining after one day of cell culture. Representative immunofluorescence imaging using Pax7 (red) and MyoD (green) myogenic markers for undifferentiated primary myoblasts. Nuclei were stained with DAPI (blue) (B).
Figure 2. Phase contrast image and immunofluorescent staining of myotubes after differentiation for 3 days. A. Representative phase contrast image of differentiated myotube morphology (20x). B. Primary myoblasts were cultured on a Matrigel-coated plate for 3 days to form fully differentiated myotubes. For immunostaining, MyoG (red) and MF20 (green) were used as myogenic markers for differentiation to visualize multinucleated myotubes. Nuclei were stained with DAPI (blue).
Notes
- The yield of primary myoblasts isolated from young mice (3-4 weeks old) is higher than that of adult mice (2 months old)
- It is imperative that hair is completely removed during dissection and washing steps as excess hair can be a source of primary culture contamination.
- Finely cutting into small pieces of muscle tissues during the mincing step (Step C4) is helpful to increase myoblast yield.
- Additional rounds of pre-plating can be performed if myoblast purity is less than 95%.
- For staining, incubation of primary antibody at room temperature for 1 h is possible, but overnight incubation at 4 °C is strongly recommended.
Recipes
- 70% ethanol
Add 70 ml of 100% ethanol in 30 ml ddH2O. Store at room temperature - Collagen type I for coating
- Dissolve 0.1% collagen type I in 0.1 M acetic acid and stir the mixture at room temperature for 1 h to make a 10x stock solution
- 10x collagen type I stock solution is diluted to 1x in 0.1 M acetic acid to obtain 0.01% collagen type I
- Store at 4 °C
- Matrigel for coating
Add 1.4 volumes DMEM medium to dilute Matrigel
For 2.4 ml solution, dilute 1 ml Matrigel with 1.4 ml DMEM medium. Store at 4 °C - Digestion Medium
Prepare digestion medium by dissolving dispase II and collagenase type II in PBS at a concentration of 2.4 U/ml and 2.9 U/ml, respectively, and supplement with 2.5 mM CaCl2
For 50 ml digestion medium, dissolve 125 mg dispase, 0.5 g collagenase type II, then add 125 μl 1 M CaCl2. Store at -20 °C - Neutralization medium
F-10 Ham’s medium is supplemented with 20% FBS, and 1% penicillin-streptomycin.
For 50 ml neutralization medium, 39.5 ml F-10 Ham’s medium will be supplemented with 10 ml FBS and 500 μl penicillin-streptomycin. Store at 4 °C - Growth medium
F-10 Ham’s medium is supplemented with 20% FBS, 4 ng/ml basic fibroblast growth factor, and 1% penicillin-streptomycin
For 50 ml growth medium, F-10 Ham’s medium will be supplemented with 10 ml FBS, 50 μl bFGF stock and 500 μl penicillin-streptomycin. Store at 4 °C - Differentiation medium
Dulbecco’s modified Eagle’s medium (DMEM) is supplemented with 2% horse serum, and 1% penicillin-streptomycin.
For 50 ml differentiation medium, DMEM will be supplemented with 1 ml horse serum and 500 μl penicillin-streptomycin. Store at 4 °C - 0.05% Trypsin
For 50 ml 0.05% trypsin, add 10 ml of 0.25% trypsin in 40 ml autoclaved PBS
Store at 4 °C - 4% Paraformaldehyde (PFA)
Dissolve 4% PFA (m/v) in PBS solution, stir the mixture at 60 °C water bath, and raise the pH to 7.4 by adding NaOH to make PFA powder more dissolvable
For 100 ml 4% PFA, take 80 ml PBS solution, add 4 g PFA powder, dissolve the PFA as described above, and adjust the volume to 100 ml with PBS solution. Store at 4 °C - 100 mM glycine solution
Dissolve glycine in PBS at a concentration of 100 mM
For 50 ml glycine solution, dissolve 3.75 g glycine in 50 ml PBS. Store at 4 °C - Blocking buffer
PBS supplemented with 5% goat serum, 2% BSA, 0.2% Triton X-100, 0.1% sodium azide
For 100 ml blocking buffer, 5 ml goat serum, 2 g BSA, 2 ml 10% Triton X-100 and 0.1 g sodium azide is added in PBS. Aliquot and store at -20 °C - bFGF stock
Prepare bFGF stock by dissolving bFGF in serum-free medium at a concentration of 4 μg/ml (1,000x)
For 4 μg/ml bFGF stock, 4 μg bFGF is dissolved in 1 ml of serum-free DMEM. Aliquot and store at -20 °C - PBS-T
Add 0.1% of Tween-20 in PBS
For 1 L PBS-T solution, 1 ml Tween-20 is added in 1 L PBS. Store at room temperature
Acknowledgments
This protocol was adapted from our previous work published in Nature Communications (Yue et al., 2017) and Elife (Jia et al., 2019). This work was supported by grants from the National Institutes of Health of USA (R01AR071649) and the National Institute of Food and Agriculture of the United States Department of Agriculture (NC-1184).
Competing interests
The authors declare no competing interests.
Ethics
All procedures performed in animal study were guided by Purdue University’s Animal Care and Use Committee (PACUC) protocol number 1112000440.
References
- Dumont, N. A. and Rudnicki, M. A. (2016). Targeting muscle stem cell intrinsic defects to treat Duchenne muscular dystrophy. NPJ Regen Med 1.
- Jia, Z., Nie, Y., Yue, F., Kong, Y., Gu, L., Gavin, T. P., Liu, X. and Kuang, S. (2019). A requirement of Polo-like kinase 1 in murine embryonic myogenesis and adult muscle regeneration. Elife 8: e47097.
- Kuang, S., Kuroda, K., Le Grand, F. and Rudnicki, M. A. (2007). Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell 129(5): 999-1010.
- Kuang, S., Gillespie, M. A. and Rudnicki, M. A. (2008). Niche regulation of muscle satellite cell self-renewal and differentiation. Cell Stem Cell 2(1): 22-31.
- Liu, L., Cheung, T. H., Charville, G. W. and Rando, T. A. (2015). Isolation of skeletal muscle stem cells by fluorescence-activated cell sorting. Nat Protoc 10(10): 1612-1624.
- Motohashi, N., Asakura, Y. and Asakura, A. (2014). Isolation, culture, and transplantation of muscle satellite cells. JOVE (86). DOI: 10.3791/50846.
- Rudnicki, M. A., Le Grand, F., McKinnell, I. and Kuang, S. (2008). The molecular regulation of muscle stem cell function. Cold Spring Harb Symp Quant Biol 73: 323-331.
- Shefer, G. and Yablonka-Reuveni, Z. (2005). Isolation and culture of skeletal muscle myofibers as a means to analyze satellite cells. Methods Mol Biol 290: 281-304.
- Yin, H., Price, F. and Rudnicki, M. A. (2013). Satellite cells and the muscle stem cell niche. Physiol Rev 93(1): 23-67.
- Yue, F., Bi, P., Wang, C., Shan, T., Nie, Y., Ratliff, T. L., Gavin, T. P. and Kuang, S. (2017). Pten is necessary for the quiescence and maintenance of adult muscle stem cells. Nat Commun 8: 14328.
Article Information
Copyright
![]() Kim et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Kim et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Kim, K. H., Qiu, J. and Kuang, S. (2020). Isolation, Culture, and Differentiation of Primary Myoblasts Derived from Muscle Satellite Cells. Bio-protocol 10(14): e3686. DOI: 10.21769/BioProtoc.3686.
- Jia, Z., Nie, Y., Yue, F., Kong, Y., Gu, L., Gavin, T. P., Liu, X. and Kuang, S. (2019). A requirement of Polo-like kinase 1 in murine embryonic myogenesis and adult muscle regeneration. Elife 8: e47097.
Category
Developmental Biology > Cell growth and fate > Regeneration
Cell Biology > Cell isolation and culture > Monolayer culture
Cell Biology > Cell isolation and culture > Cell differentiation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.