- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Preparation of Drosophila Polytene Chromosomes, Followed by Immunofluorescence Analysis of Chromatin Structure by Multi-fluorescence Correlations
Published: Vol 10, Iss 13, Jul 5, 2020 DOI: 10.21769/BioProtoc.3673 Views: 10226
Reviewed by: Imre GáspárRumen IvanovAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2019
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Drosophila larval salivary gland polytene chromosome squashes have been used for decades to analyze genome-wide protein-binding patterns, transcriptional activation processes, and changes in chromatin structure at specific genetic loci. There have been many evolutions of the squashing protocol over the years, with sub-optimal reproducibility and low sample success rate as accepted caveats. However, low sample success rates are an obvious disadvantage when polytene chromosomes are used for more high-throughput approaches, such as genetic or antibody screens, or for experiments requiring high-quality chromosome structure preservation. Here we present an exceptionally reproducible squashing and fluorescence staining protocol, which generates high-quality fluorescence images on well-spread chromosomes. This is followed by our novel, semi-automated MATLAB analysis program used to determine correlations between fluorescence signals of interest at a single site on polytene chromosomes, in a pixel-by-pixel manner. In our case, we have used this approach to assess chromatin changes at genomic sites, ectopically targeted by nuclear pore proteins. The use of our analysis program increases the ability to make unbiased conclusions on changes in chromatin structure, or in protein recruitment to chromatin, regardless of sample variation in immunofluorescence staining. As it is simply based upon differences in fluorescence intensity at a defined location, the provided analysis program is not limited to analysis of polytene chromosome, and could be applied to many different contexts where correlation between fluorescent signals at any particular location is of interest.
Keywords: Polytene chromosome squashesBackground
Polytene chromosomes from Drosophila larval salivary glands have an unusually large size (visible under a typical cell-culture microscope). This is due to altered cell cycle processes that result in ~1,000 copies of each chromosome that do not segregate from each other. The resulting large chromosomes have stereotyped and reproducible alternating bands of condensed and decondensed chromatin, as chromosomes are aligned and locus-specific chromatin structure is visible upon polytenization. The nature of these chromosomes makes the analysis of chromatin structure uniquely visible relative to diploid cell types, enabling analysis of chromatin changes throughout development, transcriptional activation, or upon perturbation. Additionally, genome-wide and site-specific chromatin binding of proteins can be visualized by immunofluorescence staining of polytene chromosomes (Paro, 2008; Cai et al., 2010), allowing for investigation of the relationships between candidate proteins and chromatin condensation, transcriptional activity, and histone modifications. In this manner, polytene chromosome staining provides a rapid and inexpensive alternative or supplement to molecular assays such as Chromatin Immunoprecipitation, coupled to sequencing (ChIP-seq).
Our squashing and fluorescence analysis pipeline was specifically designed for analysis of chromatin structure and protein recruitment/changes at single bands of interest, originally for a band of LacI-tethered protein bound to a genomic lacO repeat array (Kuhn et al., 2019). In this study, we targeted specific nuclear pore components, fused to LacI, to lacO arrays, integrated at defined sites in the genome. We then aimed to determine whether such targeting leads to recruitment of candidate interacting partners, and also to changes in target chromatin compaction. This approach involved identification of a single band of interest (harboring the lacO site) within an entire genome of spread chromosomes, which required high-quality chromosome spreading for increased visibility and analysis of chromosome banding patterns. Additionally, screening through multiple antibodies to assay for many candidate interacting partners required an extremely reproducible squashing protocol, with minimal sample failure rate. To achieve these goals, we developed this optimized polytene squashing protocol, which can be utilized to improve image quality and sample success rate. It is also especially useful for chromosome ‘mappability’ (determining genomic location by cytological mapping of polytene banding patterns) and for more high-throughput applications.
In order to reliably analyze site-specific changes in chromatin structure (i.e., changes in chromatin condensation as assessed by DNA stain fluorescence intensity), and in protein recruitment, we developed a semi-automated MATLAB function. This function, PCCcalc, calculates the Pearson Correlation Coefficients (PCC) between multiple fluorescence signals at a defined genomic site on a pixel-by-pixel basis in an unbiased manner. This greatly reduces data analysis time and subjectivity derived from manual analysis and user-based variability. Additionally, this approach circumvents the impact of slide-to-slide variability in fluorescence background or image acquisition, as the analysis focuses on the relationship between fluorescence signals more than absolute values of a given signal. This method can be used on any easily identifiable site on polytene chromosomes to determine binding correlations between multiple protein factors, or to find correlations between protein of interests and associated changes in chromatin compaction. Furthermore, we believe this analysis method can be extended to determine fluorescence correlation beyond the described use in polytene chromosomes, and can be used to analyze co-localization or co-occurrence of fluorescently labeled biological markers in many other contexts and cell types.
Materials and Reagents
- Filter paper (Fisherbrand, catalog number: 05-714-4 )
- Hammer with a rubber head (Graham Field, catalog number: 1312-1 )
- Fine sharpie
- 1.5 ml tubes (USA Scientific, catalog number: 1615-5510 )
- Dissection Well Dish/Pyrex Spot Plate (Fisher Scientific, catalog number: 13-748B )
- Dumont Fine #55 Forceps (Fine Sceince Tools, catalog number: 11295-51 )
- Coverslips (Fisherbrand, catalog number: 12-542A )
- Coplin Jar (VWR, catalog number: 74830-150 )
- Humidity chamber (such as Pyrex Lidded Glass Baking Dish)
- Poly-L-Lysine-treated Slides (Polysciences, catalog number: 22247 )
- Poly-L-Lysine (alternative to pre-treated slides above) (Sigma-Aldrich, catalog number: P8920-100 ml )
- Sigmacote (Sigma-Aldrich, catalog number: SL2-100ML )
- Phosphate Buffered Saline (PBS) reagents:
- NaCl (Fisher, catalog number: BP358-212 )
- KCl (Fisher, catalog number: BP366-500 )
- Na2HPO4 (Sigma, catalog number: S3264-500G )
- KH2PO4 (Sigma, catalog number: P9791-1KG )
- Primary Antibodies used: anti-LacI (Rockland, catalog number: 600-401-B05 ), anti-Nup98 (Capelson et al., 2010), anti-Brm (Nakayama et al., 2012)
- Tween 20 (Fisher, catalog number: BP337-500 )
- Glacial Acetic Acid (Fisher, catalog number: BP2401-212 )
- 16% Paraformaldehyde (PFA) (Alfa Aesar, catalog number: 43368 )
- Liquid nitrogen
- Kimwipes or other tissue-style wiper (Fisher Scientific, catalog number: 06-666A )
- Fluorescent Secondary Antibodies (e.g., Thermo Fisher, catalog number: A-11004 )
- Hoechst 33342 (Thermo Fisher, catalog number: H3570 ) (10 mg/ml stock)
- Prolong Gold Antifade (Thermo Fisher, catalog number: P36930 )
- PBS (see Recipes)
- PBST (see Recipes)
- Fixation solution (see Recipes)
- 45% acetic acid (see Recipes)
- Blocking solution/Antibody cocktail base (see Recipes)
Equipment
- Forceps
- Pipettes
- Chemical hood
- Dissecting Microscope (such as Leica, model: Leica S6E )
- Cell Culture/Phase Contrast Microscope (such as Leica, model: Leica DM IL )
- Widefield epifluorescent microscope (such as Leica, model: Leica DM6000 ) and/or laser scanning confocal microscope
- (Optional) Orbital shaker (VWR, catalog number:12620-938)
Software
- MATLAB (license) (necessary for analysis)
- Graphing/Statistical Analysis software, e.g., Excel, Prism (licenses) (necessary for analysis)
- (Optional) ImageJ (free)
Procedure
- Prior preparation of Sigmacote treated coverslips and poly-L-lysine treated slides
Note: Coverslips are treated with Sigmacote to prevent sticking of chromosomes and to encourage preferential adherence of chromosomes to poly-L-lysine treated slides for subsequent staining and imaging. These are best if treated prior to the day of the experiment.
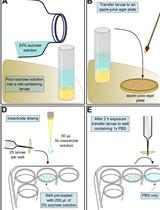
Sigmacote treated coverslips- Wrap a 1.5 ml tube rack (or similar structure) in plastic wrap and set on a larger rectangle of paper towels in a chemical hood, see Figure 1.
- Pour Sigmacote (stored at 4 °C) into the Sigmacote bottle’s cap.
- Using clean, fine forceps, dip the coverslips individually into Sigmacote, completely submerging. This is best with curved/bent tips so that the forceps are minimally touching the coverslip (see Step B7 below).
- Promptly remove the coverslip, touch the corner of the coverslip to the surface of the liquid in the Sigmacote bottle’s cap to remove excess, and place the coverslip at ~60° angle against the plastic-covered rack on top of the paper towel so any remaining Sigmacote wicks away from coverslips (see Figure 1).
- Once the desired number of coverslips is made, return the Sigmacote from the cap into the Sigmacote bottle, store at 4 °C, use within 1 year.
- Leave coverslips to dry overnight in a chemical hood (can use after ~1 h if time is limited).
- Store coverslips in 4 °C (stacked back in the original container is fine after overnight drying), best if used within ~2 months.

Figure 1. A simple lab set-up for drying coverslips after Sigmacote treatment. Coverslips to be used for polytene chromosome squashes are treated with Sigmacote and placed leaning against a 1.5 ml tube rack wrapped in plastic wrap, placed on paper towels, inside of the chemical hood, as described in A1-A4 above.
Poly-L-lysine treated slides
Poly-L-lysine slides can be purchased pretreated (we found pretreated slides to have lower autofluorescence background compared to self-treated slides), but to save costs, or if pre-treated slides are unavailable, standard frosted microscope slides can be treated with poly-L-lysine similarly to treating coverslips with Sigmacote:- Pour poly-L-lysine into a glass Coplin jar.
- Insert frosted microscope slides so that the non-frosted glass part of the slide (i.e., excluding frosted label area) is submerged in the Poly-L-lysine.
- Once the Coplin jar is filled with slides, remove the slides in original order in which they were treated, and place at a ~60° angle against a vertical surface (such as conical rack) on paper towels.
- Let slides dry overnight, minimum ~2 h if in a rush.
- Store at 4 °C (stacked back in original slide box is fine after overnight drying), best if used within ~2 months.
- Gland Dissection and Chromosome squashing
Before beginning dissections, gather all materials, make fixation solutions, and fill an appropriate container with liquid nitrogen.- Pick ideal 3rd instar wandering larvae and place up to 4 in a glass dissection well dish in PBS.
Note: Picking larvae is the most important step for getting highest quality squashes! The 3rd instar wandering larvae that you use for harvesting salivary glands should be:- From un-crowded vials/bottles–ideal larvae have had low competition for nutrients and are fat and healthy.
- Late wandering 3rd instar, minimally moving, but not yet in pre-pupa stage, as older larvae (primarily moving only mouth) will have larger chromosomes and give the best squashes. 1)Larvae still quickly moving around vial/bottle walls will be younger and have had fewer polytene chromosome replications and therefore smaller chromosomes.2)Larvae too close to pupation/pre-pupa stage will however have too much salivary gland "glue" protein production, and this can lead to difficult spreading and increased immunofluorescence background. To avoid this, large "glassy" or more transparent salivary glands (which are full of the glue-like substance that releases upon puncturing), (see Figure 2) should be skipped for best quality squashes. Never take larvae with anterior spiracles extended, as they will almost always have these types of glands.
- From the very first wave of wandering larvae that usually show up 6-7 days after the vial/cross has been set up or flipped (if reared at 21-22 °C room temperature).
Quality squashes can come from larvae taken on the second day of wandering in uncrowded conditions, but salivary glands and chromosomes are generally largest from larvae from the very first wave, so success rate/quality will be highest from first-wave larvae. - Females (unless interested in sex-specific differences). Female larvae tend to have larger salivary glands and polytene chromosomes than male larvae. Female sex of 3rd instar larvae is determined by the absence of large, transparent, spherical gonads visible on either side of the interior of larvae, about 1/3 up from the posterior tip of the larvae. These large gonads are instead indicative of males.
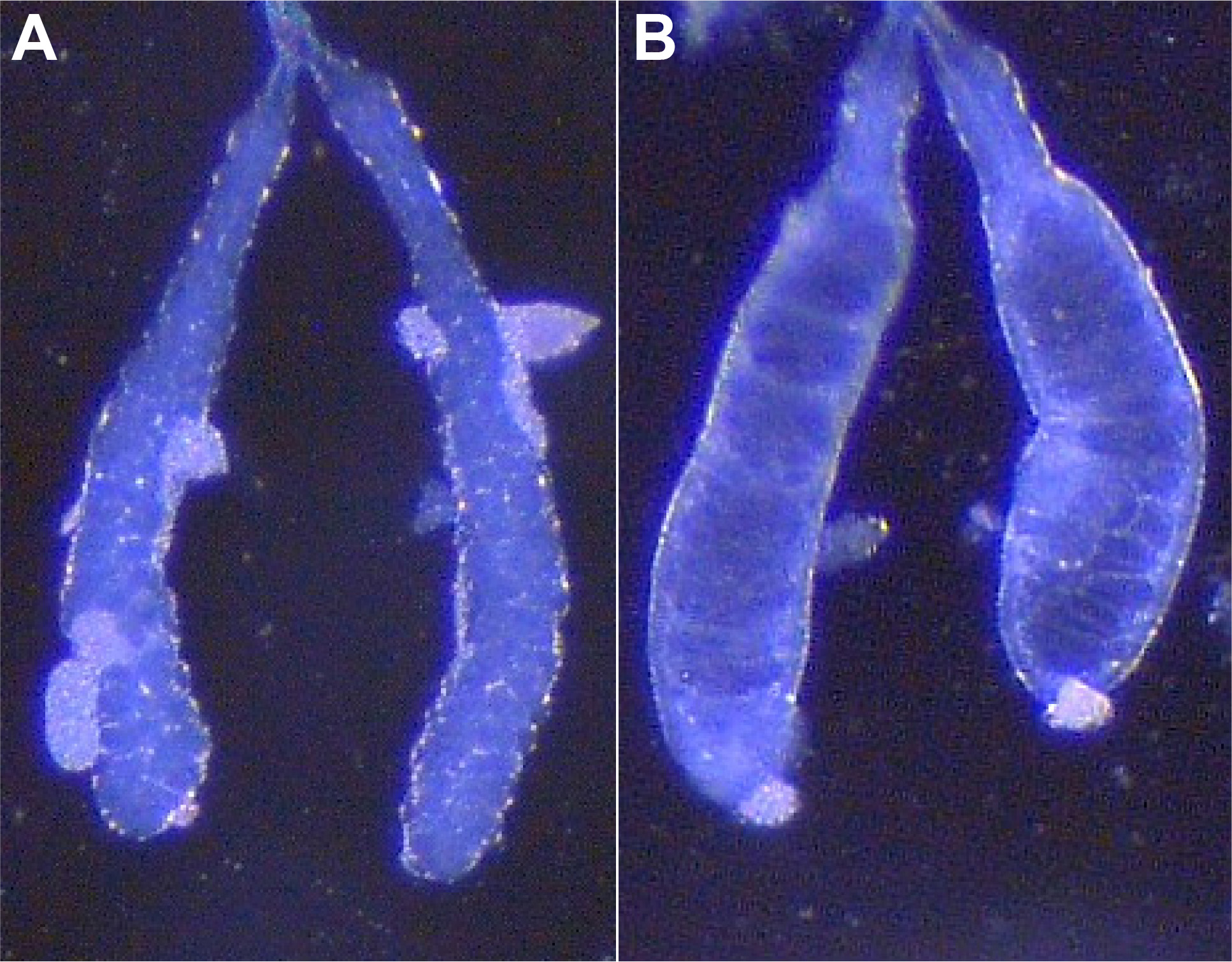
Figure 2. Different types of salivary glands isolated from 3rd instar wandering larvae. A. Typical set of salivary glands, usually opaque/“milky” in appearance, with well-defined cells visible. B. Mature glands to be avoided, taken from larva close to pupation, appearing much more clear/transparent/”glassy” due to increased glue substance, usually with less visible cellular borders. White pieces attached to glands are remaining fat body.
- Transfer one larva into a new well with PBS for dissection. Replace the PBS from this well after every 2-3 dissections.
- Dissect out salivary glands by holding larval mouth hooks with one pair of forceps and gently pulling larva in the other direction from about halfway down their body with another pair of forceps. (For demonstration of salivary gland dissections from Drosophila larvae, there are a number of public videos available, available here)
- Peel off the fat bodies (white strips of tissue) from the sides of glands, as this can contribute to fluorescence background in downstream imaging; however don’t spend more than a couple of minutes removing these if they are persistent, it is better to proceed to fixation sooner. Dissections should not proceed longer than ~5 min before fixation.
- Transfer the glands to a different dissection dish well containing ~100-200 μl of freshly made fixation solution and leave for precisely 1 min.
- Glands may be held by the “wishbone”/stem area where they are adjoined to transfer to fixative solution, as this prevents damaging gland cells.
- Transferring glands here can be made much easier by the use of a pair of forceps with one bent/curved tip, described in Step B7 below.
- Acetic acid serves here to disrupt nuclear membranes and aid in chromosome spreading, however not all antibodies work well under these conditions, and some may require optimization in different fixation lengths or solutions, see Note in Section C.
- In our hands, all acetic acid solutions must be made fresh every 30-45 min from 100% acetic acid to ensure proper chromosome spreading, as acetic acid dilutions go bad quickly and will make chromosomes much more difficult to spread.
- Additionally, once opened, it is critical that 16% PFA is stored at 4 °C (generally transferred from glass ampule to 15 ml conical tube) and ONLY used for 3 weeks for optimal chromosome spreading and structure preservation.
- Important! From this point forward, work quickly, yet carefully, as fixation will continue until slide is frozen in liquid nitrogen (below). Best to prepare the coverslip and label the slide during the fixation incubation above.
- Wipe any residue from the Sigmacote-treated coverslip using a kimwipe or other tissue-style wiper, add 7 μl of fresh 45% acetic acid onto the coverslip, and gently transfer the fragile glands into the drop of acetic acid.
- Glands become incredibly fragile after fixation and will require finesse to transfer.
- Highly recommended for this step is a pair of forceps with one bent tip, such that it forms a curved or hooked shape at the end facing toward the other tip, preventing complete closure of the forceps. These can pick up the glands in a small drop of liquid at the tip, and prevents glands from being squished and sticking to the forceps. These can be used for all transferring steps, but are especially necessary after fixation when glands become fragile.
- Lower the poly-L-lysine treated slide gently onto the drop of acetic acid so that the glands end up approximately in the middle of the slide.
- Flip over the slide and, using a gloved index finger, apply pressure on the coverslip to simultaneously push the coverslip “into” the slide while also moving the coverslip around in a circle, ~1 cm in each direction in a clockwise motion, 3 times around, to break apart the tissue and spread the cells and chromosomes (see Video 1 for Steps B9-B14).
- It is important to always go in a circle in one direction to prevent chromosome tangling.
- The coverslip should slide relatively smoothly and not get stuck. If sticking occurs, see the following Note.
- Optimization and practice will be required at this step for each individual to learn the appropriate movements and amount of finger-pressure to get their best spreads. Figure 3A shows an example of what chromosomes look like when more pressure is needed from this step.
Things that may help prevent coverslip sticking:- Avoid clear, “gluey” glands (as discussed above in Step B1, these appear glass-like/transparent) (see Figure 2).
- Use freshly made, completely dry (best overnight), and fully wiped-off Sigmacote-treated coverslips, made with fresh Sigmacote (the same applies to poly-L-lysine treated slides if you are treating them yourself).
- Use fresh acetic acid solutions made within 30-45 min of use.
 Video 1. Polytene Chromosome Squash
Video 1. Polytene Chromosome Squash
- Flip the slide over so the coverslip is on the bottom and carefully insert the slide into a folded piece of thick filter paper.
- Hold two fingers on either side of where the coverslip is located to secure the slide and prevent lateral movement between slide and coverslip.
- Tap the coverslip area with a rubber hammer with medium force ~30 times. This step will take practice and the amount of force used will need to be optimized for each individual.
- Push straight down on the slide with two thumbs firmly on either side of the coverslip, without letting the coverslip slip relative to the slide (see Video 1), in order to flatten the chromosomes and prevent 3D/out-of-focus chromosome structure when imaging (Figures 3A and 3C).
- Remove the slide out from filter paper, and mark the location of the corners of the coverslip on the back of the slide with fine sharpie in order to locate the chromosome area in future antibody incubations and mounting.
- Check chromosome spreading quality under a cell culture/phase contrast microscope, under 20x or 40x objective. If spreading is satisfactory (Figures 3D-3E), proceed. Otherwise, throw the slide away and repeat with the next larva, making sure to change out PBS in the dissection well after every 2-3 larvae.
Note: Quality control of chromosome spreads:- A good chromosome spread should have chromosomes that fan out, are somewhat separated, are intact and not stretched/torn, and are flat (Figures 3D-E).
- If slides show most of the chromosomes torn/stretched (Figure 3B), chromosomes very tangled together (Figures A-B), or nuclei not adequately broken with chromosomes still in a tight ball (Figure 3A), it is usually very difficult to get useable data from these and it is most often a waste of time and resources to proceed with substandard slides.
- With this protocol, once one has practiced, we generally have a much higher success rate than with previously attempted protocols, up to 80-90% for experienced individuals starting with ideal larvae. However, you may still throw several slides away at this stage. Because of the innate variability in squashing and staining, you want to end up with a minimum of 3 quality slides per condition (genotype/antibody/etc.).
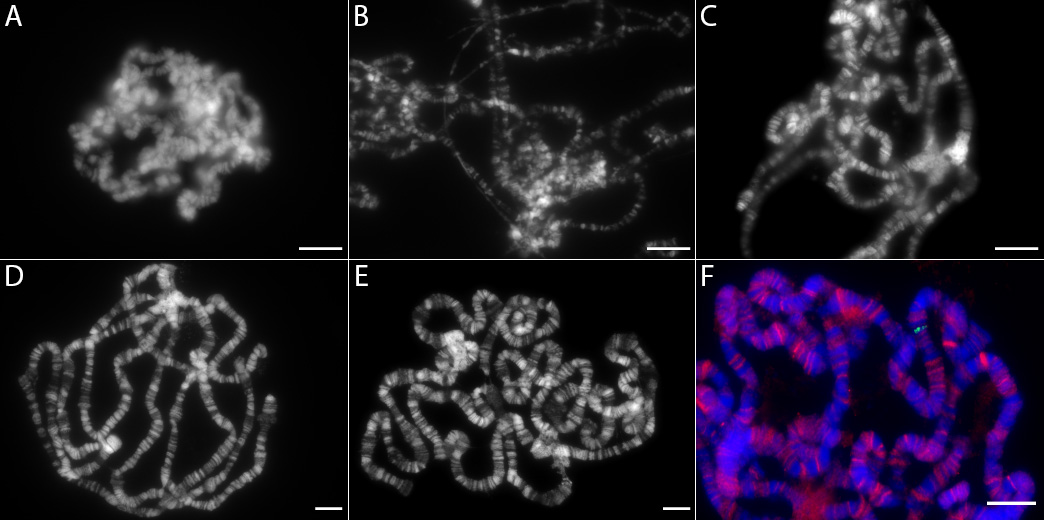
Figure 3. Examples of poor and high quality polytene chromosome squashes. A. Under-spread squash, evident by the lack of chromosomes spreading out from the original round nuclear shape, and under-flattened squash, evident by chromosomes present in multiple focal planes, i.e., blurry and in-focus chromosomes visible simultaneously. Common fixes: To increase chromosome spreading, more pressure must be used during coverslip movement in Step B9; to decrease “blurriness,” make sure to push the coverslip into the slide with more pressure in Step B13 (avoiding lateral movement of coverslip relative to slide). Scale bar = 10 μm. Chromosomes are stained with Hoechst (shown as white), in A-E. B. Over-stretched chromosomes, visible by thin strands with poorly defined chromatin bands. Chromatin is also broken apart in places, without bands appearing in their stereotyped, organized order. Common fix: Hit the slide less forcefully with the hammer in Step B12, and also make sure to avoid any lateral movement of coverslip relative to the slide throughout the entire squashing and staining process, from hammering to final coverslip mounting. Scale bar = 10 μm. C. Well-spread, but under-flattened chromosomes, evident by chromosomes present in multiple focal planes, i.e., blurry and in-focus chromosomes visible simultaneously. Common fix: See Figure 3A legend regarding “blurriness”. Scale bar = 10 μm. D-E. Ideal Squashes. Characteristics include: flat chromosomes all visible in same focal plane, chromosomes are well spread from each other, with chromosome ends often visible for identification/cytological mapping; however chromosomes are not over-stretched and are roughly even in thickness/width throughout spread. Scale bar = 10 μm. F. Ideal squash and stain. Example includes lacO integration site at cytological site 96C, staining with antibodies against Nup98 (red, originally described in Capelson et al., 2010) and LacI (green, Rockland 600-401-B05 ), recognizing LacI-Sec13 fusion protein bound to lacO 96C, and Hoechst staining (blue). Scale bar = 10 μm. - If the squash is of good quality, proceed by dipping the slide into liquid nitrogen while holding the slide with large blunt forceps (not fine dissection forceps).
- Wait until fizzling/boiling in liquid nitrogen stops, and then remove slide.
- Carefully flip off coverslip with razorblade, taking care to not slide razorblade too far into the area containing the chromosomes as they can be scraped off the slide.
- Immediately place the slide in a Coplin jar containing Wash Solution.
- Collect multiple slides at this step.
- If you are not able to collect all larvae on the same day because of differences in genotype emergence timing etc., slides can instead be kept in a Coplin jar in PBS without Tween at 4 °C overnight (this appears to be compatible with most antibodies).
- When finished dissecting and squashing all slides, incubate the slides in Blocking Solution for 30-60 min.
- This can be done by exchanging the Wash Solution for 50 ml of the Blocking Solution in the Coplin jar containing the slides, or by adding the Blocking Solution directly to the slides, in the same manner as the antibody cocktails, as described below (if trying to conserve BSA or if experimenting with different blocking conditions containing precious reagents etc.)
- Optimize the blocking time for different antibodies if necessary.
- Blocking overnight is not advised, and if storing slides overnight before antibody treatment is necessary, it is preferable to stop at the prior step in PBS, without Tween, as described above.
- Pick ideal 3rd instar wandering larvae and place up to 4 in a glass dissection well dish in PBS.
- Primary antibody treatment
- Collect Blocking Solution from the Coplin jar to make primary antibody cocktail for next step (or use fresh), and save 1 ml (or more if necessary) in 1.5 ml tube at 4 °C overnight for secondary antibody cocktail(s) for the following day.
- Make antibody cocktail(s), using 30 μl per slide (and making enough for one additional slide to account for pipetting inaccuracies), with primary antibodies generally starting at a 1/100 (volume/volume) dilution in Blocking Solution.
- Antibody dilutions for polytene chromosome spreads are generally 10x more concentrated relative to Western Blot dilutions.
- Extreme dilutions for particularly weak or strong antibodies can get up to 1/20 or down to 1/500, respectively.
- If there appears to be much nonspecific interband signal, try diluting the antibody.
- Antibody cocktail and other volumes would need to be adjusted if different sized coverslips are used.
Notes:- Fixation time in the standard fixation solution can vary between 30-120 s and may need to be optimized if 1 min fixation leads to antibody staining that is not adequate.
- For some antibodies, especially those against antigens that may be somewhat extracted from chromatin by this acetic acid-based fixation method (such as histone proteins, for instance), a different fixation strategy may be required in order to better preserve the target epitope, such as the following: 1)Glands are first pre-fixed for 30 s in 2% PFA alone.2)Glands are then transferred to 2% PFA/45% Acetic Acid for 2 min.3)Squash/spread glands in a drop of 45% acetic acid as described previously.4)After freezing slide in liquid nitrogen and removing coverslip, place directly into cold 70% ethanol at -20 °C for a minimum of 30 min.
For many antibodies, glands can be left overnight in ethanol and staining will be unaffected.5)Once all slides are collected and have been in ethanol at least 30 min, wash twice with PBST at room temperature to completely remove ethanol and proceed with staining normally.6)The timing of these steps, as well as solution strengths, may need to be optimized for various antibodies.
- Remove the slide from the Coplin jar, carefully remove excess PBST from the slide in the area outside the sharpie-outlined square by using a kimwipe or other tissue-style wiper, and pipette 30 μl of the antibody cocktail onto the outlined area.
- Carefully lower down an untreated coverslip onto the outlined area containing the antibody cocktail, using a pair of fine forceps, starting with coverslip at an angle at one edge of the outlined area and slowly lowering down/flattening the coverslip out, avoiding bubbles.
- Gently place slide on top of a slightly wet (but not soaking) paper towel in a closeable humidity chamber.
- Once all slides have been treated with primary antibodies, covered with coverslips, and placed in the humidity chamber, carefully close the chamber and gently place somewhere flat and undisturbed at 4 °C overnight.
a.Drying out of the tissue should be avoided at all costs as it leads to high background and poor staining.
b.Some antibodies can be incubated at room temperature for 3 h instead of overnight at 4 °C.
- Washing and secondary antibody treatment
- Carefully slide coverslips from slides using a gentle movement with a gloved finger in a direction parallel to the slide, and wash slides by putting them in a Coplin Jar containing Wash Solution.
Note: If coverslips do not easily slide off with minimal friction and effort, the antibody cocktail may have dried out and that slide may not be useable, so take note and compare to other slides under same conditions when imaging to ensure quality. - Wash the slides 10 min in Wash Solution, 3 times, either stationary on a bench top, or gently mixing on an orbital shaker at 100 rpm.
- Introduce fluorescently conjugated secondary antibodies (1/300 is a good working dilution, adjust as needed) in the same manner as with the primary antibodies, and incubate in a closed humidity chamber for 1h at room temperature in the dark (a bench drawer works well, just close and open slowly).
- Carefully slide coverslips from slides using a gentle movement with a gloved finger in a direction parallel to the slide, and wash slides by putting them in a Coplin Jar containing Wash Solution.
- Hoechst/DAPI treatment and coverslip mounting
- Carefully remove the coverslips and wash the slides again with Wash Solution 3 x 10min in the dark.
- Stain slides with Hoechst or DAPI (1/1,000 in PBS) for 2 min by adding 100 μl of the Hoechst/PBS cocktail onto the outlined area (do not cover with coverslip) and protecting slides from light, for example by covering with a tube rack cover lined with foil.
- Wash slides in PBS without Tween for 10 min in the dark.
- Remove excess liquid from around the outlined area of each slide with a wipe, and add 5-10 μl of Prolong Gold Anti-fade (or similar mounting medium) in one quadrant of outlined area.
- Carefully lower untreated coverslip onto the outlined area, and avoid bubbles by angling the coverslip so it contacts the quadrant with the mounting medium first, and then slowly lower to the rest of outlined area until flat.
- Carefully place nail polish on the coverslip corners to secure the slide, cover the coverslips, and wait ~5 min to allow the nail polish to dry.
Note: Any forceful movement of the coverslip at this stage, or at any point after the chromosome fixation and flash freezing, will lead to chromosome stretching and tears, and should be avoided at all costs. - When the nail polish at the coverslip corners is dry and the coverslip does not move, carefully seal the coverslip completely along the edges with the nail polish.
Note: If time is not a concern, letting slide dry for 30 min will allow the mounting medium (if a hardening medium) to dry and make coverslips even more secure before completely sealing. - Let the nail polish dry for a minimum of ~10 min (covered) and proceed to imaging, or store at 4 °C.
- Quality of slides will be best if imaged within one week (some antibodies will persist longer but the fluorescence signal of secondary antibodies will diminish over time).
- Imaging
- Image slides using a 60x objective lens for a large field of view, or a 100x objective lens with 1.6x magnification for the greatest zoom, using exposure levels or laser intensities low enough that no channels are overexposed, but such that brightest pixels are still bright.
- Image all slides under the same acquisition conditions, determined by setting the imaging settings on the brightest samples.
- Imaging on widefield microscopes is sufficient for most purposes, and will be less likely to photobleach the samples than using a laser scanning confocal will; however fine-scale chromatin structure, and also signal from some weaker antibodies, are better resolved using confocal imaging.
- Collect images of 10+ different sets of chromosomes per slide, and image 3 slides per condition/genotype.
- If saving files in proprietary formats such as .lif (Leica) or .czi (Zeiss), make sure to split the channels and save into tiff files prior to analysis in MATLAB by using a program such as ImageJ etc.
Note: Otherwise, save tiff images as separate channels directly using image acquisition software before concluding image collection.
Data analysis
Here we present instructions for running the custom MATLAB program PCCcalc (see Supplement File 1) to analyze fluorescence intensity correlations. The program calculates the Pearson Correlation Coefficient (PCC) on a pixel-by-pixel basis between each pair of channels in three-channel images of chromosome squashes. In these examples, channels are Hoechst (or DAPI), a LacI-tethered protein at a single lacO integration site (the “tester” channel), and a “target” channel representing a protein or complex whose enrichment at the lacO integration site is being assessed (Figure 3F). The LacI-tethered protein forms a bright band on the polytene chromosome. PCCcalc uses the LacI signal to create a threshold image mask. The pixels defined by the mask are used to assess the correlation of fluorescence intensities. Files/images of the individual channels of the composite images used as an example are included as Supplement Files 2, 3, 4 [which are images of immunofluorescent staining with Hoechst (blue), Lac-I (green), and Brm (red, originally described in Nakayama et al., 2012)] for input into the PCCcalc function of MATLAB to follow along with the protocol referenced in this section.
PCCcalc is designed to be executable by a novice. It can be used to measure intensity correlations in any three-channel data where a mask covering a region of interest can be defined by a single threshold intensity in the tester image. The current code includes the use of the DAPI/Hoechst channel to define the mask, in the event that the tester signal spreads off of chromatin/across multiple bands.
- Add the PCCcalc file to the MATLAB path so MATLAB can access the program, from here on referred to as ‘function’
- The MATLAB path can be determined by inputting “userpath” into the MATLAB command line.
- The name of the PCCcalc file must not differ from the function name (denoted by the word following the = sign within the first line of the function file), or MATLAB will not be able to find and run the function.
- Run the function by inputting into the MATLAB command line the function name, “pearsonCorr = PCCcalc” (without quotations).
- A dialog box will appear, to enable navigation to and selection of a folder containing the image data to be analyzed. The target folder must contain .tif(f) files of images containing individual fluorescence channels, with names containing separate channel designations, for example ch00, ch01, ch02.
- When prompted, enter the channel designations/”strings” corresponding to channel identifiers in the file names.
e.g., ch00 for blue, ch01 for green (tester), ch02 for red. - An RGB image will appear showing Hoechst in blue and the tester in green, with crosshairs. Click on the tester band/band to be analyzed.
- A BW (Black and White) image of the Hoechst (DNA) stain should appear. The displayed image is from a smaller, cropped form generated to focus on the band of interest, and only objects visible in this cropped image will be available for data analysis proceeding forward. (The size of this "plaquette" is hard-coded at 105 x 105 pixels but can be adjusted based on individual needs).
- Press ‘t’ to toggle between this image and a BW image of the tester/green channel.
- Look for the chromatin band in the Hoechst stain corresponding to the chromatin band containing the tester/green signal.
- When ready, press ‘s’ to bring up the Hoechst image and crosshairs.
- Click a spot along the edge of the target Hoechst band, and this will select a pixel whose intensity will be used to generate an initial Hoechst mask.
- A window, the Mask Selection Figure, with four images will appear (Figure 4). The upper images are BW images upon which are superimposed the outlines of the Hoechst (left) and green tester (right) masks. The lower left image shows the Hoechst mask (magenta), the tester mask (green), and the overlap between the two (white). The lower right image is an overlay of the images from the Hoechst (magenta) and tester (green) channels, with the overlapping Hoechst and tester masks outlined in cyan. The region within this cyan boundary is the final candidate region for calculating the correlation coefficient between the fluorescence signals therein. The inclusion of the Hoechst mask ensures that no signals, if any, present off of the DNA will be incorporated in the calculations.
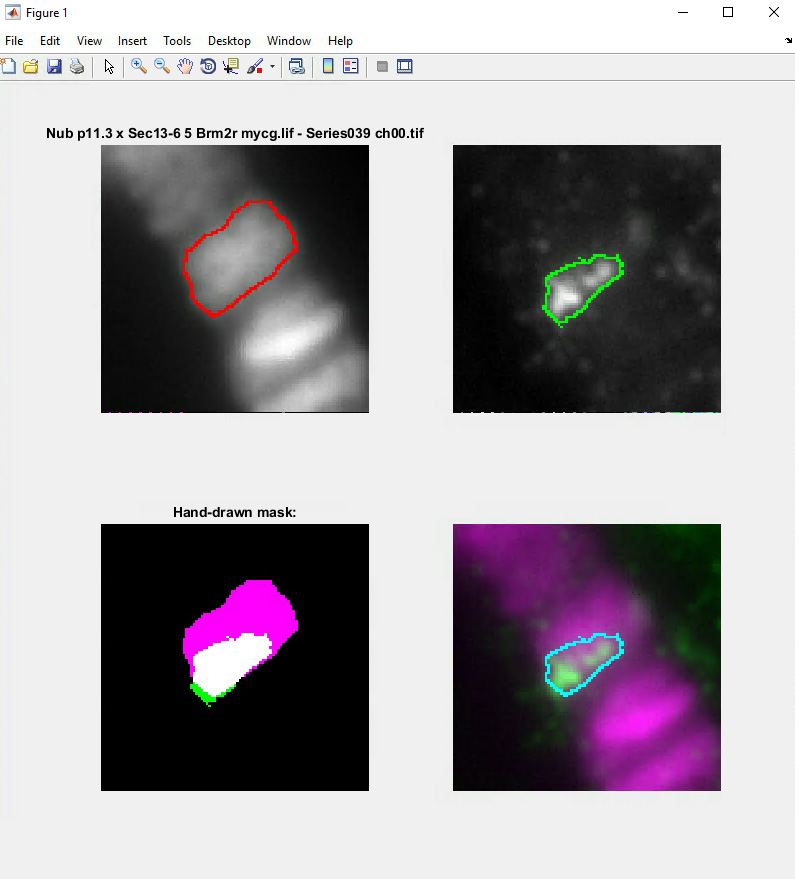
Figure 4. Mask Selection Figure. An example image from the MATLAB data analysis program “PCCcalc” (Supplement File 1) generated during the mask creation procedure used to designate the fluorescence area to analyze, with the mask defined by the band of interest (e.g., LacI fusion protein) and the Hoechst DNA stain for chromatin boundaries. - The Hoechst and tester masks can be manually adjusted using the following commands from the keyboard:
key command
a decrease current Hoechst mask by erosion
s increase current Hoechst mask by dilation
d decrease current Hoechst mask using higher threshold
f increase current Hoechst mask using lower threshold
u restart selection of Hoechst mask using candidate mask selected in Step 6d. above
z decrease current tester mask by erosion
x increase current tester mask by dilation
c decrease current tester mask using higher threshold
v increase current tester mask using lower threshold
j restart selection of tester mask using initial candidate mask
k restart process and return to Step 6d above
g draw a polygon by hand corresponding to the Hoechst region of interest
i skip this image
r save masks and proceed to calculate correlation coefficient - If the manipulations of the Hoechst mask do not adequately limit the defined overlap to the band of interest present on DNA, press g to draw a Hoechst mask in manual mode. A new window will open with a BW image of the Hoechst channel.
- Use the crosshairs to draw a polygon around the DAPI band containing the tester band of interest, clicking at positions around the DAPI band corresponding to points of the desired polygon.
- Click back on the first point when finished. Individual points can be adjusted by dragging with the mouse, and the entire polygon can be moved by clicking within the shape and dragging.
- When finished, right-click inside the polygon and select “Create mask” from the popup menu. The window closes and the new Hoechst mask appears in the Mask Selection Figure (referred to in Step 7 above).
- Now use the following commands to compare and accept or reject the hand-drawn mask:
key command
t toggle between the new hand-drawn mask and the previous automatically computed mask, or previously generated hand-drawn mask if multiple are attempted.
p redraw the mask.
g accept the hand-drawn mask, exit manual mode, and return to step H command options.
l reject the hand-drawn mask, use the previous mask and return to manual adjustments above.
- Once the masks are acceptable, press ‘r’.
If masks cannot satisfactorily be made that accurately represent the band of interest, the current image can be skipped by pressing ‘i'. - If 'r' has been pressed, then the Mask Selection Figure closes and a new figure with five panels, the Analysis Results Figure (Figure 5), opens.
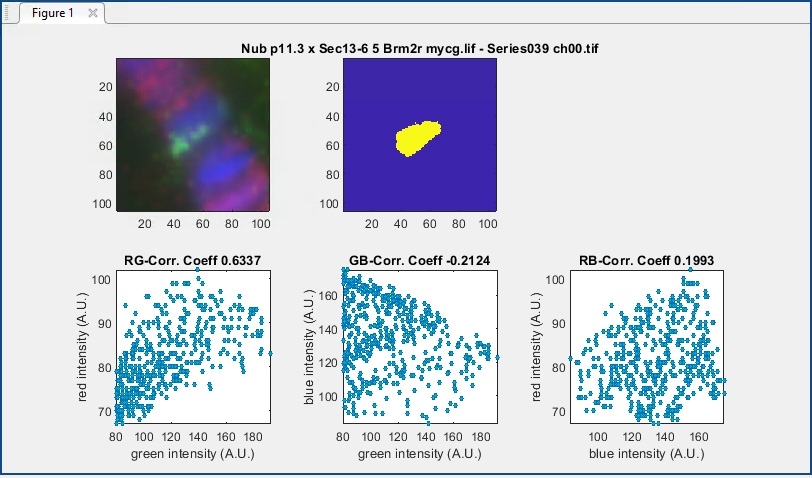
Figure 5. Analysis Results Figure. Found in MATLAB data analysis after mask creation is completed, this window displays the image being analyzed, the mask used for analysis, and graphs relating fluorescence intensity values of signals from two fluorescence channels at a time [Red-Green(RG), Green-Blue(GB), and Red-Blue(RB)], where each data point represents one pixel. The resulting Pearson Correlation Coefficients (“Corr Coeff”) derived from those relationships are displayed on the top of each graph. The immunofluorescence staining in the top left image includes Lac-I (green), Brm (red), and Hoechst (blue).- The top row displays an RGB image (left) of all three channels (as defined in the first step, e.g., red = target, green = tester, blue = Hoechst) and the final overlap mask (right).
- The lower row shows three scatter plots with each dot representing an individual pixel within the mask, with its x and y values corresponding to the intensities of the respective fluorescence signals. The overall PCC for each pair of channels, calculated from the relationship between all the points in each plot, is then calculated, and displayed above each plot.
- The cropped images, the results figure, and the correlation coefficients are all automatically saved in a subfolder called “autocrop” generated in the directory folder.
- Press any key to continue to repeat with the next image/set of channels (starting with step 5).
- Repeat until all images in the folder have been analyzed, and then a table will show up the in MATLAB workspace called pearsonCorr, and will be automatically saved as a file in the “autocrop” subfolder (titled “pearsonCCs.mat”) with the correlation values for that set of images.
- Each column corresponds to the correlation values for one image, and each row a different pair of channels, in order of image analysis.
row 1: red-green correlations
row 2: green-blue correlations
row 3: red-blue correlations - These values should then be copied into a file in your preferred graphing/statistical analysis software (e.g., Excel, Prism etc.) for later processing before proceeding to another data set or completing analysis, as the generated pearsonCCs.mat file with the correlation values is MATLAB-specific and will not be accessible on a computer without MATLAB software and license.
- We found it is best to use a minimum of 10 images per slide, 3 slides (larvae) per genotype/condition.
- To compare the distributions of correlation coefficients between conditions/genotypes, perform a Fisher z-transformation of the correlation coefficients r using the following:
z = 0.5 ln [(1 + r)/(1 - r)]
The distributions of the transformed correlations z can then be compared using a t-test or one-way ANOVA. If using Prism, a one-way ANOVA test is a robust statistical analysis for these data with 3+ conditions/genotypes, using Tukey’s multiple comparisons post-test.
- Each column corresponds to the correlation values for one image, and each row a different pair of channels, in order of image analysis.
Recipes
- PBS (Cold Spring Harbor Labs Recipe)
Reagent Amount for 1x Final 1x Concentration Amount for 10x Stock Final 10x Concentration
NaCl 8 g 137 mM 80 g 1.37 M
KCl 0.2 g 2.7 mM 2 g 27 mM
Na2HPO4 1.44 g 10 mM 14.4 g 100 mM
KH2PO4 0.24 g 1.8 mM 2.4 g 18 mM
Note: pH to 7.4 at 1x with HCl. - PBST
PBS + 0.1% Tween 20 diluted from 20% storage solution - Fixation Solution: 2% PFA/45% Acetic Acid
16% PFA diluted to 2% and 100% Acetic Acid diluted to 45% together in Milli-Q H2O
**Fixation solution freshly made every 30 min of dissection
***16% PFA stored at 4 °C after opening the ampule and used within 3 weeks or chromosome structure preservation will be impaired - 45% acetic acid
100% Acetic acid diluted to 45% with Milli-Q H2O
**Freshly made every 30 min of dissection - Blocking solution/Antibody cocktail base
PBS + 0.1% Tween 20 + 3% BSA (BSA dilution is mass per volume, so grams per ml)
Acknowledgments
This protocol was originally published in a research manuscript by Kuhn et al. (2019). We thank members of the Capelson and Little labs for their input on the research project that prompted development of this method, and J. Aleman for assistance in photo collection. M.C. is supported by the Research Scholar Grant RSG-15-159-01-CSM from the American Cancer Society and by NIH R01GM124143.
Competing interests
Authors declare no competing interests.
References
- Cai, W., Jin, Y., Girton, J., Johansen, J. and Johansen, K. M. (2010). Preparation of Drosophila polytene chromosome squashes for antibody labeling. J Vis Exp (36). DOI: 10.3791/1748.
- Kuhn, T. M., Pascual-Garcia, P., Gozalo, A., Little, S. C. and Capelson, M. (2019). Chromatin targeting of nuclear pore proteins induces chromatin decondensation. J Cell Biol 218(9): 2945-2961.
- Paro, R. (2008). Mapping protein distributions on polytene chromosomes by immunostaining. CSH protocols 2008: pdb.prot4714.
- Capelson, M., Liang, Y., Schulte, R., Mair, W., Wagner, U. and Hetzer, M. W. (2010). Chromatin-bound nuclear pore components regulate gene expression in higher eukaryotes. Cell 140(3): 372-383.
- Nakayama, T., Shimojima, T. and Hirose, S. (2012). The PBAP remodeling complex is required for histone H3.3 replacement at chromatin boundaries and for boundary functions. Development 139(24): 4582-4590.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Kuhn, T. M., Little, S. C. and Capelson, M. (2020). Preparation of Drosophila Polytene Chromosomes, Followed by Immunofluorescence Analysis of Chromatin Structure by Multi-fluorescence Correlations. Bio-protocol 10(13): e3673. DOI: 10.21769/BioProtoc.3673.
- Kuhn, T. M., Pascual-Garcia, P., Gozalo, A., Little, S. C. and Capelson, M. (2019). Chromatin targeting of nuclear pore proteins induces chromatin decondensation. J Cell Biol 218(9): 2945-2961.
Category
Biochemistry > Protein > Fluorescence
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.