- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

HoSeIn: A Workflow for Integrating Various Homology Search Results from Metagenomic and Metatranscriptomic Sequence Datasets

Published: Vol 10, Iss 14, Jul 20, 2020 DOI: 10.21769/BioProtoc.3679 Views: 4912
Reviewed by: Prashanth N SuravajhalaPaola Monica TaliaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2013

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Data generated by metagenomic and metatranscriptomic experiments is both enormous and inherently noisy. When using taxonomy-dependent alignment-based methods to classify and label reads, the first step consists in performing homology searches against sequence databases. To obtain the most information from the samples, nucleotide sequences are usually compared to various databases (nucleotide and protein) using local sequence aligners such as BLASTN and BLASTX. Nevertheless, the analysis and integration of these results can be problematic because the outputs from these searches usually show inconsistencies, which can be notorious when working with RNA-seq. Moreover, and to the best of our knowledge, existing tools do not criss-cross and integrate information from the different homology searches, but provide the results of each analysis separately. We developed the HoSeIn workflow to intersect the information from these homology searches, and then determine the taxonomic and functional profile of the sample using this integrated information. The workflow is based on the assumption that the sequences that correspond to a certain taxon are composed of:
1)sequences that were assigned to the same taxon by both homology searches;
2)sequences that were assigned to that taxon by one of the homology searches but returned no hits in the other one.
Background
The microbiome can be characterised and its potential function inferred using metagenomics, whereas metatranscriptomics provides a snapshot of the active functional (and taxonomic) profile of the microbial community by analysing the collection of expressed RNAs through high-throughput sequencing of the corresponding cDNAs (Marchesi and Ravel, 2015). Data generated by metagenomic and metatranscriptomic experiments is both enormous and inherently noisy (Wooley et al., 2010). The pipelines used to analyse this kind of data normally include three main steps: (1) pre-processing and (2) processing of the reads, and (3) downstream analyses (Aguiar-Pulido et al., 2016). Pre-processing mainly involves removing adapters, filtering by quality and length, and preparing data for subsequent analysis (Aguiar-Pulido et al., 2016). After pre-processing the reads, the next step (processing) consists in classifying each read according to the organism with the highest probability of being the origin of that read. This classification and labelling can be either taxonomy-dependent or independent. Taxonomy-dependent methods use reference databases, and these can be further classified as alignment-based, composition-based, or hybrid. Alignment-based methods usually give the highest accuracy but are limited by the reference database and the alignment parameters used, and are generally computation and memory intensive. Composition-based methods have not yet achieved the accuracy of alignment-based approaches, but require fewer computational resources because they use compact models instead of whole genomes (Aguiar-Pulido et al., 2016). Taxonomy-independent methods do not require a priori knowledge because they separate reads based on certain properties (distance, k-mers, abundance levels, and frequencies) (Aguiar-Pulido et al., 2016).
Once the reads have been classified or labelled as best as possible, downstream analyses (step 3) attempt to extract useful knowledge from the data, such as the potential (metagenomics) or active (metatranscriptomics) functional profile. There are various useful resources for the functional annotation of the genes to which the reads are mapped, such as functional databases–gene ontology (GO) (Ashburner et al., 2000; Blake et al., 2015), Kyoto Encyclopaedia of Genes and Genomes (KEGG) (Ogata et al., 1999; Kotera et al., 2015), Clusters of Orthologous Groups (COG) (Tatusov, 2000), InterPRO (Finn et al., 2017), SPARCLE (Marchler-Bauer et al., 2017), and SEED (Overbeek et al., 2014)–and other tools that can also be used to obtain functional profiles. Among the latter, some are web-based, such as MG-RAST (Glass and Meyer, 2011) and IMG/M (Markowitz et al., 2012), and others are standalone programs, like MEGAN (Huson et al., 2007). MEGAN uses the NCBI taxonomy to classify the results from the homology searches, and uses reference InterPRO (Finn et al., 2017), EggNOG (Powell et al., 2012), KEGG (Ogata et al., 1999) and SEED (Overbeek et al., 2014) databases to perform functional assignment.
The same suite of tools can be used to perform taxonomic assignments of metagenomic and metatranscriptomic data. Nevertheless, in both cases the same limitations are encountered, including algorithms that have to process large volumes of data (short reads), and the paucity of reference sequences in the databases. Additionally, most of these tools only use a subset of available genomes or focus on certain organisms, and many do not include eukaryotes. On the other hand, there are major differences in how each workflow determines the taxonomic profile, because some perform searches against protein databases, whereas others do so in a nucleotide space (a review can be found in Shakya et al., 2019). Our HoSeIn workflow (from Homology Search Integration) centres on the processing and downstream analyses steps, and we developed it for using with taxonomy-dependent alignment-based methods (Video 1). As we already mentioned, the latter use homology searches against sequence databases as the first step to classify and label reads. To obtain as much information as possible from the samples, the nucleotide datasets are compared to nucleotide and protein databases using local sequence aligners such as BLAST (Altschul et al., 1990) or FASTA (Pearson, 2004). Nevertheless, once the homology searches are complete, the analysis and integration of these results can be problematic because the outputs from these searches usually show differences and inconsistencies, which can be particularly notorious when working with RNA-seq (Video 1 and Figure 1). On one hand, amino acid- based searches can detect organisms distantly related to those in the reference database but are prone to false discovery. In contrast, nucleotide searches are more specific but are unable to identify insufficiently conserved sequences. Consequently, taxonomic and functional profiles should be carefully interpreted when they are assigned using one or the other. For example, assignments using searches against nucleotide databases, especially for protein coding genes, are likely to be less effective if no near neighbours exist in the reference databases. In this respect, and to the best of our knowledge, existing tools do not intersect information from the different homology searches to integrate the different results, but provide the results of each analysis separately. We developed the HoSeIn workflow to criss-cross the information from both homology search results (nucleotide and protein) and then perform final assignments on the basis of this integrated information. Sequences are assigned to a certain taxon if they were assigned to that taxon by both homology searches, and if they were assigned to that taxon by one of the homology searches but returned no hits in the other one (Video 1 and Figure 1). Specifically, our workflow extracts all the available information for each sequence from the different tools that were used to process the dataset (homology searches and whatever method was used to classify and label the sequences, for example MEGAN [Huson et al., 2007]), and uses it to build a local database. The data for each sequence is then intersected to define the taxonomic profile of the sample following the above-mentioned criteria. Consequently, the main novelty of our workflow is that final assignments integrate results from both homology searches, capitalising on their strengths and thus making them more robust and reliable (Video 1). For metatranscriptomics in particular, where results are difficult to interpret, this represents a very useful tool.

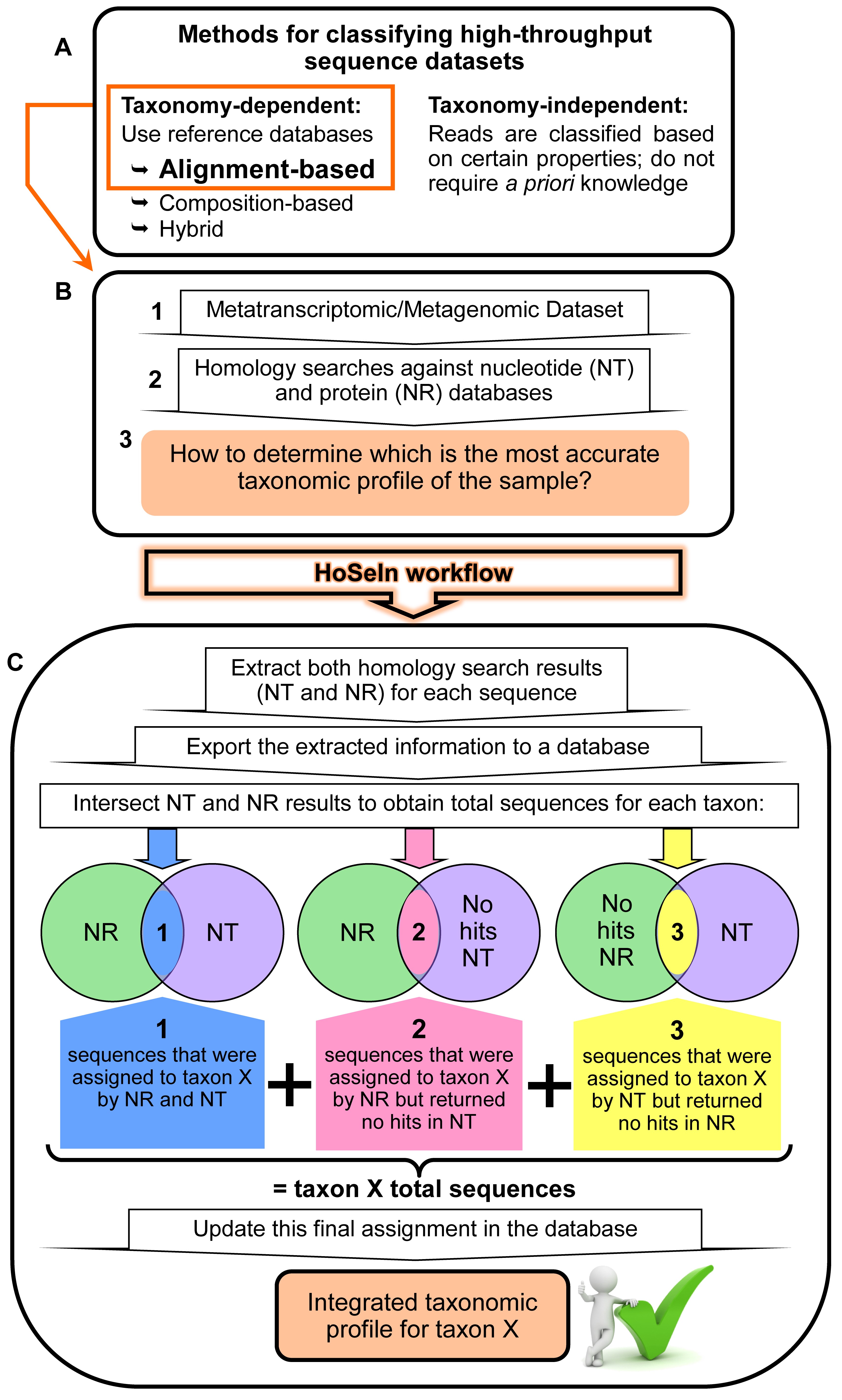
Figure 1. Rationale behind the HoSeIn workflow. A. There are various methods for determining the taxonomic profile of a microbiome in a sequencing-based analysis, and these can be taxonomy dependent or independent (see text for details). B. When using taxonomy-dependent alignment-based methods to analyse metagenomic or metatranscriptomic datasets (1), these are usually compared to nucleotide and protein databases using local sequence aligners such as BLAST (Altschul et al., 1990) or FASTA (Pearson, 2004) (2). Nevertheless, the analysis and integration of these results can be problematic because the outputs from these searches usually show inconsistencies (3). C. The HoSeIn workflow intersects the information from both homology search results and final assignments are determined on the basis of this integrated information. In this way, sequences are assigned to a certain taxon if they were assigned to that taxon by both homology searches (1), and if they were assigned to that taxon by one of the homology searches but returned no hits in the other one (2 and 3).
Equipment
- Desktop computer with an Intel Core i7 2600 processor (3,40 Ghz, 8 Mb, 4 Cores, 8 Threads, video and Turboboost); Intel DH67BL Motherboard, LGA 1155 socket; with 7.1 + 2 sound; 1 Gb network; RAID 0,1,5 y 10; and four Kingston 1.333 Mhz DDR3 4 GB memories
Software
- Ubuntu 18.04.3 LTS (Ubuntu, https://ubuntu.com/#download), last accessed on 11/9/2019
- BLAST (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/), blast-2.2.25+ last accessed 2/7/2013
Note: FASTA programs (FASTA DNA:DNA and FASTX) can also be used for the homology searches. Nevertheless, BLAST and FASTA programs represent a major computational bottleneck when aligning high-throughput datasets against protein databases, and different tools have recently been developed to improve performance. In particular, DIAMOND is an open-source sequence aligner for protein and translated DNA searches which performs at 500x-20,000x the speed of BLAST, is suitable for running on standard desktops and laptops, and offers various output formats as well as taxonomic classification (Buchfink et al., 2015). Thus, when aligning large datasets against protein databases with limited computational resources, we recommend using Diamond. - MEGAN6 (http://ab.inf.uni-tuebingen.de/software/megan6/); MEGAN_Community_windows-x64_6_17_0 version last accessed on 18/9/2019
In this tutorial MEGAN is used to process the homology search output files and then extract the taxonomic and functional information. For downloading and installing this software:- Go to the MEGAN website (http://ab.inf.uni-tuebingen.de/software/megan6) and download the MEGAN6 version that matches your Operating System, as well as the corresponding mapping files
- Run the installer
- Go to the MEGAN website (http://ab.inf.uni-tuebingen.de/software/megan6) and download the MEGAN6 version that matches your Operating System, as well as the corresponding mapping files
- DB Browser for SQLite (DB4S) (https://sqlitebrowser.org/); DB.Browser.for.SQLite-3.11.2-win64 version last accessed on 5/9/2019
DB Browser for SQLite (DB4S) is a high quality, visual, open source tool used to create, design, and edit database files compatible with SQLite. It uses a familiar spreadsheet-like interface, and does not require learning complicated SQL commands. In our workflow we use DB4S to create a local database that includes all the available information for each sequence from the dataset. All this data is then used to define the taxonomic and functional profile of the sample. For downloading and installing this software:- Download the DB4S version that matches your Operating System from the website (https://sqlitebrowser.org/)
- Run the installer
- Download the DB4S version that matches your Operating System from the website (https://sqlitebrowser.org/)
Procedure
Note: This tutorial describes the global procedure for analysing high-throughput metatranscriptomic sequences from an environmental sample, and focuses on how to define its taxonomic and functional profile in a robust and reliable way.
It does not include a detailed description of the pre-processing of high-throughput sequences obtained from an environmental sample (for this, see Kim et al., 2013; Aguiar-Pulido et al., 2016), nor on how to use MEGAN (for this, see Huson et al. [2007 and 2011] and the MEGAN user manual).
Below we provide a detailed tutorial to show how HoSeIn works, exemplifying with one of our samples, a sequence dataset obtained from the gut of a lepidopteran larva. The analysis of the metatranscriptomic part of this dataset was recently accepted for publication (Rozadilla et al., 2020). As this type of analysis is often dictated by the goals of the experiment (Shakya et al., 2019), a few remarks follow to explain certain distinctive features of this particular sample and its subsequent analysis. Spodoptera frugiperda (Lepidoptera: Noctuidae) is an economically important agricultural pest native to the American continent. The purpose for analysing this pest was to describe the taxonomic and functional profile of the larval gut transcriptome and associated metatranscriptome to identify new pest control targets. For this, total RNA was extracted from fifth instar larval guts, submitted to a one-step reverse transcription and PCR sequence-independent amplification procedure, and then pyrosequenced (McCarthy et al., 2015); the high-throughput reads were later assembled into contigs (Rozadilla et al., 2020). As we were interested in identifying, differentiating and characterising both the host (S. frugiperda) gut transcriptome and its associated metatranscriptome, we downloaded the following NCBI databases to perform the homology searches locally (ftp://ftp.ncbi.nlm.nih.gov/blast/db/) (download_db.mp4, a video tutorial that shows how to download different types of database files from NCBI, and download_db.sh, a bash script that automatically downloads these database files one by one, are provided as Supplementary Material 1):
–“Non-redundant” nucleotide sequence (nt)
–16S rRNA gene (16S)
–Lepidopteran whole genome shotgun (Lep) projects completed at the time of the analysis.
Sequences from nt, 16S and Lep, were then combined in a single database (DB:nt16SLep) using the appropriate BLAST+ applications (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/) (blast_tutorial.mp4, a video tutorial that shows how to build and combine different databases and how to run a homology search locally with BLAST, and blast_commands.txt, which contains the commands used in the tutorial, are provided as Supplementary Material 2). The Lep sequences in the combined nucleotide database simplified the identification of host sequences (which represented the majority), and the nt and 16S databases enabled the identification of the associated metatranscriptome (and of host sequences).
–non-redundant protein sequence (nr)
- Analyse sequences with local sequence aligners: Contigs are compared locally to the combined nucleotide database (nt16SLep) using BLASTN (Altschul et al., 1990) with a 1e-50 cutoff E-value, and to the protein database (nr) using BLASTX (Altschul et al., 1990) with a 1e-17 cutoff E-value (Supplementary Material 2).
Note: Here we use BLASTX because the dataset we are querying is small (the aforementioned assembled reads, namely 737 contigs); but for large datasets and limited computational resources we recommend using Diamond (Buchfink et al., 2015). - Process the homology search results:
Step A (the stepA.mp4 video tutorial guides you through step-by-step): The output files from both homology searches are then processed with MEGAN, a software which performs taxonomic binning and assigns sequences to taxa using the Lowest Common Ancestor (LCA)-assignment algorithm (Huson et al., 2007). Taxonomic and functional assignments performed by MEGAN for each contig are then exported using a MEGAN functionality.
Note: MEGAN computes a “species profile” by finding the lowest node in the NCBI taxonomy that encompasses the set of hit taxa and assigns the sequence to the taxon represented by that lowest node. With this approach, every sequence is assigned to some taxon; if the sequence aligns very specifically only to a single taxon, then it is assigned to that taxon; the less specifically a sequence hits taxa, the higher up in the taxonomy it is placed (see the “MEGAN User Manual” for a detailed explanation). We chose MEGAN because this software uses the LCA-assignment algorithm and has a straightforward functionality for exporting the taxonomic and functional information for each sequence from the dataset (i.e., the “species profile” for each sequence can easily be accessed and downloaded). Nevertheless, any other tool or platform that provides this same functionality (i.e., exporting the taxonomic/functional assignment for each sequence from the dataset) can also be used.
Step B (the stepB.mp4 video tutorial guides you through step-by-step): The output files from both homology searches are also processed with a custom bash script. This script parses the homology search output files and generates two files (one for each homology search) containing the name of each contig, its best hit (or no hit) and the corresponding E-value. - Create local database: Step C (the stepC.mp4 video tutorial found in Supplementary Material Step C guides you through step-by-step): All this information (from the exported MEGAN files and from the bash script output files) is then used to create a local SQLite database which includes all the available information for each contig (from both homology searches).
- Analyse the local database: Step D (the stepD.mp4 video tutorial found in Supplementary Material Step D guides you through step-by-step): Final taxonomic assignments are then performed by criss-crossing and comparing all this information using different SQLite commands. Step E (the stepE.mp4 video tutorial found in Supplementary Material Step E guides you through step-by-step): Transcript assignment is achieved by executing certain SQLite commands to group transcripts that correspond to mRNA, rRNA, those that cannot be assigned (not assigned), and those that have to be revised manually (Revise). Step F (the stepF.mp4 video tutorial found in Supplementary Material Step F guides you through step-by-step): Finally, functional assignments of transcripts that were classified by the functional databases are integrated in a single column by executing certain SQLite commands. Only transcripts in the “mRNA” and “Revise” categories can putatively be classified by the functional databases, but because the information in these reference databases is still considerably limited, only around a third of these are assigned a function. Functional assignment of the rest of these transcripts can be done manually on the basis of the homology search results (which are included in the local SQLite database) (see Data analysis).
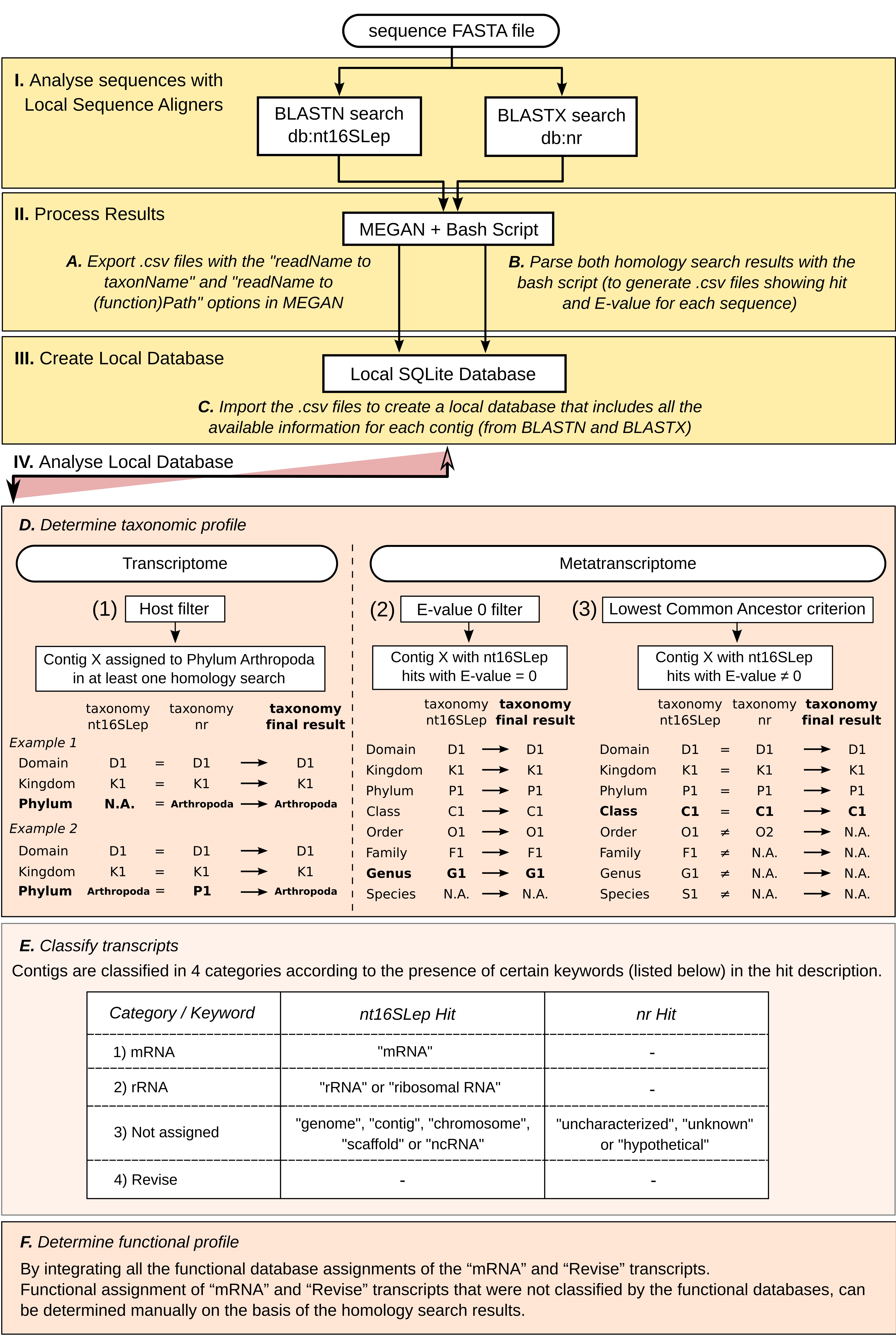
Figure 2. HoSeIn (Homology Search Integration) workflow. This figure shows an overview of the main steps that make up the workflow (see the tutorial for details): I. Analyse sequences with local sequence aligners: Sequences are submitted to homology searches against nucleotide (nt16SLep) and protein (nr) databases. II. Process results: Results from both homology searches are processed with MEGAN and with a custom bash script. Step A: Files containing the MEGAN taxonomic and functional assignments are exported using a MEGAN functionality. Step B: The custom bash script generates files containing the name of each sequence, its best hit (or no hit) and the corresponding E-value, for both homology searches. III. Create local database: Step C: The MEGAN and bash script files are then used to create a local SQLite database which includes all available data for each sequence. IV. Analyse local database: Final taxonomic and functional assignments are then performed by criss-crossing and comparing all this information using different SQLite commands. Step D: Taxonomic assignment is defined by using certain criteria and filters: 1) The “host filter” determines which sequences correspond to the S. frugiperda gut transcriptome; 2) the “E-value = 0 filter” for nt16SLep hits, groups those unequivocal hits; 3) the “Lowest Common Ancestor criterion” for sequences with nt16SLep hits showing E-value ≠ 0, defines the identity of the rest of the contigs by criss-crossing the results from both homology searches and retaining the lowest common ancestor assignments. Step E: Transcript assignment is achieved by searching for certain keywords in the hit description. In this way, it is possible to group transcripts that correspond to mRNA, rRNA, those that cannot be assigned (not assigned), and those that have to be revised manually (revise). Step F: Finally, functional annotation of all the sequences that correspond to mRNA (or “Revise”) is then integrated in a single column. N.A., Not assigned.
We provide various files as Supplementary Material for the reader to be able to go through the tutorial and reproduce the same results we show below:
–A FASTA file containing the assembled sequences (Sf_TV_contigs.fasta) and a text file (coverage.csv) containing the assembly information for each contig (i.e., contig name, number of reads used to assemble each contig, read length and contig coverage), are provided as Supplementary Material 3;
–The output files from both homology searches in BLAST pairwise format (blastn_nt16SLep_total-contigs_Sf-TV.txt and blastx_nr_total-contigs_Sf-TV.txt) are provided as Supplementary Material 4;
–Two custom scripts written in bash that process the homology search results (search_parser.sh and analyser_blast.sh) are provided as Supplementary Material 5;
–The "RMA" files generated by MEGAN6 after processing the homology search output files (blastn_nt16sLep_total-contigs.rma6 and blastx_nr_total-contigs.rma6) are provided as Supplementary Material 6;
–text files containing different commands to intersect, assign and analyse the data in the local SQLite database: step_C_creating_taxonomy.txt found in Supplementary Material Step C, step_D_crisscrossing_taxonomy.txt found in Supplementary Material Step D, step_E_assigning transcripts.txt found in Supplementary Material Step E, step_F_functional_assignment.txt found in Supplementary Material Step F, and analysing_taxonomy.txt.
HoSeIn Tutorial (also see Figure 2):
I. Analyse sequences with local sequence aligners: As mentioned previously, homology searches were performed locally using BLASTN and BLASTX (Altschul et al., 1990) against the combined nucleotide (nt16SLep) and protein (nr) databases with 1e-50 and 1e-17 cutoff E-values, respectively. The homology search results are found in the blastn_nt16SLep_total-contigs_Sf-TV.txt and blastx_nr_total-contigs_Sf-TV.txt files (Supplementary Material 4). The video tutorial download_db.mp4 shows how to download different types of database files from NCBI, and the bash script download_db.sh automatically downloads these database files one by one (Supplementary Material 1). The video tutorial blast_tutorial.mp4 shows how to build and combine different databases, and how to run a homology search locally with BLAST; the commands used in this video can be found in blast_commands.txt (Supplementary Material 2).
II. Process the homology search results: The output files from both homology searches were processed with MEGAN and saved as blastn_nt16sLep_total-contigs.rma6 and blastx_nr_total-contigs.rma6 (Supplementary Material 6).
- Export the taxonomic and functional assignments performed by MEGAN for both homology searches (Figures 3-9, StepA.mp4 video tutorial and Supplementary Figure S1):
- Use MEGAN to open the provided RMA files (blastn_nt16sLep_total-contigs.rma6 and blastx_nr_total-contigs.rma6 found in Supplementary Material 6). To open the RMA files, select File > Open and then browse to the desired file (Figure 3). The main window is used to display the taxonomy and to control the program using the main menus. Once a dataset has been processed, the taxonomy induced by that dataset is shown. The size of the nodes indicates the number of sequences that have been assigned to the nodes (see “MEGAN User Manual” for a detailed explanation).
- To extract the taxonomic information from MEGAN, the taxonomic tree must be progressively expanded from Domain to species, selecting all the leaves, and extracting the text files in csv (comma-separated values) format. Choose the taxonomic level that you wish to extract (from Domain to Species): “Tree” > “Rank” (Figure 4).
- To select the leaves: “Select” > “All Leaves” (Figure 5).
- Without deselecting the leaves, export the file to csv format: “File” > “Export” > “CSV Format” (Figure 6).
- Choose what data you want to export and in what way it will be tabbed in the csv file (choose “summarized” so it exports the sequences contained in the chosen taxonomic level as well as all the lower levels) (Figure 7). We recommend naming the file with a representative name indicating the type of homology search and the taxonomic level, for example “nucl_domain”.
- In this way a csv text file is obtained (which can be viewed in a basic word processor such as WordPad). Each of these files has two fields, one with the sequence name and another with the corresponding assigned taxonomic level (Domain, Phylum, etc.) (Figure 8).
- Repeat this procedure for each taxonomic level, and for the other homology search. In this way, 14 files are obtained (7 files for each homology search), each one corresponding to a particular taxonomic level (Figure 9).
- The procedure for extracting the functional information is very similar (see Supplementary Figure S1 and StepA.mp4 video tutorial): 1) Choose the functional tree you want to visualise, e.g., InterPro2GO (Figure S1A.1); 2) Uncollapse all nodes: “Tree” > “Uncollapse All” (Figure S1A.2); 3) “Select” > “All Leaves” (Figure S1A.3); 4) Without deselecting the leaves, export the file to csv format: “File” > “Export” > “CSV Format” (Figure S1A.4); 5) Choose what data will be exported: “readName_to_interpro2goPath” (Figure S1A.5). Repeat the procedure for all the functional trees you want to include in the final analysis; for this tutorial we also exported the EggNOG assignments (Figure S1B).
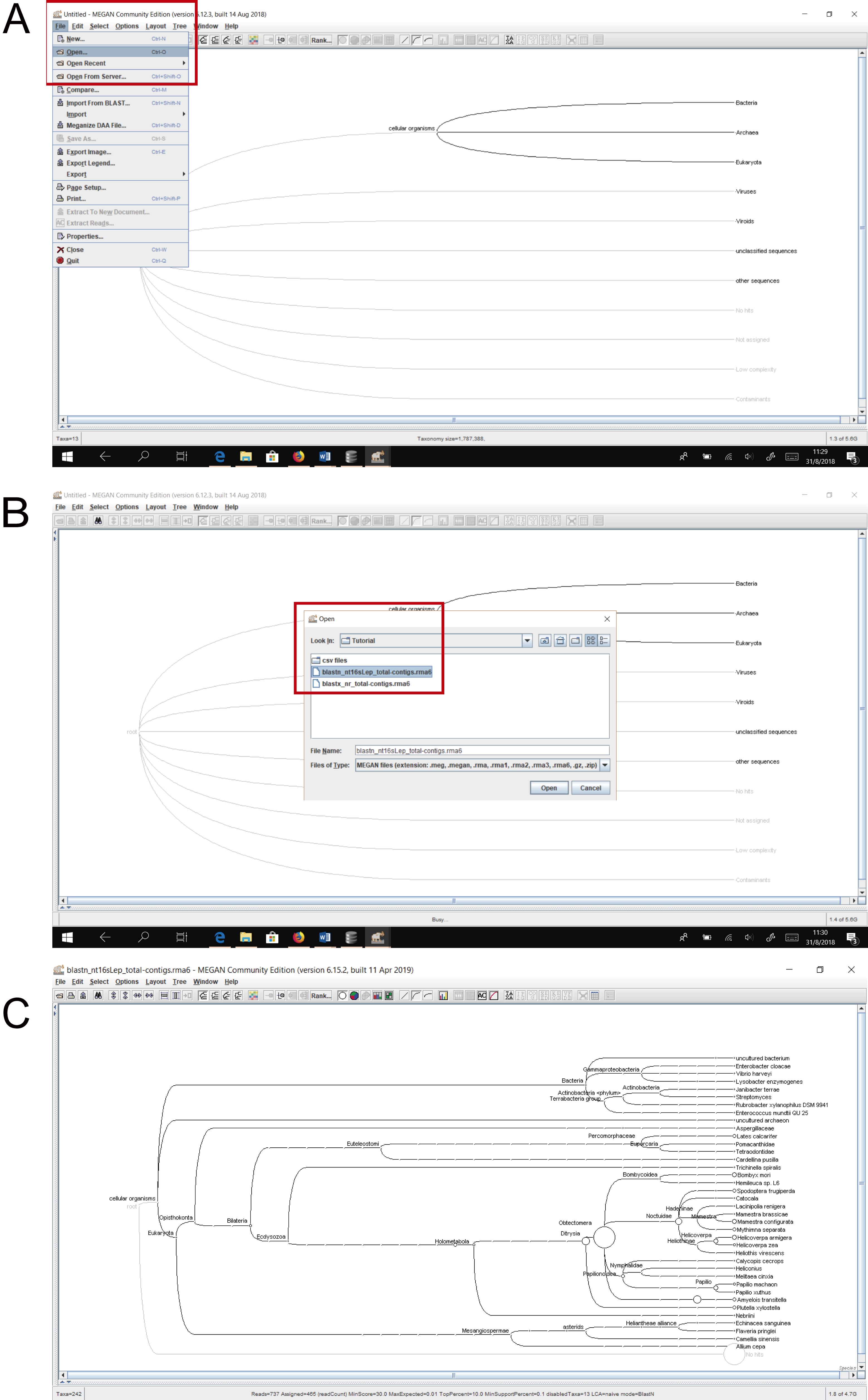
Figure 3. Opening the provided .rma6 files in MEGAN. A. Select “Open” from the “File” leaf (red rectangle). B. Select one of the provided .rma6 files from the location where you saved it (blastn_nt16sLep_total-contigs.rma6 was selected here; red rectangle). C. Appearance of blastn_nt16sLep_total-contigs.rma6 collapsed to “species” taxonomic rank.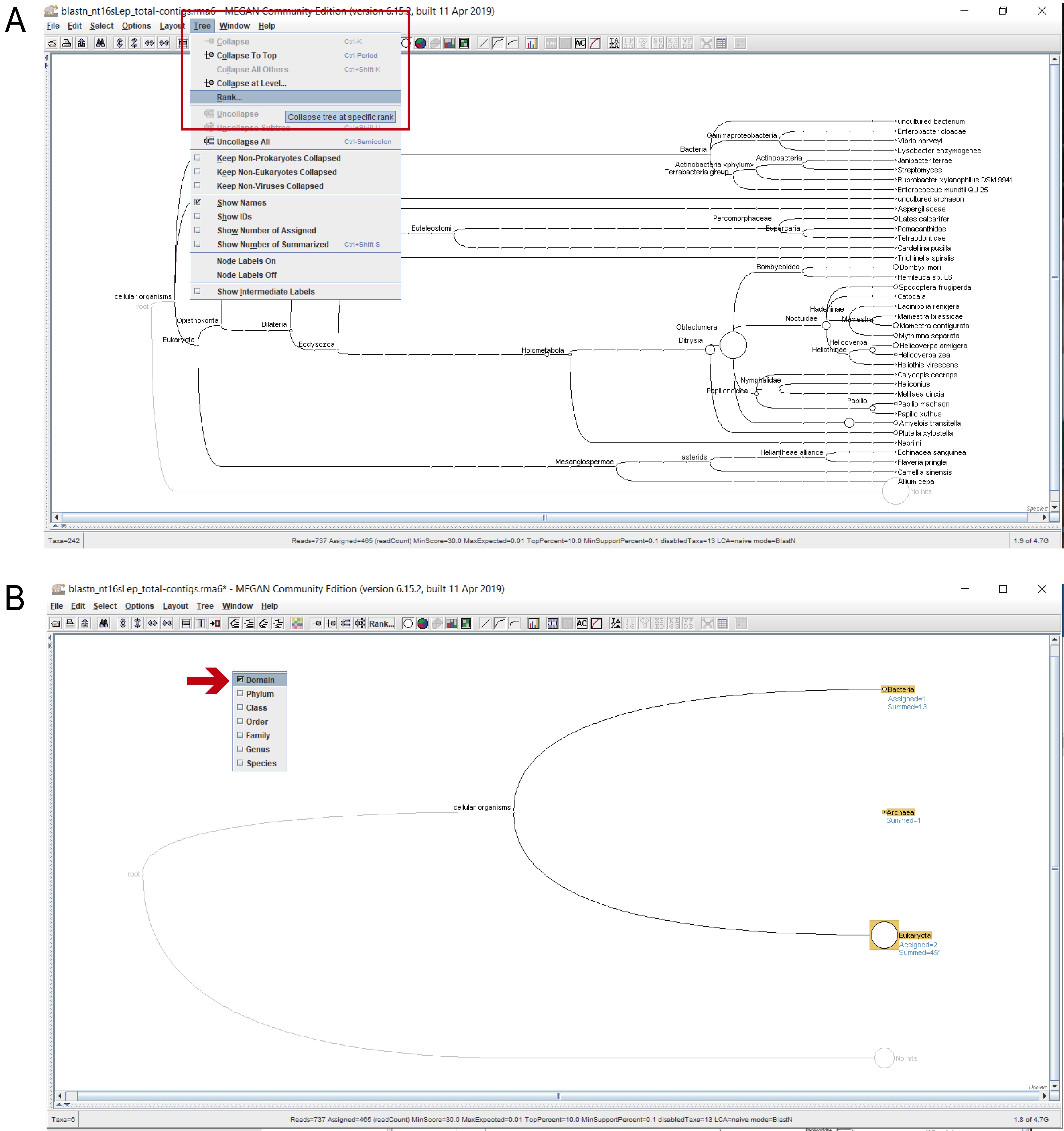
Figure 4. Selecting a taxonomic rank. A. Select “Rank” from the “Tree” leaf (red rectangle). B. Tick the desired rank from the dropdown menu (the red arrow indicates that “Domain” was selected here).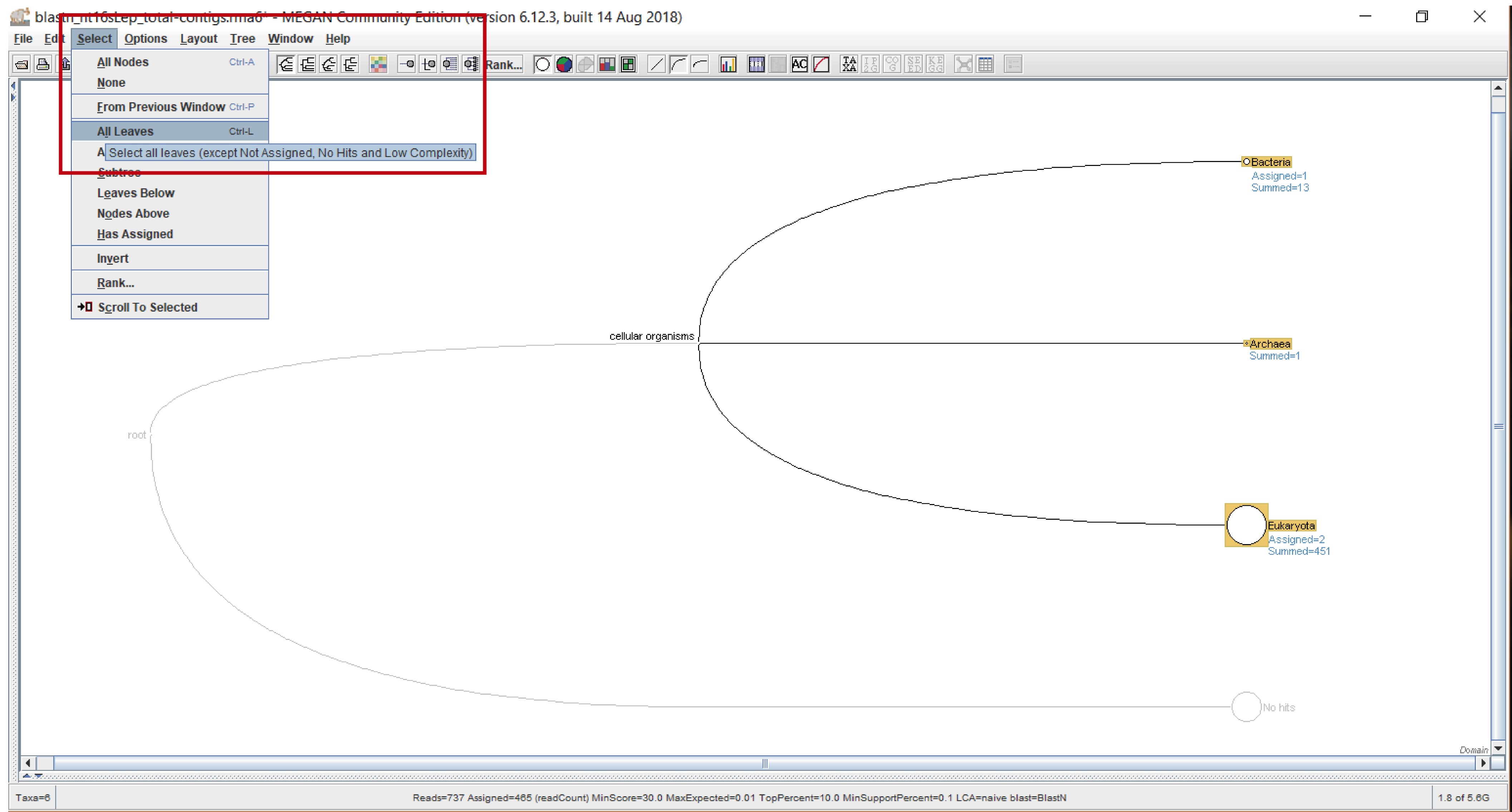
Figure 5. Selecting all the leaves from the chosen taxonomic rank. Select “All Leaves” from the “Select” leaf (red rectangle).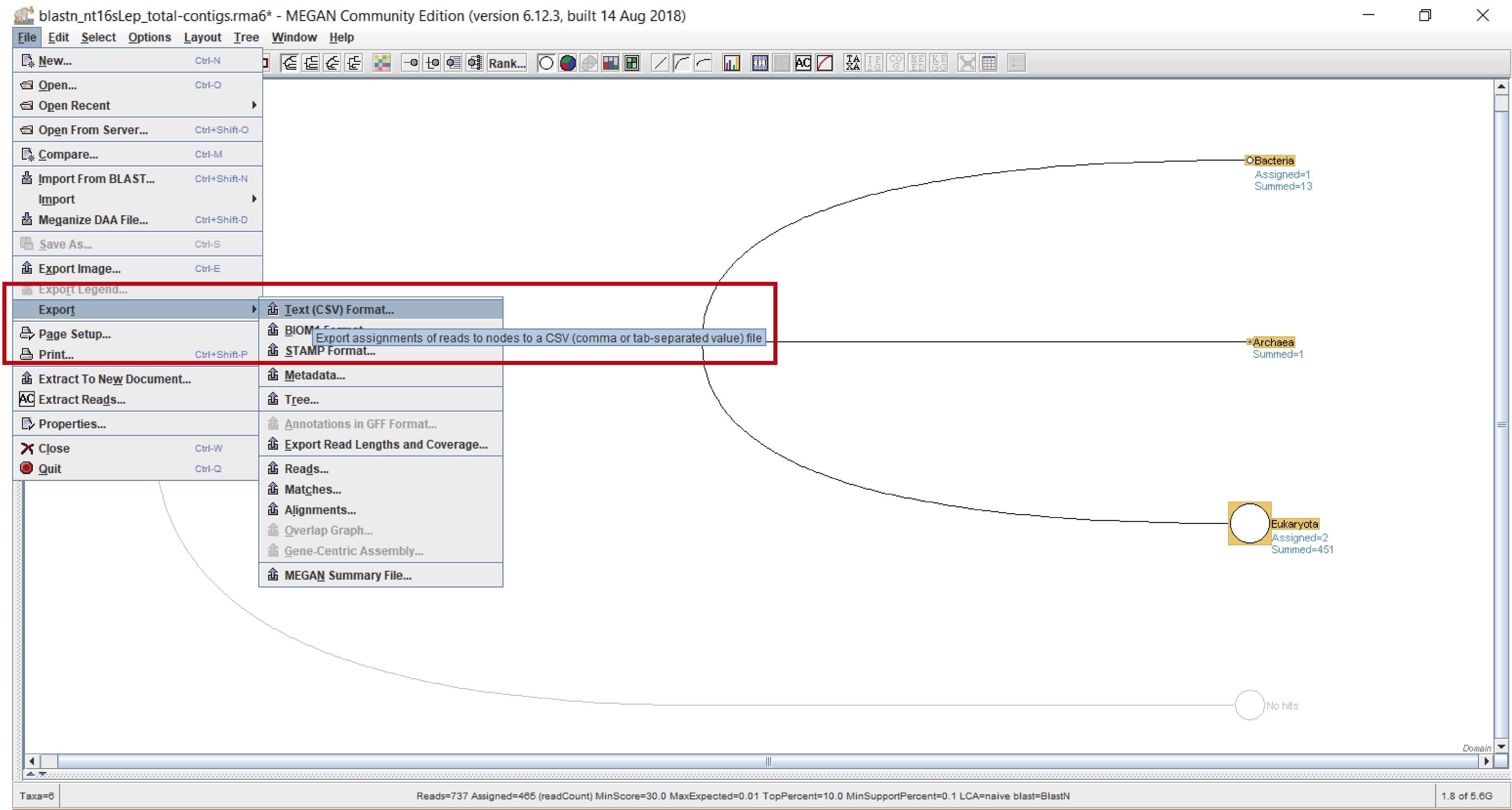
Figure 6. Exporting in csv format. Select “Export” from the “File” leaf, and choose “Text (CSV) format” from the dropdown menu (red rectangle).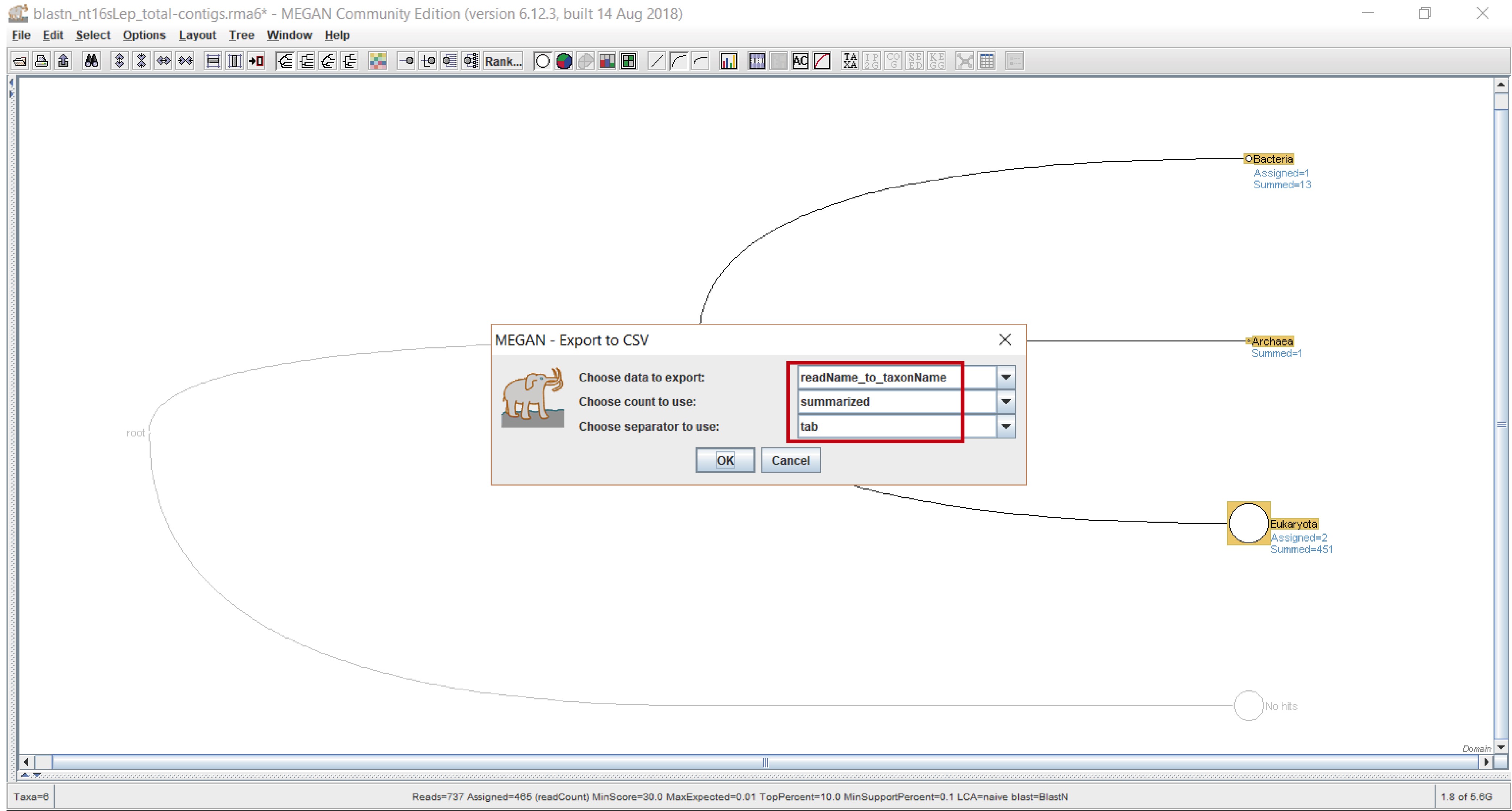
Figure 7. Selecting the way in which data will be exported. Select “readName_to_taxonName” from the “Choose data to export” dropdown menu, “summarized” from the “Choose count to use” dropdown menu, and “tab” from the “Choose separator to use” dropdown menu (red rectangle).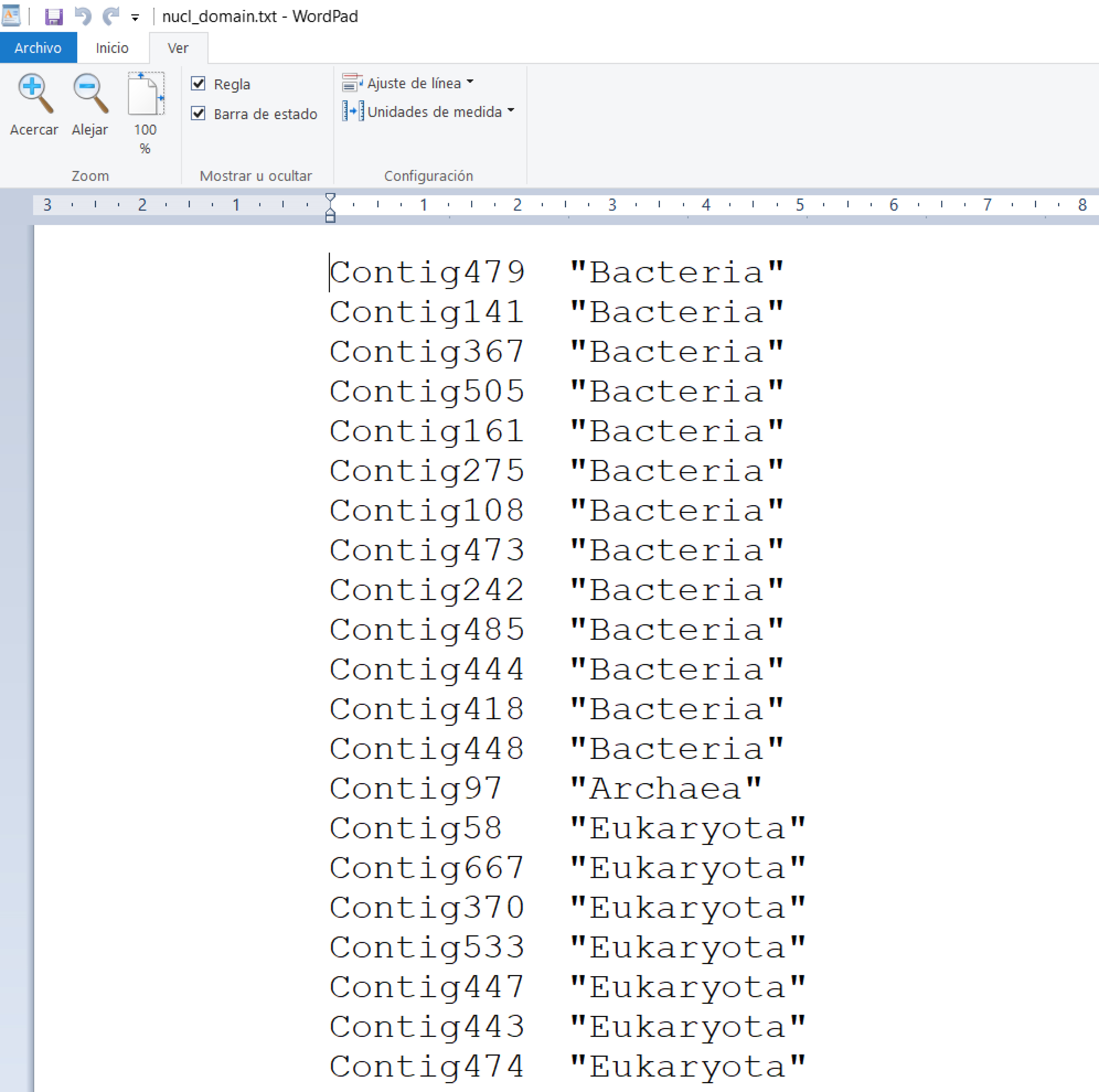
Figure 8. Partial view of the exported “nucl_domain.csv” file. The first column contains the sequence name (e.g., Contig479), and the second column the taxonomic rank assigned to each sequence (in this figure “Domain”, e.g., “Bacteria”).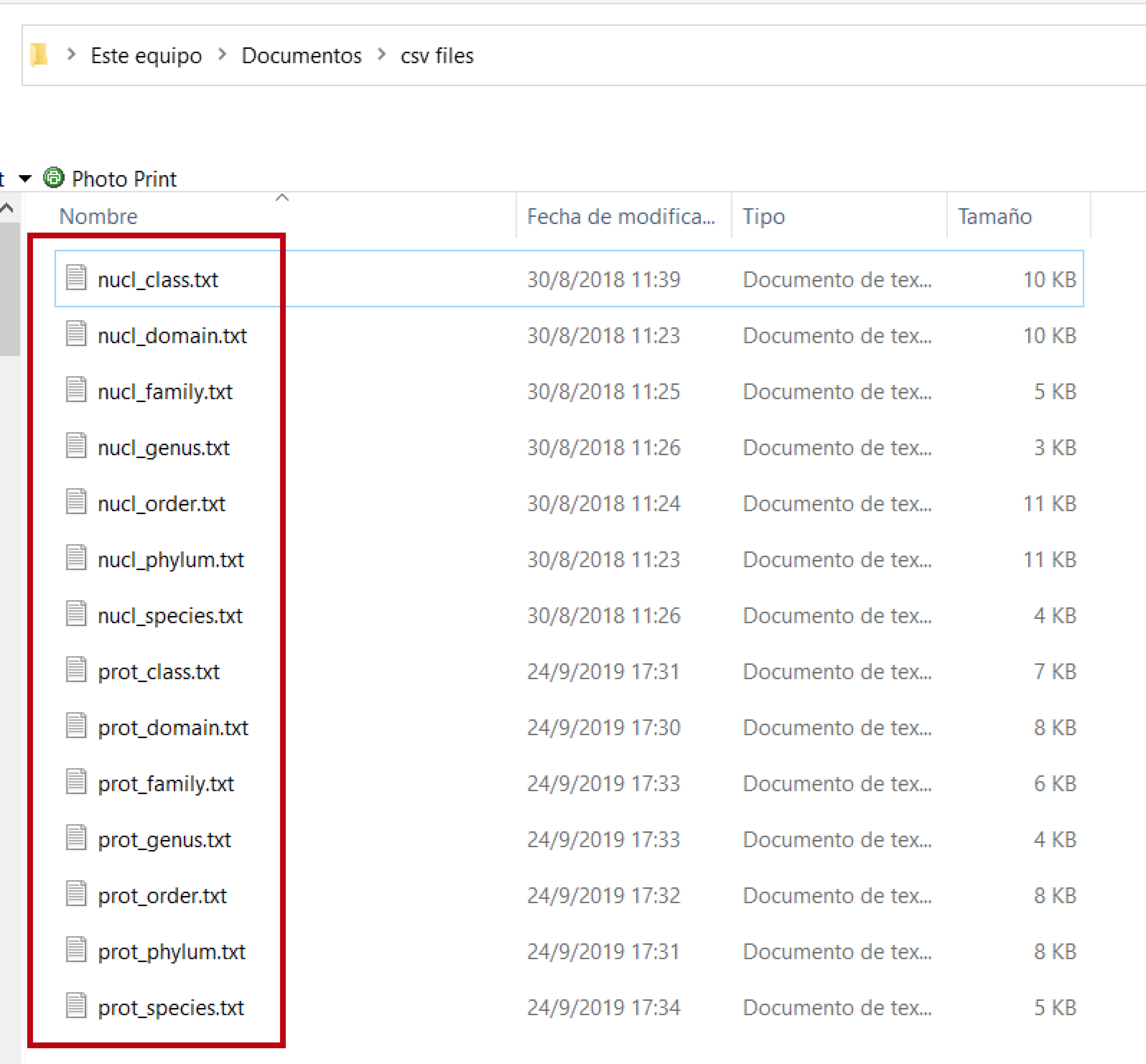
Figure 9. Exported MEGAN files. Folder containing all 14 files exported from MEGAN, named according to the corresponding homology search and taxonomic level (red rectangle).
- Parse the output files from the homology searches (Figure 10 and Supplementary Material stepB.mp4 video tutorial):
This step must be carried out in a Linux Operating System because the scripts that parse the homology search results were written in bash. The scripts process the FASTA file (containing the query sequences) and the homology search output files:- The provided scripts (search_parser.sh and analyser_blast.sh from Supplementary Material 5), FASTA file (Sf_TV_contigs.fasta from Supplementary Material 3) and output files from the homology searches (blastn_nt16SLep_total-contigs_Sf-TV.txt and blastx_nr_total-contigs_Sf-TV.txt from Supplementary Material 4), must all be placed in the same folder (Figure 10A).
- Of the two bash scripts we provide, only execute search_parser.sh. This script works by executing various analyser_blast.sh scripts simultaneously to speed up the process. Open a terminal in the same folder that contains the files and execute the search_parser.sh bash script strictly in the following order (also see example below and Figures 10B-10C):
bash search_parser.sh (name and file extension of the query fasta) (name and file extension of the homology search result) (chosen name and file extension for the output file)
bash search_parser.sh Sf_TV_contigs.fasta blastn_nt16Slep_total-contigs.txt nucl_hits.csv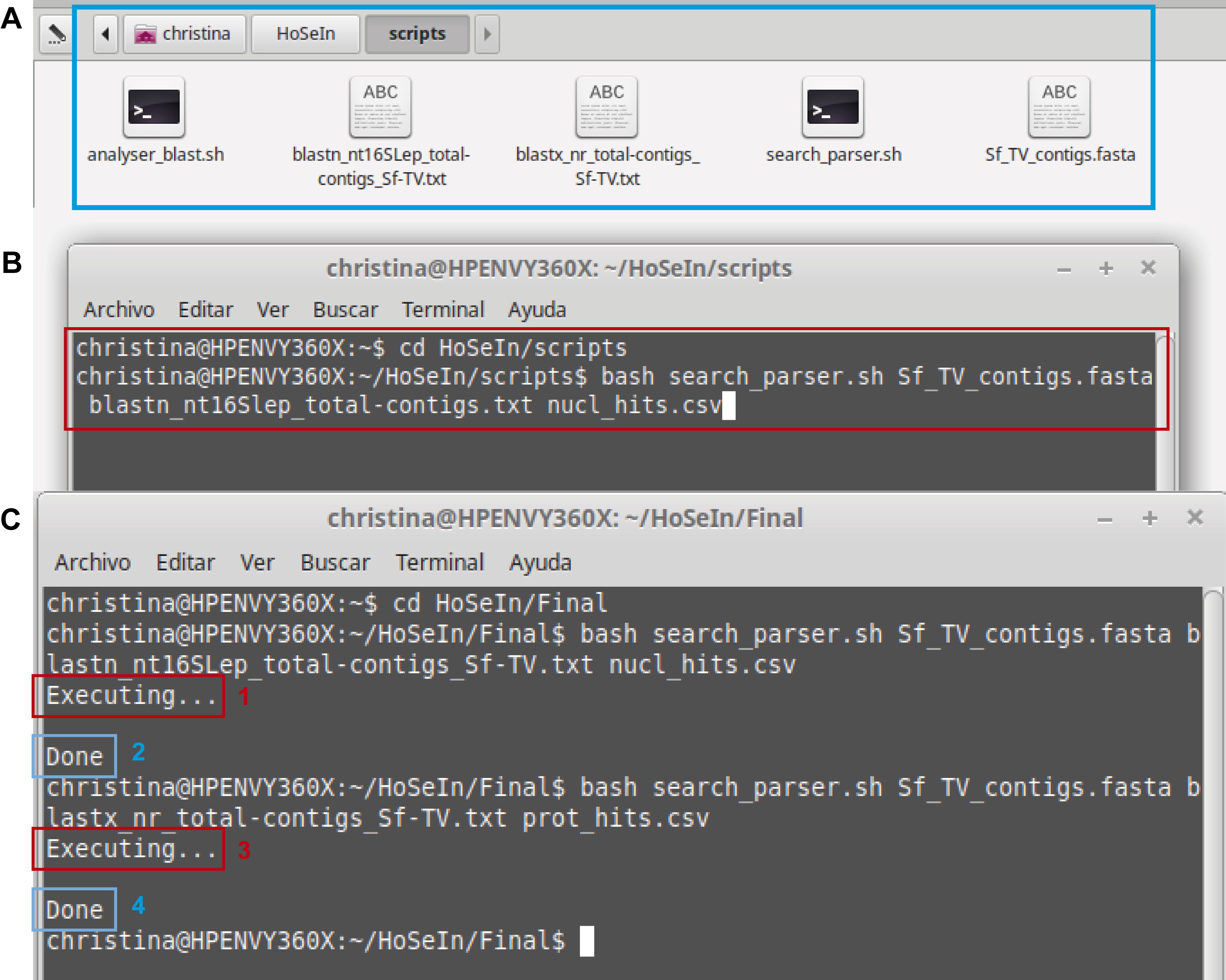
Figure 10. Using the bash script. A. The pale blue rectangle indicates the folder containing all the necessary files for the scripts to work correctly. B. The red rectangle indicates how to execute the script to process the BLASTN output file (homology search against the nucleotide database). C. The image shows the messages that appear in the terminal after processing the output files from the BLASTN homology search (1 and 2) and from the BLASTX homology search (3 and 4). - The script generates a csv file with three fields separated by “%”: the first one shows the name of each sequence, the second shows its best hit (or nothing if there is no hit), and the third its corresponding E-value (or nothing if there is no hit) (Figure 11).
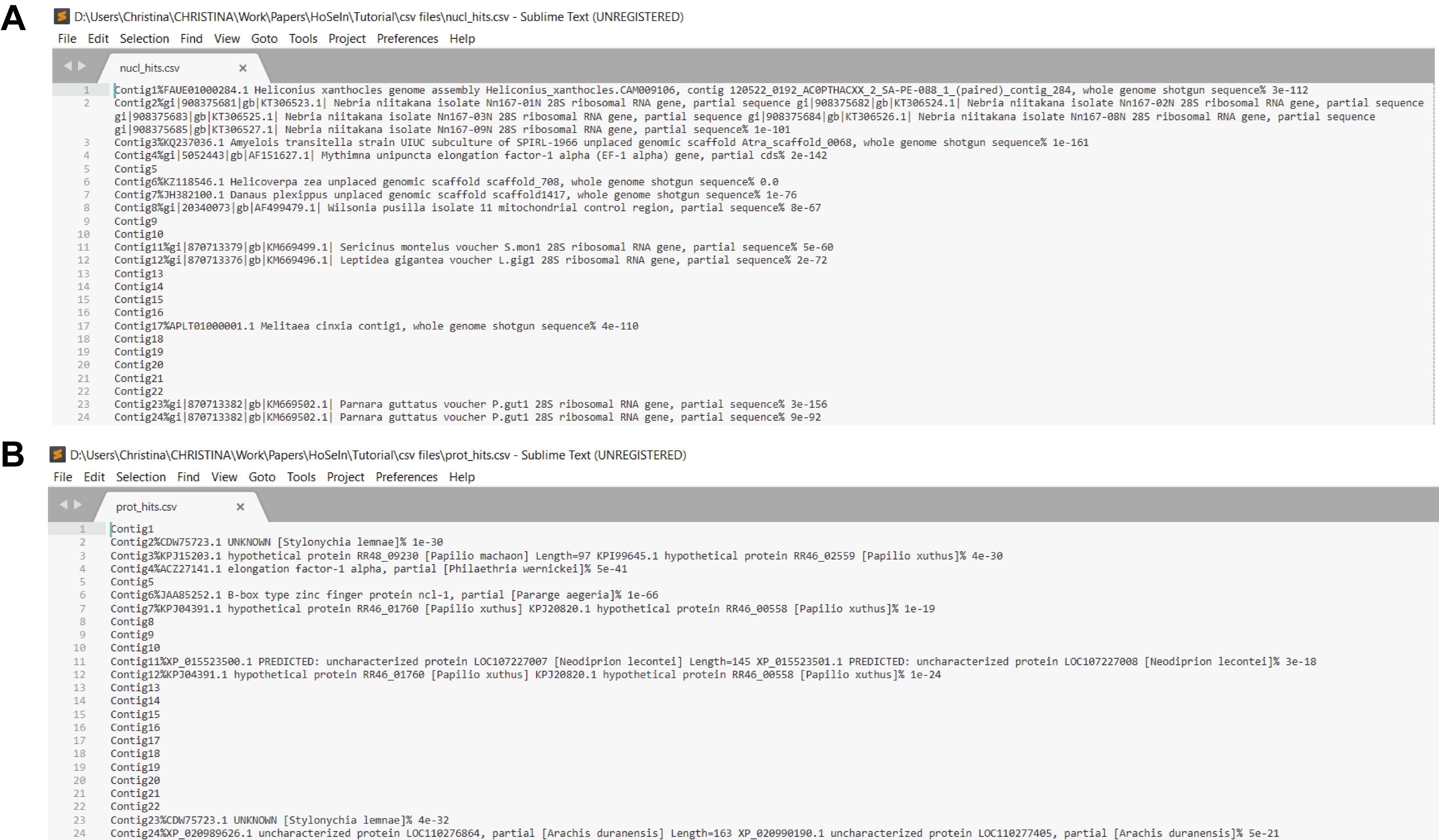
Figure 11. Partial images of the csv files generated by the script. Files showing the listed BLASTN (A) and BLASTX hits (B). In both files, fields showing the sequence name, its best hit and the corresponding E-value, are separated by “%”.
- Create the database in DB4S (Figures 12-18, and step_C_creating_taxonomy.txt and stepC.mp4 video tutorial found in Supplementary Material Step C):
The file containing all the information for the assembled reads (coverage.csv found in Supplementary Material 3; includes contig name, number of reads used to assemble each contig, read length, and contig coverage), the exported MEGAN files (14 taxonomic files and 2 functional files from Step A) and the bash script output files (2 files from Step B), will now be used to create a local database with DB4S which will include all the available information for each contig (from both homology searches, BLASTN and BLASTX). To do this, first each csv file must be imported individually to DB4S:- Create a new database clicking on “New Database” and choosing where to save it (Figure 12).
- Individually import the csv files that were exported from MEGAN (16 files), those that were created by the bash script (2 files), and the file containing the assembly information for the contigs (1 file): “File” > “Import” > “Table from CSV file” (Figure 13).
- Choose a name for the table and indicate field separator (for the files imported from MEGAN, Tab; for the files generated by the bash script, Other > %) (Figure 14). Only for coverage.csv, check the “Column names in first line” box, which will automatically give the columns their correct name (Figure 14C).
- We recommend renaming table columns with representative names as indicated in Figure 15 to be able to use the commands we provide for Steps C6, D, E and F (and also to simplify interpretation of the commands and avoid mistakes).
- Figure 16 shows what the list of tables should look like after renaming them.
- The next step consists in integrating the information from all the imported files in a single new table, as indicated in Figures 17 and 18. The set of commands in step_C_creating_taxonomy.txt from Supplementary Material Step C, creates a new table named “taxonomy” that unifies the columns from all the imported csv files, and adds empty columns (final_domain, final_phylum, etc.) to be filled with the result of the subsequent taxonomic criss-crossing (Step D). It also adds auxiliary columns “state_taxo” (to indicate if the contig was taxonomically assigned or not and to avoid multiple assignments; Step D), “rna_type” (to indicate if the transcripts correspond to mRNA or rRNA; Step E), “state_function” (to indicate whether the transcripts were assigned or need to be revised; Step E), and “function_type” (to integrate the functional assignment of those transcripts that were classified by the functional databases; Step F).
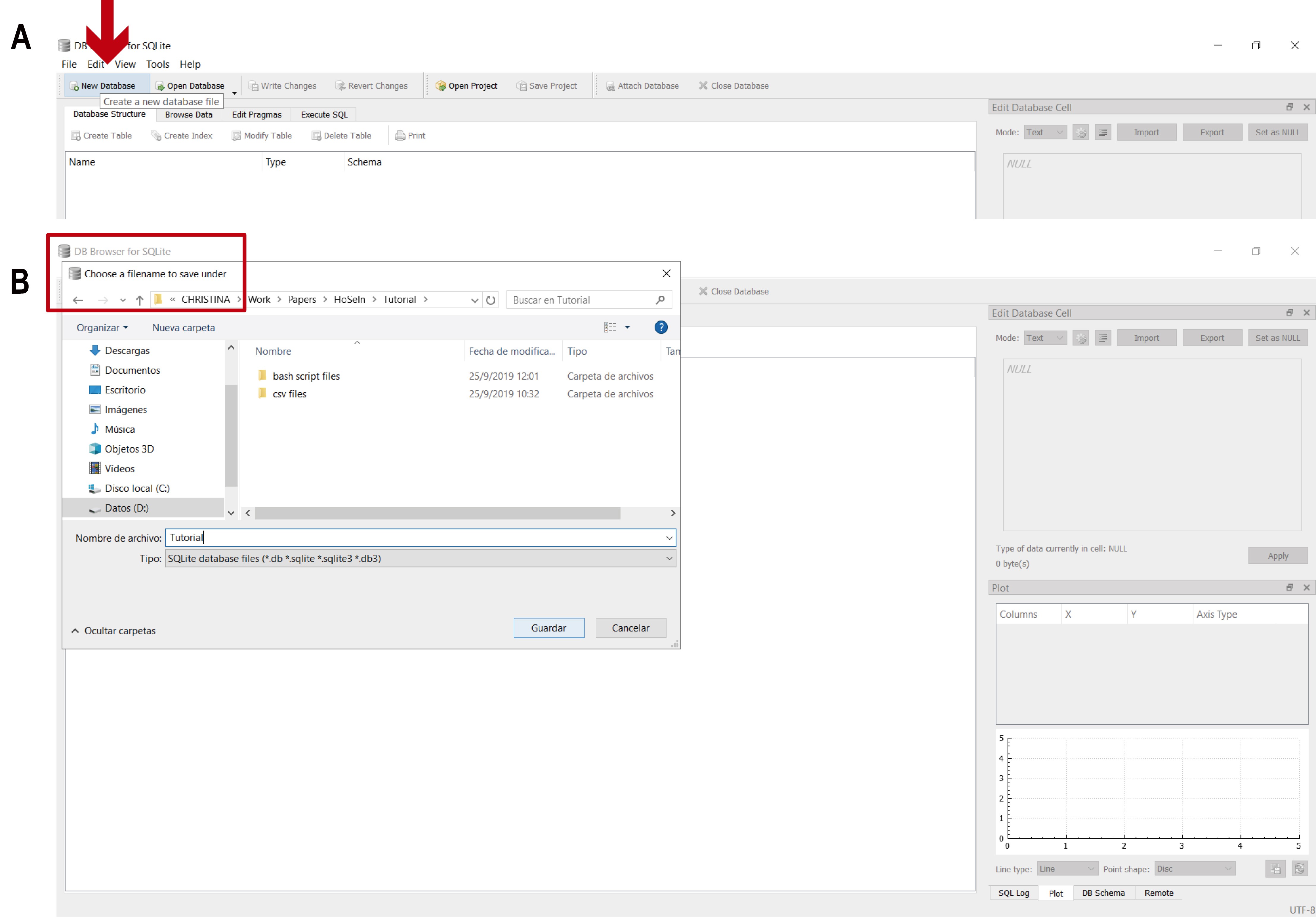
Figure 12. Creating a new database in DB4S. A. Click on “New Database” (red arrow). B. A window will appear requesting you to choose a filename and location to save the new database (red rectangle).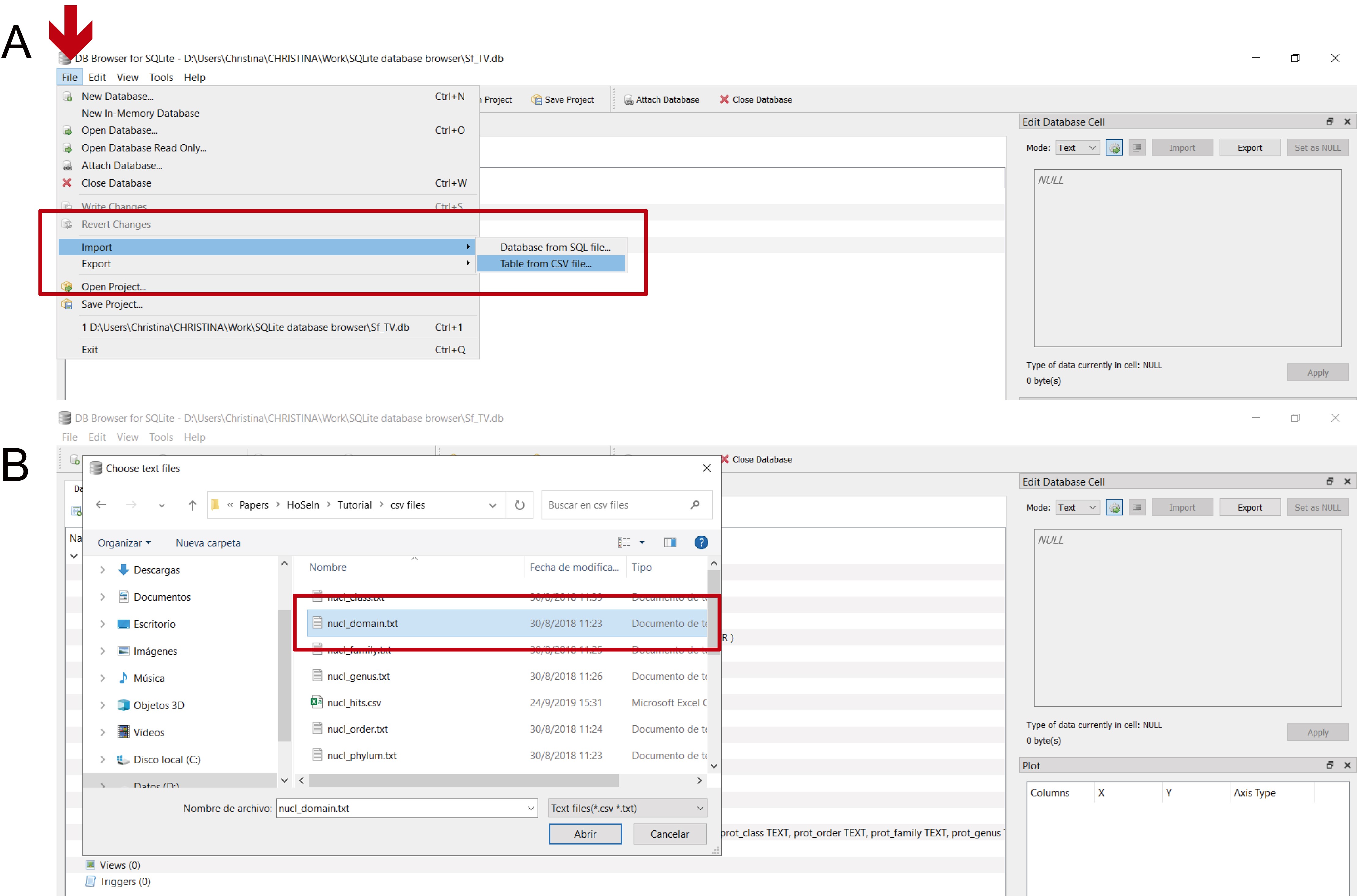
Figure 13. Importing the csv files. A. Click on the “File” leaf (red arrow) and select “Import” and “Table from CSV file” from the dropdown menus (red rectangle). B. Select the csv file that you want to import (“nucl_domain.txt” in the figure; red rectangle).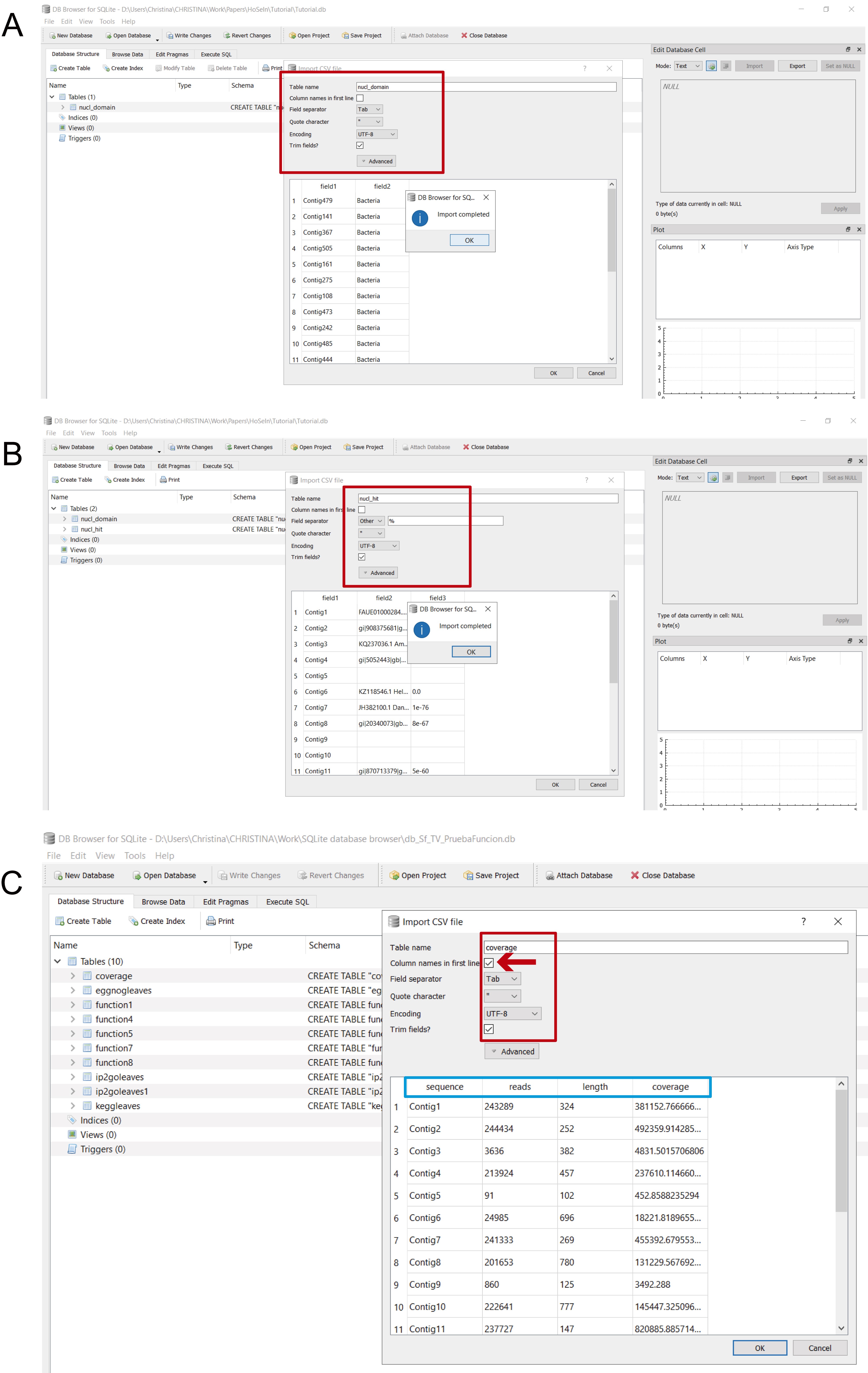
Figure 14. Naming the imported tables. Once you have selected a csv file, a new window will appear in which you have to name the table and indicate the field separator. A. For files imported from MEGAN the field separator is “Tab”; this table was named “nucl_domain” (red rectangle). B. For files generated by the bash script, the field separator is “Other” > “%”; this table was named “nucl_hit” (red rectangle). C. For the “coverage” file, check the “Column names in first line” box (indicated by the red arrow) and choose “Tab” as field separator; the pale blue rectangle indicates the assigned column names.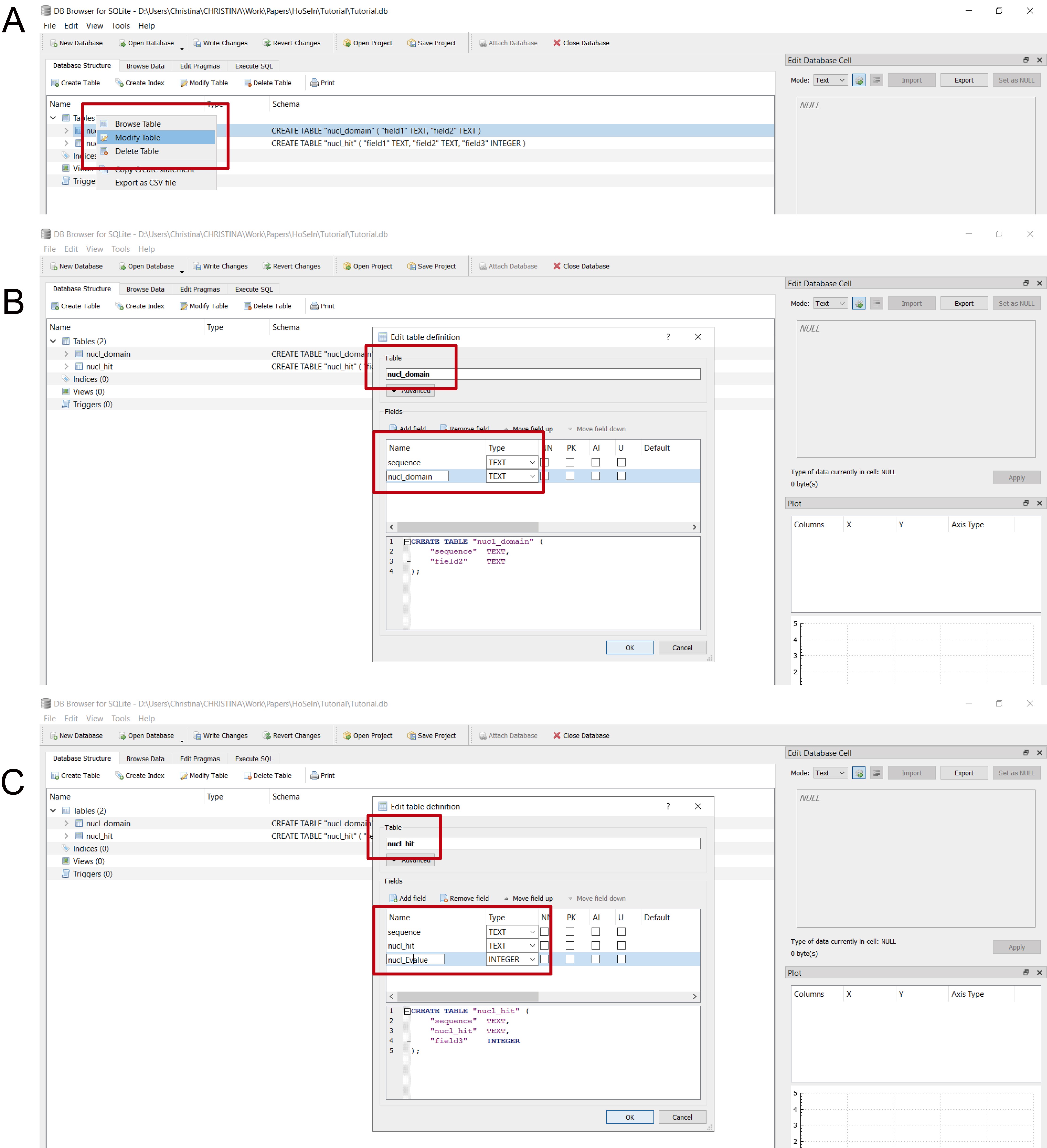
Figure 15. Renaming table columns. A. In the main window, select the table that you want to modify with the mouse´s right button, and click on “Modify table” (red rectangle). B. A new window will open where the fields (which correspond to the column names) appear; double click on each to rename. For the tables obtained from the MEGAN files, rename “field1” as “sequence”, name the table and “field2” according to the database which was used for the homology search (nucl or prot) followed by the appropriate taxonomic rank (e.g., domain); in the figure, “nucl_domain” (red rectangles). C. The tables obtained from the files generated by the bash script have three columns: rename “field1” as “sequence”; rename “field2” as “nucl_hit” for the BLASTN hits, and as “prot_hit” for the BLASTX hits; rename “field3” as “nucl_Evalue” for the BLASTN hits, and as “prot_Evalue” for the BLASTX hits (red rectangles).
Note: Columns from the “coverage” file were named in the previous step so they do not need to be modified.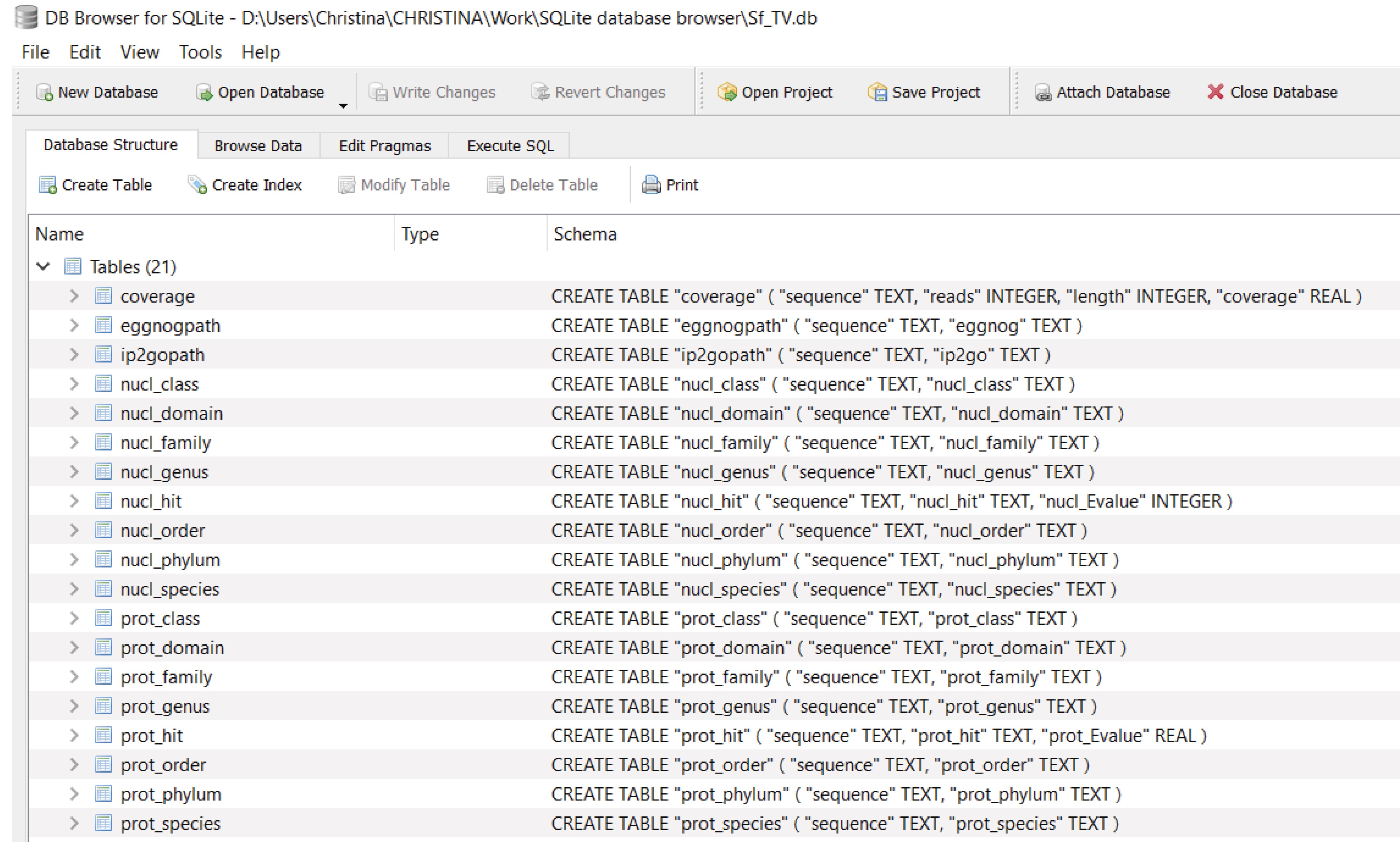
Figure 16. “Database Structure” window in DB4S listing all the imported and renamed tables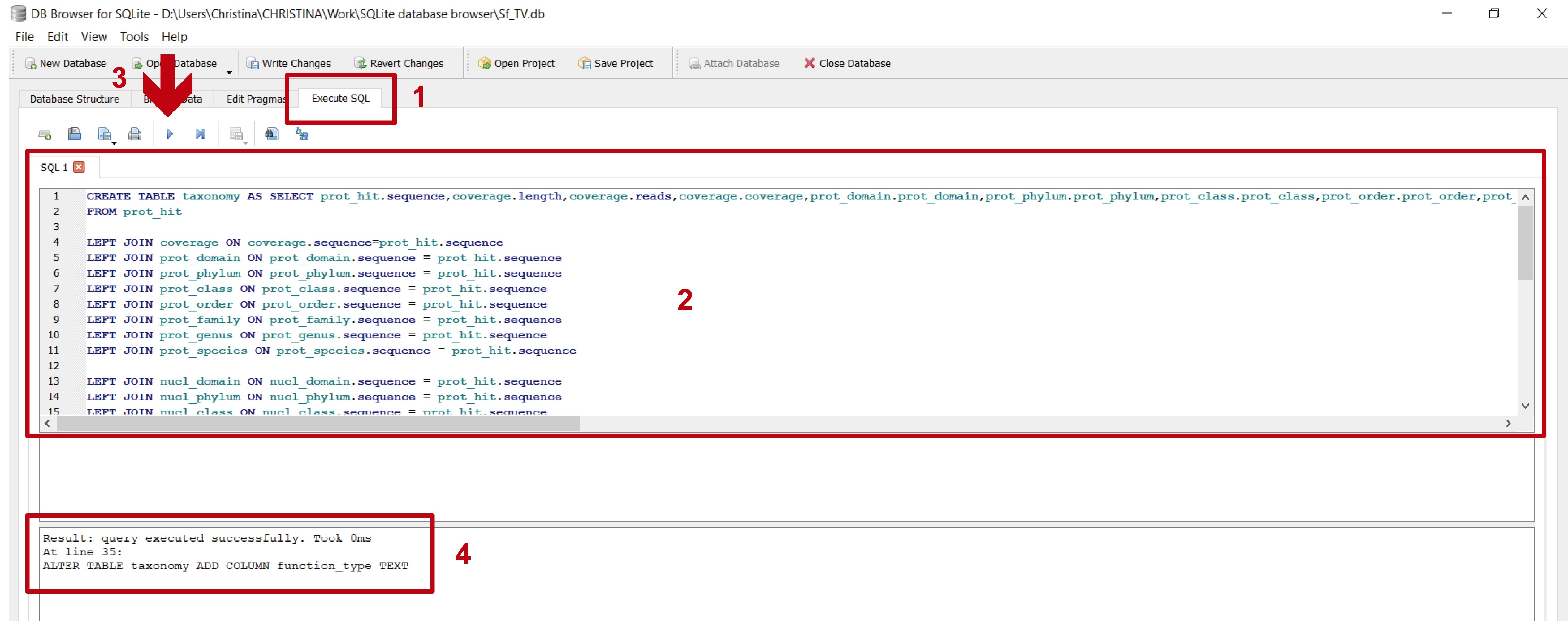
Figure 17. Unifying the information from all the imported files to create a single table. 1) In the “Execute SQL” leaf, 2) paste the commands contained in step_C_creating_taxonomy.txt and 3) execute them with “Play”; 4) the lower panel indicates if the commands were executed successfully.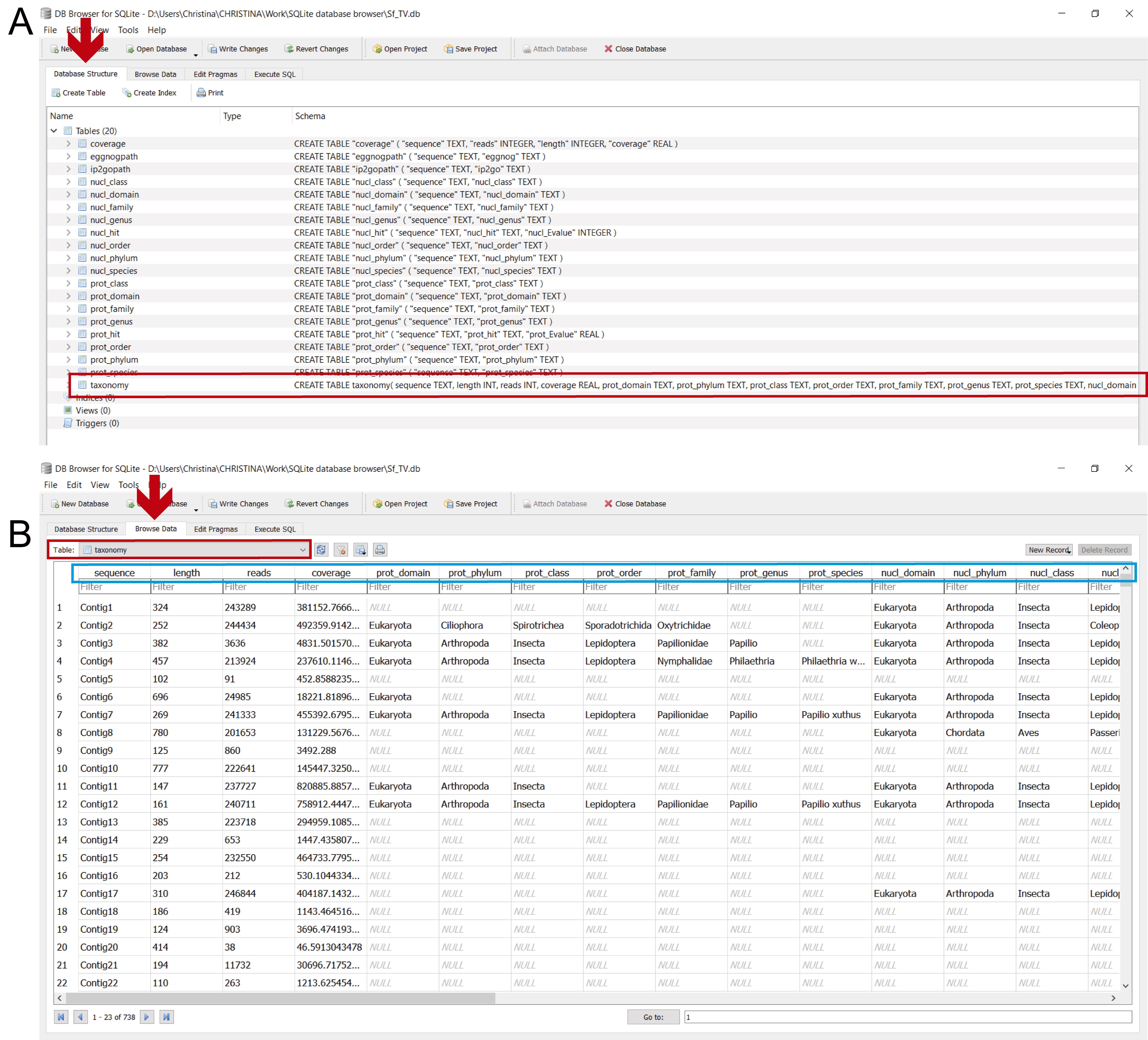
Figure 18. View of the “taxonomy” table created in the previous step. A. In the “Database Structure” leaf (red arrow) a new “taxonomy” table will appear (red rectangle). B. To browse the new “taxonomy” table, select it from the dropdown menu in the “Browse Data” leaf (indicated by the red arrow and rectangle); the light blue rectangle shows a partial view of the columns that were created in “taxonomy”.
- Determining the taxonomic profile (Figure 19, and step_D_crisscrossing_taxonomy.txt and stepD.mp4 video tutorial found in Supplementary Material Step D):
The next step consists in intersecting all the information contained in the “taxonomy” table to elucidate the taxonomic profile of the sample, based on the following criteria:- Contigs that were assigned to Arthropoda in at least one of the homology searches are assigned to the host transcriptome.
- Contigs that have hits with E-value = 0 in the nt16SLep search, are directly assigned to that taxon.
- The rest of the contigs are assigned by comparing the MEGAN assignments from both homology searches according to the LCA logic; i.e., the level of taxonomic assignment for a contig is the one found in common for both results, or for the only result if it returns no hits in the other homology search.
- Contigs that were not assigned to any taxon by MEGAN in any of the homology searches are considered as “not assigned”; contigs that returned no hits in both homology searches are considered as “no hits”.
These assignments are carried out by executing the commands in step_D_crisscrossing_taxonomy.txt in DB4S (Figure 19 and stepD.mp4 video tutorial from Supplementary Material Step D).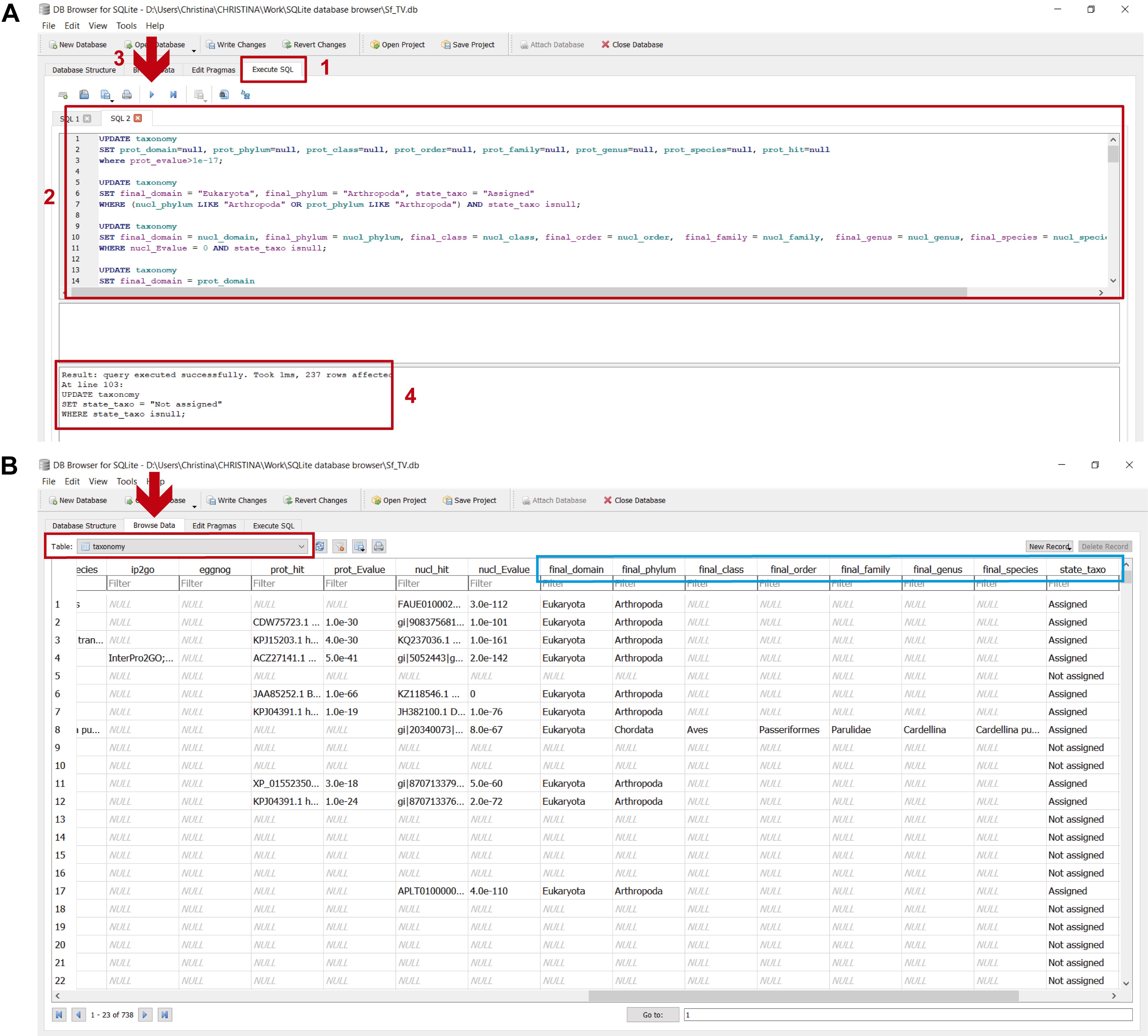
Figure 19. Determining final taxonomic assignments in the “taxonomy” table. A. To perform these assignments: 1) in the “Execute SQL” leaf, 2) paste the commands contained in step_D_crisscrossing_taxonomy.txt and 3) execute them with “Play”; 4) the lower panel indicates if the commands were executed successfully. B. To view the updated “taxonomy” table, select it from the dropdown menu in the “Browse Data” leaf (indicated by the red arrow and rectangle); the light blue rectangle indicates the columns with the final taxonomic assignments after criss-crossing the taxonomic information from both homology searches (final_domain, final_phylum, etc.).
- Classifying the transcripts (Figure 20, and step_E_assigning transcripts.txt and stepE.mp4 video tutorial found in Supplementary Material Step E):
As this sample was obtained from total RNA, the sequences can be further classified as either messenger or ribosomal RNA using the information contained in the hit description from the homology searches (“nucl_hit” and “prot_hit” columns), based on the following criteria:- Contigs are assigned to mRNA if they show the word “mRNA” in the “nucl_hit” column.
- Contigs are assigned to rRNA if they show the words “rRNA/ribosomal RNA” in the “nucl_hit” column.
- Contigs are considered as functionally “Not assigned” if they show the words "genome/chromosome/scaffold/contig" or "uncharacterized/hypothetical/unknown/ncRNA" in the “nucl_hit” and “prot_hit” columns, respectively.
- All the rest of the contigs are included in a “Revise” category to be manually revised.
These assignments are carried out by executing the commands found in step_E_assigning transcripts.txt in DB4S (Figure 20 and stepE.mp4 video tutorial from Supplementary Material Step E).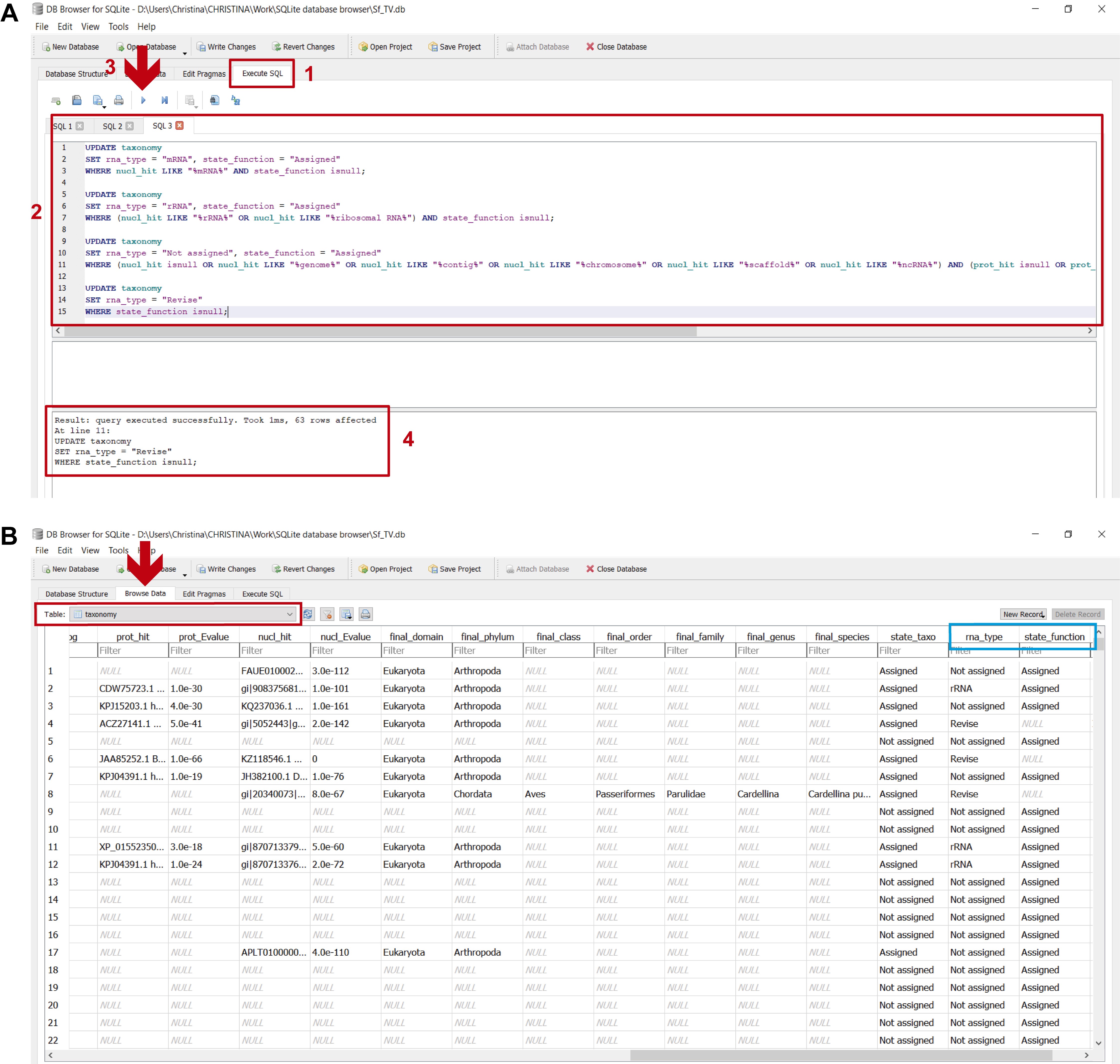
Figure 20. Determining transcript assignments in the “taxonomy” table. A. To perform these assignments: 1) in the “Execute SQL” leaf, 2) paste the commands contained in step_E_assigning transcripts.txt and 3) execute them with “Play”; 4) the lower panel indicates if the commands were executed successfully. B. To view the updated “taxonomy” table, select it from the dropdown menu in the “Browse Data” leaf (indicated by the red arrow and rectangle); the light blue rectangle indicates the columns with the final transcript assignments: “rna_type” and “state_function”. The “rna_type” column indicates what category the transcript was assigned to (i.e., “mRNA”, “rRNA”, “Not assigned” or “Revise”). An “Assigned” status in the “state_function” column appears when the transcripts are assigned to either “mRNA”, “rRNA” or “Not assigned”; if the transcript was included in a “Revise” category, it is “NULL”.
- Functional assignment (Figure 21, and step_F_functional assignment.txt and stepF.mp4 video tutorial found in Supplementary Material Step F):
This step integrates all the functional assignments of those transcripts that were classified by the functional databases in a single column (“function_type”). This is done by executing the commands found in step_F_functional assignment.txt in DB4S (Figure 21 and stepF.mp4 video tutorial from Supplementary Material Step F). As can be deduced from the previous step, only transcripts in the “mRNA” and “Revise” categories can putatively be classified by the functional databases. Nevertheless, only about a third are assigned a function because the information in these reference databases is still considerably limited (see “Data analysis”).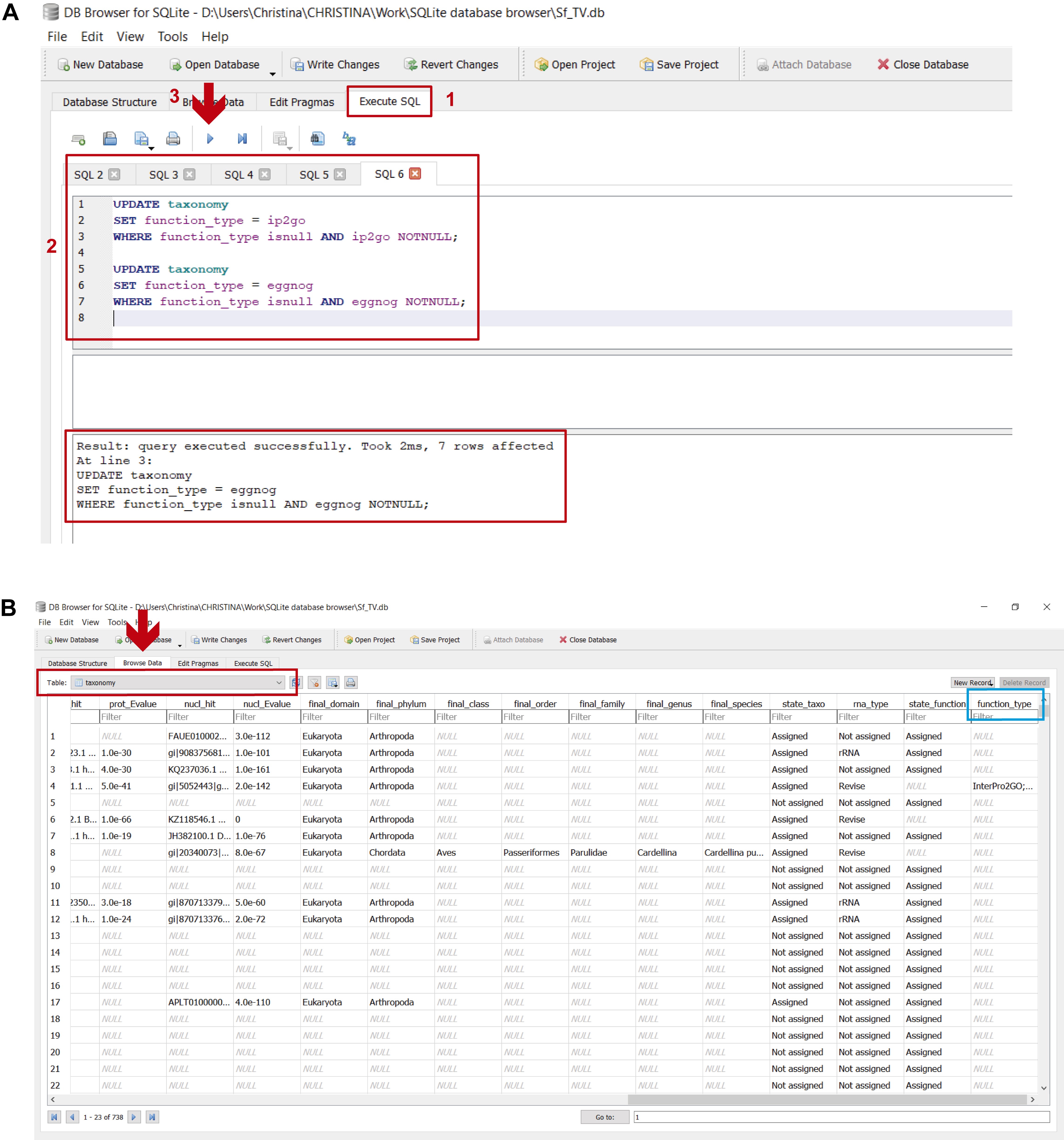
Figure 21. Integrating functional assignments in the “taxonomy” table. A. 1) in the “Execute SQL” leaf, 2) paste the commands contained in step_F_functional assignment.txt and 3) execute them with “Play”; 4) the lower panel indicates if the commands were executed successfully. B. To view the updated “taxonomy” table, select it from the dropdown menu in the “Browse Data” leaf (indicated by the red arrow and rectangle); the light blue rectangle indicates the “function_type” column showing the integrated functional assignments. Transcripts that were not classified by the functional databases appear as “NULL”.
Data analysis
Browsing the “taxonomy” table (Supplementary Figures S2-S5 and analysing_taxonomy.txt):
We are finally ready to browse and analyse the updated “taxonomy” table in the “Browse Data” leaf. One or more filters can be applied to facilitate data analysis. Further, some commands can be executed to visualise, for example, the taxonomic distribution of the sample (Supplementary Figure S2), the distribution by transcript type (Supplementary Figure S3), or a non-redundant list of the hits obtained in the homology searches (Supplementary Figure S4).
To analyse the functional profile in more detail, all the “mRNA” and “Revise” transcripts that were classified by the functional databases, and those that were not, can be listed (Supplementary Figure S5). As we mentioned before, because the reference databases are not comprehensive, we found that only around 30% of all the transcripts that could putatively be classified by the functional databases (“mRNA” and “Revise” categories), were actually classified (49 contigs; Supplementary Figure S5A). To determine the functional profile of the remaining 70% (122 contigs; Supplementary Figure S5B), functional assignment of these transcripts can be determined individually on the basis of the homology search results and then entered manually in the database. To determine an order of priorities and help reduce this considerable workload, contigs can be viewed according to coverage (Supplementary Figure S5B).
All these commands are included in analysing_taxonomy.txt and can be adapted to cover other interests.
Notes
- Due to a question of file size we exemplified the use of our workflow with the assembled reads (737 contigs), but we have also used HoSeIn to analyse our reads (~300,000) and it works seamlessly.
- This workflow was originally developed to analyse high-throughput metatranscriptomic sequences, but we have also used it to analyse high-throughput metagenomic sequences. Moreover, we validated our workflow by analysing a mock metagenome (BMock12) (Sevim et al., 2019) and comparing the results we obtained with those reported for the synthetic metagenome (Sevim et al., 2019). This validation was included in the study in which we presented the analysis of the dataset used for this tutorial, which was recently accepted for publication (Rozadilla et al., 2020), and is included here as a Supplementary Analysis (Supplementary Analysis_mock metagenome.docx). In summary, we contrasted our results with those reported by Sevim et al. (2019) (Table S1) and found that our workflow not only identified all the members of the mock metagenome, but also that the number of contigs that we identified per community member was greater (or the same, but never lower) than what the authors reported (Table S1). In conclusion, our workflow enabled us to identify all the community members of the mock metagenome with greater sensitivity than what was previously reported.
- Even though our workflow has quite a few manual steps, these are comparable to the number of steps used by taxonomy-dependent alignment-based methods to classify and label reads from metatranscriptomic/metagenomic datasets. There are bioinformatic workflows for metatranscriptomic datasets which aim to streamline some of this complexity by connecting multiple individual tools into a workflow that can take raw sequencing reads, process them and provide data files with taxonomic identities, functional genes, and/or differentially expressed transcripts (Shakya et al., 2019). Nevertheless, to define the taxonomic and functional assignments, these platforms perform their sequence-based searches against either protein or nucleotide databases, not both (Shakya et al., 2019). As has already been mentioned, searches against protein databases enable the detection of distantly related organisms but are liable to false discovery, whereas searches against nucleotide databases are more specific but are unable to identify insufficiently conserved sequences. For this reason, analyses of metatranscriptomes using these streamlined workflows must be carefully interpreted. Another major drawback is that several of these workflows assign taxonomy by searching against databases that are designed for functional characterisation (Shakya et al., 2019).
- Summary of the unique innovations in the HoSeIn workflow:
- All the available information for each sequence is assembled and integrated in a local database, from both homology searches and from whatever method was used to classify and label the sequences, and it can be easily viewed and analysed.
- The taxonomic profile of the sample is defined by comparing the taxonomic assignments from both homology searches for each sequence following the LCA logic; i.e., the taxonomic assignment level of a sequence is the one found in common for both homology search results, or for the only result if it returns no hits in the other homology search.
- Consequently, the novelty of our workflow is that final assignments integrate results from both homology searches, capitalising on their strengths, and thus making them more robust and reliable. For metatranscriptomics in particular, where results are difficult to interpret, this represents a very useful tool.
- The functional profile is defined by first assigning transcripts and then integrating all the functional information in a single column (in the local database). What we have observed is that functional databases currently are only able to classify ~30% of all the transcripts that can putatively be functionally classified. To the best of our knowledge, the functional information for the remaining two thirds of those transcripts remains unresolved in other existing tools. In contrast, with our workflow the functional assignment of these transcripts can be determined based on the homology search results (which are included in the local database), thus providing a much more complete and detailed functional profile.
- All the available information for each sequence is assembled and integrated in a local database, from both homology searches and from whatever method was used to classify and label the sequences, and it can be easily viewed and analysed.
Acknowledgments
This research was supported by Agencia Nacional de Promoción Científica y Tecnológica (PICT PRH 112 and PICT CABBIO 3632), and Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) (PIP 0294) grants to CBM. CBM is a member of the CONICET research career. GR and JMC are the recipients of CONICET fellowships. This paper was derived from McCarthy et al. (2013) and Rozadilla et al. (2020).
Competing interests
The authors declare no competing interests.
References
- Aguiar-Pulido, V., Huang, W., Suarez-Ulloa, V., Cickovski, T., Mathee, K. and Narasimhan, G. (2016). Metagenomics, metatranscriptomics, and metabolomics approaches for microbiome analysis. Evol Bioinform Online 12(Suppl 1): 5-16.
- Altschul, S. F., Gish, W., Miller, W., Myers, E. W. and Lipman, D. J. (1990). Basic local alignment search tool. J Mol Biol 215(3): 403-410.
- Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., Davis, A. P., Dolinski, K., Dwight, S. S., Eppig, J. T., Harris, M. A., Hill, D. P., Issel-Tarver, L., Kasarskis, A., Lewis, S., Matese, J. C., Richardson, J. E., Ringwald, M., Rubin, G. M. and Sherlock, G. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25(1): 25-29.
- Blake, J. A., Christie, K. R., Dolan, M. E., Drabkin, H. J., Hill, D. P., Ni, L., Sitnikov, D., et al. (2015). Gene Ontology Consortium: going forward. Nucleic Acids Res 43(Database issue): D1049-1056.
- Buchfink, B., Xie, C. and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nature Methods 12(1): 59-60.
- Finn, R. D., Attwood, T. K., Babbitt, P. C., Bateman, A., Bork, P., Bridge, A. J., Chang, H. Y., Dosztanyi, Z., El-Gebali, S., Fraser, M., Gough, J., Haft, D., Holliday, G. L., Huang, H., Huang, X., Letunic, I., Lopez, R., Lu, S., Marchler-Bauer, A., Mi, H., Mistry, J., Natale, D. A., Necci, M., Nuka, G., Orengo, C. A., Park, Y., Pesseat, S., Piovesan, D., Potter, S. C., Rawlings, N. D., Redaschi, N., Richardson, L., Rivoire, C., Sangrador-Vegas, A., Sigrist, C., Sillitoe, I., Smithers, B., Squizzato, S., Sutton, G., Thanki, N., Thomas, P. D., Tosatto, S. C., Wu, C. H., Xenarios, I., Yeh, L. S., Young, S. Y. and Mitchell, A. L. (2017). InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res 45(D1): D190-D199.
- Glass, E.M. and Meyer, F. (2011). The metagenomics RAST server: A public resource for the automatic phylogenetic and functional analysis of metagenomes. In: Handbook of Molecular Microbial Ecology I: Metagenomics and Complementary Approaches, BioMed Central 9(1): 325-331.
- Huson, D. H., Auch, A. F., Qi, J. and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res 17(3): 377-386.
- Huson, D. H., Mitra, S., Ruscheweyh, H. J., Weber, N. and Schuster, S. C. (2011). Integrative analysis of environmental sequences using MEGAN4. Genome Res 21(9): 1552-1560.
- Kim, M., Lee, K. H., Yoon, S. W., Kim, B. S., Chun, J. and Yi, H. (2013). Analytical tools and databases for metagenomics in the next-generation sequencing era. Genomics Inform 11(3): 102-113.
- Kotera, M., Moriya, Y., Tokimatsu, T., Kanehisa, M. and Goto, S. (2015). KEGG and GenomeNet, New Developments, Metagenomic Analysis. In: Encyclopedia of Metagenomics: Genes, Genomes and Metagenomes: Basics, Methods, Databases and Tools. Nelson. K. E. (Ed.). Boston, MA, Springer US: 329-339.
- Marchesi, J. R. and Ravel, J. (2015). The vocabulary of microbiome research: a proposal. Microbiome 3: 31.
- Marchler-Bauer, A., Bo, Y., Han, L., He, J., Lanczycki, C. J., Lu, S., Chitsaz, F., Derbyshire, M. K., Geer, R. C., Gonzales, N. R., Gwadz, M., Hurwitz, D. I., Lu, F., Marchler, G. H., Song, J. S., Thanki, N., Wang, Z., Yamashita, R. A., Zhang, D., Zheng, C., Geer, L. Y. and Bryant, S. H. (2017). CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res 45(D1): D200-D203.
- Markowitz, V. M., Chen, I. M., Palaniappan, K., Chu, K., Szeto, E., Grechkin, Y., Ratner, A., Jacob, B., Huang, J., Williams, P., Huntemann, M., Anderson, I., Mavromatis, K., Ivanova, N. N. and Kyrpides, N. C. (2012). IMG: the Integrated Microbial Genomes database and comparative analysis system. Nucleic Acids Res 40(Database issue): D115-122.
- McCarthy, C. B., Santini, M. S., Pimenta, P. F. and Diambra, L. A. (2013). First comparative transcriptomic analysis of wild adult male and female Lutzomyia longipalpis, vector of visceral leishmaniasis. PLoS One 8(3): e58645.
- McCarthy, C. B., Cabrera, N. A. and Virla, E. G. (2015). Metatranscriptomic Analysis of Larval Guts from Field-Collected and Laboratory-Reared Spodoptera frugiperda from the South American Subtropical Region. Genome Announc 3(4): e00777-15.
- Ogata, H., Goto, S., Sato, K., Fujibuchi, W., Bono, H. and Kanehisa, M. (1999). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 27(1): 29-34.
- Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., Edwards, R. A., Gerdes, S., Parrello, B., Shukla, M., Vonstein, V., Wattam, A. R., Xia, F. and Stevens, R. (2014). The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res 42(Database issue): D206-214.
- Pearson, W. (2004). Finding protein and nucleotide similarities with FASTA. Curr Protoc Bioinformatics Chapter 3: Unit3 9.
- Powell, S., Szklarczyk, D., Trachana, K., Roth, A., Kuhn, M., Muller, J., Arnold, R., Rattei, T., Letunic, I., Doerks, T., Jensen, L. J., von Mering, C. and Bork, P. (2012). eggNOG v3.0: orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res 40(Database issue): D284-289.
- Rozadilla, G., Cabrera, N. A., Virla, E. G., Greco, N. M. and McCarthy, C. B. (2020). Gut microbiota of Spodoptera frugiperda (J.E. Smith) larvae as revealed by metatranscriptomic analysis. Journal of Applied Entomology n/a(n/a). doi.org/10.1111/jen.12742.
- Sevim, V., Lee, J., Egan, R., Clum, A., Hundley, H., Lee, J., Everroad, R. C., Detweiler, A. M., Bebout, B. M., Pett-Ridge, J., Goker, M., Murray, A. E., Lindemann, S. R., Klenk, H. P., O'Malley, R., Zane, M., Cheng, J. F., Copeland, A., Daum, C., Singer, E. and Woyke, T. (2019). Shotgun metagenome data of a defined mock community using Oxford Nanopore, PacBio and Illumina technologies. Sci Data 6(1): 285.
- Shakya, M., Lo, C. C. and Chain, P. S. G. (2019). Advances and challenges in metatranscriptomic analysis. Front Genet 10: 904.
- Tatusov, R. L., Galperin, M. Y., Natale, D. A. and Koonin, E. V. (2000). The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28(1): 33-36.
- Wooley, J. C., Godzik, A. and Friedberg, I. (2010). A primer on metagenomics. PLoS Comput Biol 6(2): e1000667.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Rozadilla, G., Moreiras Clemente, J. and McCarthy, C. B. (2020). HoSeIn: A Workflow for Integrating Various Homology Search Results from Metagenomic and Metatranscriptomic Sequence Datasets. Bio-protocol 10(14): e3679. DOI: 10.21769/BioProtoc.3679.
Category
Systems Biology > Genomics > Phylogenetics
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.