- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Observing Nutrient Gradients, Gene Expression and Growth Variation Using the "Yeast Machine" Microfluidic Device
Published: Vol 10, Iss 13, Jul 5, 2020 DOI: 10.21769/BioProtoc.3668 Views: 5278
Reviewed by: Samantha E. R. DundonJulie WeidnerAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The natural environment of microbial cells like bacteria and yeast is often a complex community in which growth and internal organization reflect morphogenetic processes and interactions that are dependent on spatial position and time. While most of research is performed in simple homogeneous environments (e.g., bulk liquid cultures), which cannot capture full spatiotemporal community dynamics, studying biofilms or colonies is complex and usually does not give access to the spatiotemporal dynamics at single cell level. Here, we detail a protocol for generation of a microfluidic device, the “yeast machine”, with arrays of long monolayers of yeast colonies to advance the global understanding of how intercellular metabolic interactions affect the internal structure of colonies within defined and customizable spatial dimensions. With Saccharomyces cerevisiae as a model yeast system we used the “yeast machine” to demonstrate the emergence of glucose gradients by following expression of fluorescently labelled hexose transporters. We further quantified the expression spatial patterns with intra-colony growth rates and expression of other genes regulated by glucose availability. In addition to this, we showed that gradients of amino acids also form within a colony, potentially opening similar approaches to study spatiotemporal formation of gradients of many other nutrients and metabolic waste products. This approach could be used in the future to decipher the interplay between long-range metabolic interactions, cellular development, and morphogenesis in other same species or more complex multi-species systems at single-cell resolution and timescales relevant to ecology and evolution.
Background
The majority of fundamental research in microbiology to date relies on reduction of complexity in order to increase experimental control and to facilitate precise quantification of observed phenomena. This underlies the strong focus on studies of single cells in homogeneous environments or populations of cells grown in batch or continuous liquid cultures, despite the fact that in many natural niches cells form structured heterogeneous environments which behave as complex temporal and spatial dynamic systems. Cells of same or different species within multicellular assemblies absorb and metabolize nutrients while producing and secreting metabolites hence creating spatial gradients of nutrients and metabolites. This in turn has an effect on cellular physiology, linking it tightly to position and moment in space and time, resulting in areas that are well perfused with nutrients and areas that are scarce. Many multicellular systems exhibit variations in cellular physiology–from bacterial and yeast colonies (Cap et al., 2012; Vulin et al., 2014) to biofilms (Nadell et al., 2016) and tumors (Carmona-Fontaine et al., 2013 and 2017; Delarue et al., 2014). These variations are often a product of emergence of long-range metabolic interactions between cells. Namely, cellular microenvironment at one position depends on the nutrient uptake rate and metabolite production at other positions. Such multicellular communities (Shapiro, 1998; Shou et al., 2007; Xavier and Foster, 2007) exhibit many adaptive benefits, including higher cell proliferation, improved access to resources and niches (Koschwanez et al., 2011), and collective defence (Nadell et al., 2016). This results in optimization of population survival when confronted with adverse physical, chemical, nutritional or biological challenges (Palková and Váchová, 2006). Hence the importance to understand the emergence and maintenance of complex spatial multicellular structures from ecological (Gonzalez et al., 2012; Widder et al., 2016; Antwis et al., 2017), medical (Bryers, 2008; Estrela and Brown, 2018; Gilbert et al., 2018) and evolutionary (Kim et al., 2008; Nadell et al., 2010; Ratcliff et al., 2012) perspectives.
Numerous calls in recent reviews to study its nutrient sensing, signaling, and related growth and development control within the natural colony context (Broach, 2012; Horák, 2013; Conrad et al., 2014). While there have recently been some advances in observing complex bacterial communities (Liu et al., 2015; Cooper et al., 2017; Dal Co et al., 2019; Hornung et al., 2018), technological advances involving the widely used eukaryotic model organism Saccharomyces cerevisiae are lagging behind. The reason lies in the complexity of designing an experimental system that would allow monitoring, over long time periods and at single cell resolution, the development of a spatially defined extended multicellular assembly. Current direct observations of three-dimensional colonies are cumbersome and often constrained by existing technologies (Nadell et al., 2016). In yeast, two-photon microscopy of sliced agarose-encapsulated yeast colonies was required to show that yeast cells may adopt different physiologies–and possibly different cell types–depending on their position within a colony (Cap et al., 2012). All mentioned approaches have some of the following limitations: small device dimensions (< 100 µm), necessary use of low nutrient concentrations (< 1 mM), limited scope of nutrient types (carbon-rich media with glucose rarely used), inability to access single cell level measurements, and inability to observe cells at high temporal resolution. For these reasons, it is challenging to apply such approaches to the general study of a large monolayer of cells in standard range and scope of nutrients, often used in biological research in liquid cultures. We designed the “yeast machine” in a way that overcomes all these limitations (Figure 1). It is of large enough dimensions (800 µm long) to observe formation of gradients and depletion of nutrients up to glucose concentrations of 444 mM, it can be used to observe gradients of many carbon sources and other nutrients like amino acids, and it is easy to segment and track cells at single cell level in real-time over many days with time-lapse fluorescence microscopy. With this device we demonstrated the emergence of heterogeneous microenvironments and quantified spatial variation in cellular growth rate and the formation of gene expression landscapes for key metabolic genes involved in glucose transport and utilization. The application of this approach could be extended to study how the microenvironment dynamically changes when external conditions are altered in frequency-dependent manner. This is an uncharted territory at the scale of a multicellular assembly that is central to the understanding of microbial ecosystem resistance to stress, environmental fluctuations and adaptation. In addition, similar approaches could be used to study aging, cooperation and competition, cell memory, bet hedging, evolutionary dynamics, as well as quantitative characterization of (synthetic) ecological systems and mixtures of cells relevant to ecology and chemical biology.
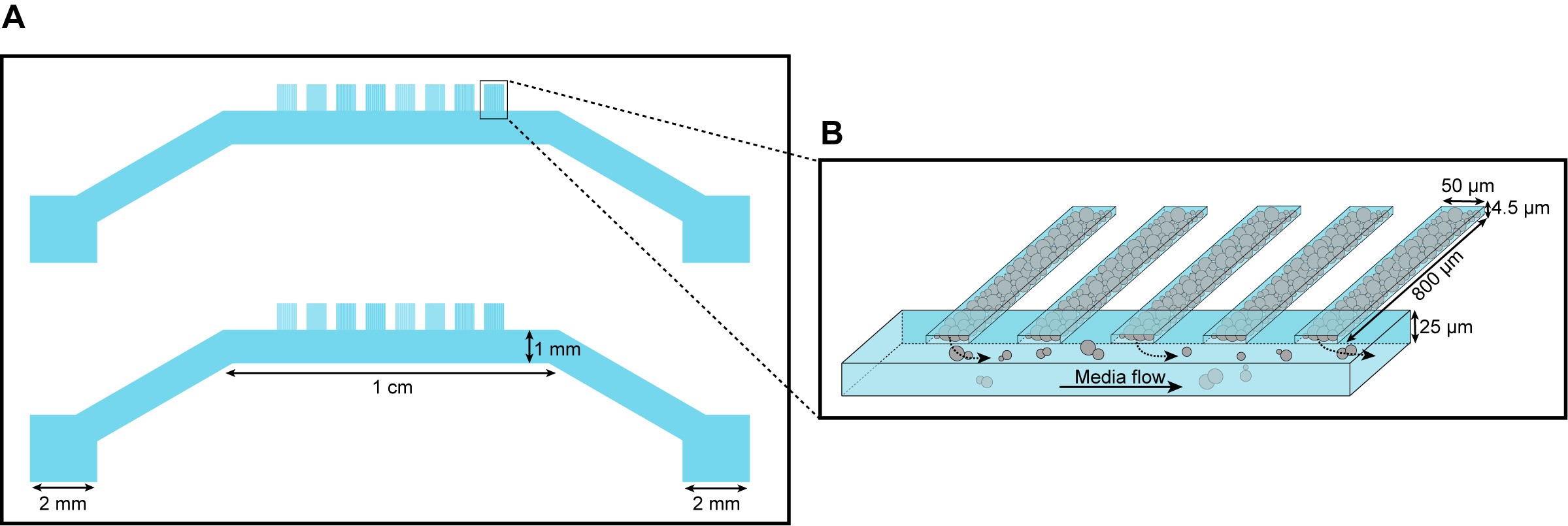
Figure 1. Microfluidic device design. A. Each single “yeast machine” has two sets of cell chambers of various widths (5 µm, 10 µm, 25 µm and 50 µm). The cell chambers are connected perpendicularly to a large flow channel (1 mm wide, 25 µm high). This design facilitates adaptation for different model systems (e.g., bacteria, yeast, mammalian cells) and high-throughput depending on the length, width and height dimensions. B. A close-up sketch of a set of cell chambers used in our experiments. They are 800 µm long, 50 µm wide, and 4.5 µm high. A single cell chamber fits a monolayer of up to 2,500 yeast cells. This figure is modified from the original article (Marinkovic et al., 2019).
Materials and Reagents
- Fluorinated ethylene-propylene (FEP) tubing 1/16" OD x .020" ID (IDEX Health & Science, catalog number: 1548L )
- Polyetheretherketone (PEEK) tubing blue 1/32" OD x .010" ID (IDEX Health & Science, catalog number: 1581 )
- Steel tubing 23 G (Darwin Microfluidics, view details)
- Plastic weighing dish (Fisher Scientific, catalog number: 08-732-113 )
- Glass coverslips, 24 x 50 mm #1.5 (VWR, Menzel-Gläser, catalog number: 631-9430 )
- Petri dishes 100 mm (Fisher Scientific, catalog number: FB0875713 )
- Microcentrifuge tubes 1.5 ml (Fisher Scientific, catalog number: 05-408-129 )
- Falcon round bottom tubes 14 ml (Fisher Scientific, catalog number: 14-959-1B )
- Falcon conical centrifuge tubes 50 ml (Fisher Scientific, catalog number: 14-432-22 )
- Glass media bottles (Fisher Scientific, catalog number: FB800500 )
- Pipet tips, standard lab autoclaved, various sizes (Genesee Scientific)
- Chrome photomask 5’’ x 5’’ .09 (Front Range PhotoMask, https://www.frontrangephotomask.com/)
- Plastic transfer pipets (Fisher Scientific, catalog number: 13-680-50 )
- Scalpel (Fisher Scientific, catalog number: 08-920B )
- Razor blade (Fisher Scientific, catalog number: 12-640 )
- Cutting mat (Fisher Scientific, catalog number: NC0671274 )
- Wash bottles (Fisher Scientific, catalog number: 02-897-10 )
- Scotch magic tape (ScotchTM Brand)
- Yeast strains
- Compressed air (in-house compressed air line)
- Polydimethylsiloxane (PDMS, Dow Corning Sylgard 184 silicone elastomer kit) (Fisher Scientific, catalog number: NC9285739 )
- 70% ethanol (Fisher Scientific, catalog number: NC9663244 )
- Isopropanol (Fisher Scientific, catalog number: A416-500 )
- Acetone (Fisher Scientific, catalog number: A18-500 )
- Distilled water
- Yeast nitrogen base without amino acids (Fisher Scientific, catalog number: DF0919-15-3 )
- Complete supplement mixture (CSM) of amino acids (Fisher Scientific, catalog number: MP114500012 )
- D-(+)-Glucose (Sigma, catalog number: G7021 )
- Aluminum foil (Fisher Scientific, catalog number: 01-213-100 )
- SU-8 2005 photoresist (Microchem)
- SU-8 2025 photoresist (Microchem)
- SU-8 developer (Microchem)
- 95% (3-Mercaptopropyl) trimethoxysilane (Sigma, catalog number: 175617 )
- Pluronic F-127 (Sigma, catalog number: P2443 )
- Compressed N2 (Airgas, catalog number: NI 300 )
- Synthetic complete (SC) medium + 2% w/vol glucose (1 L) (see Recipes)
Equipment
- Glass Erlenmyer flasks 250 ml (Fisher Scientific, catalog number: FB500250 )
- Timer (Fisher Scientific, catalog number: 06-664-252 )
- Silicon wafers (University Wafers, catalog number: 783 )
- Biopsy punch 1 mm (VWR, catalog number: 95039-092 )
- Motorized inverted epifluorescence microscope (Olympus, model: IX81 )
- EMCCD camera (Photometrics, model: Evolve 512 )
- X-Cite exacte fluorescence light source (Lumen Dynamics)
- Optical filter ET-EGFP (Chroma Technology Corporation, catalog number: U-N49002 ; Ex 470/40 nm Di495 Em 525/50 nm)
- Optical filter ET-DsRed (Chroma Technology Corporation, catalog number: U-N49005 ; Ex 545/30nm Di570 Em620/60 nm)
- Objective 10× (Olympus, Plan 10x/0.25 NA)
- Objective 60× (Olympus, PlanApo N 60x/1.42 NA Oil)
- Objective 100× (Olympus, UPlanFL N 100x/1.3 NA Oil)
- Microscope temperature control system (Life Imaging Services, The Cube & The Box)
- Mid to high end computer for the control of microscope and pressure-based microfluidic flow control system, as well as image analysis
- Pressure-based microfluidic flow control system (Fluigent, MFCS)
- Flow rate platform (Fluigent, Flowboard)
- Flow rate control module (Fluigent, Flow unit M)
- Vacuum desiccator (SP Scienceware, catalog number: F42020-0000 )
- O2 plasma cleaner (Harrick plasma, model: PDC-002 )
- Vacuum pump (Harrick plasma, model: PDC-VPE-2 )
- Laboratory oven (Fisher Scientific, catalog number: 51-028-113H )
- Balance (Mettler Toledo, catalog number: ML4002T )
- Benchtop centrifuge (Eppendorf, catalog number: 5405000042 , model: 5425 )
- 3D printed holder (printing files available at https://github.com/Lab513/contrib)
- Spin coater (Laurell, model: WS-650 ), used for loading cells with special 3D printed holder
- Shaking incubator (Thermo Fisher Scientific, catalog number: SHKE4000-1CE )
- Spectrophotometer (Eppendorf, catalog number: 6133000010 )
- Hot plate (Fisher, catalog number: S50462CH )
- Tweezers (Fisher Scientific, catalog number: 16-100-103 )
- Bunsen burner (Fisher Scientific, catalog number: 03-902Q )
- Pipetman, various sizes (Gilson)
- Chemical hood
- Photomask aligner (Süss Microtec, model: MJB4 )
- Mecanic surface profilometer (Veeco, Dektak 150)
- Optical microscope (Nikon, model: LV150 )
- Photoresist spin coater (Brewer Science, Cee Model 200x-F)
- 2 Hot plates (Fisher Scientific, catalog number: 90-691-11 )
Software
- ImageJ (Schneider et al., 2012, https://imagej.nih.gov/ij/)
- µManager (Edelstein et al., 2014, https://micro-manager.org/)
- Matlab R2016b (MathWorks, https://www.mathworks.com/products/matlab.html)
- Adobe Illustrator 2015 (Adobe, https://www.adobe.com/products/illustrator.html)
- L-Edit software (Tanner, https://www.mentor.com/tannereda/l-edit)
- All-in-One (A-i-O, Fluigent, https://www.fluigent.com/)
Procedure
- Master wafer microfabrication
Microfabrication can be tedious and complex process for which training is often required by the clean room staff. Each clean room is likely equipped with different instruments that have unique settings and operating procedures that may differ from the following microfabrication protocol. Use caution and adjust microfabrication protocol according to your local conditions. Once the process is mastered, the master wafer of “yeast machine” complexity can be produced in 4 h (Figure 2).- Turn on the mask aligner, gas and vacuum lines, and the UV lamp.
- Set hot plates to 65 °C and 95 °C.
- Cover the surface of spin coater with aluminum foil to minimize SU-8 contamination.
- Rinse the polished side of the silicon wafer with acetone, isopropanol, and water sequentially to remove any dirt. Blow both sides of the wafer with compressed N2 after the rinse with each solution until the wafer is completely dry to the eye.
- Clean and activate the wafer surface with O2 plasma cleaner at 6 mbar for 6 min.
- Center the wafer on the chuck of a spin coater with polished side up.
- Open the vacuum line to hold the wafer in place and make sure that it is perfectly centered, otherwise SU-8 will not spread evenly.
- Program the spin coater according to the SU-8 manual. We used 500 rpm for 10 s and 6,000 rpm for 30 s. These settings are not necessarily consistent in time as they depend on SU-8 age and environmental conditions.
- Spread SU-8 2000 epoxy-based resin of known thickness on the silicon wafer. There are twelve variants of the SU-8 2000 resin that differ in their viscosities. With them resin layer of 0.5 µm to 200 µm thickness through a single coating process can be made. Since the “yeast machine” is composed of two layers that overlap, one 4.5 µm thick that serves for cell chambers and the other 25 µm thick that serves for nutrient channel, start with the thinner one. For 4.5 µm thickness use SU-8 2005 resin.
- Pour 1 ml/in of wafer diameter (4 ml for 4-inch wafer) in the center and try to avoid bubbles by pouring SU-8 resin close to the wafer surface.
- Wait 3-5 min for gas and bubbles to rise.
- Run the pre-prepared program of the coat spinner.
- Once finished, release vacuum and remove the wafer from the chuck. If SU-8 layer is not evenly spread, clean the wafer with isopropanol and repeat the procedure from Step A4.
- Transfer the wafer with tweezers directly to 95 °C hot plate and “soft-bake” it for 2 min.
- Transfer the wafer with tweezers to the mask aligner. Epoxy-based resin is photosensitive and it solidifies through cross-linking once it is exposed to high-energy photons from the UV light. The chrome mask is placed above the silicon wafer covered with resin and the silicon wafer is exposed to UV light only through the pattern on the mask. Use the exposure duration of 4.5 s with the vacuum contact between the mask and the wafer. The exposure energy of the UV lamp we use is close to 20 mJ cm-2 s-1.
- Remove the wafer with tweezers from the aligner and place it on a hot plate for 3 min at 95 °C for a post exposure bake.
- Take the wafer with tweezers off the hot plate and let it cool down to room temperature in the chemical hood. Transferring the wafer directly into the developer can cause cracking.
- Pour SU-8 developer in a glass dish making sure there is enough of it to cover the wafer completely.
- Submerge the wafer into SU-8 developer. This will remove the parts of the resin that did not solidify during the exposure to UV light.
- Agitate the wafer for 2 min by hand.
- Rinse the wafer gently with isopropanol wash bottle and dry it carefully with compressed N2.
- Check the patterns under optical microscope. If some microcracks (submicron thin dark lines) are visible, put the wafer on 95 °C hot plate for 5-10 min. This will completely cure microcracks.
- Repeat the same procedure (Steps A6-A22) with adjusted parameters for the 25 µm thick nutrient channel layer as follows in Steps A24-A32.
- Spin SU-8 2025 in two steps, 500 rpm for 10 s and 4,000 rpm for 30 s.
- “Soft bake” the wafer for 3 min at 65 °C and 6 min at 95 °C.
- Transfer the wafer with tweezers to the mask aligner and be careful to align the patterns of the nutrient channel on the mask properly with the existing cell chamber pattern on the wafer. To do this, use the alignment crosses on the mask.
- Expose the wafer to UV light for 8 s with the vacuum contact between the mask and the wafer.
- Remove the wafer with tweezers from the aligner and place it on a hot plate for 1 min at 65 °C and 6 min at 95 °C for post exposure bake.
- Take the wafer with tweezers off the hot plate and let it cool down to room temperature in the chemical hood.
- Submerge the wafer into SU-8 developer and agitate for 5 min by hand.
- Rinse the wafer gently with isopropanol wash bottle and dry it carefully with compressed N2.
- Check the patterns under optical microscope. If some microcracks (submicron thin dark lines) are visible, put the wafer on 95 °C hot plate for 5-10 min. This will completely cure microcracks.
- Check the dimensions of the patterns on the master wafer using Dektak 150 surface profiler. If the dimensions are not correct (up to 10% errors are fine), make new master wafer.
- Finally, put the master wafer in a Petri dish and treat it with (3-Mercaptopropyl)trimethoxysilane 95% for 1 h in vapour phase by placing the wafer close to a plastic cup with a few drops of silane in a vacuumed dessicator. Be careful to do this in a chemical hood, as silane is a toxic compound.
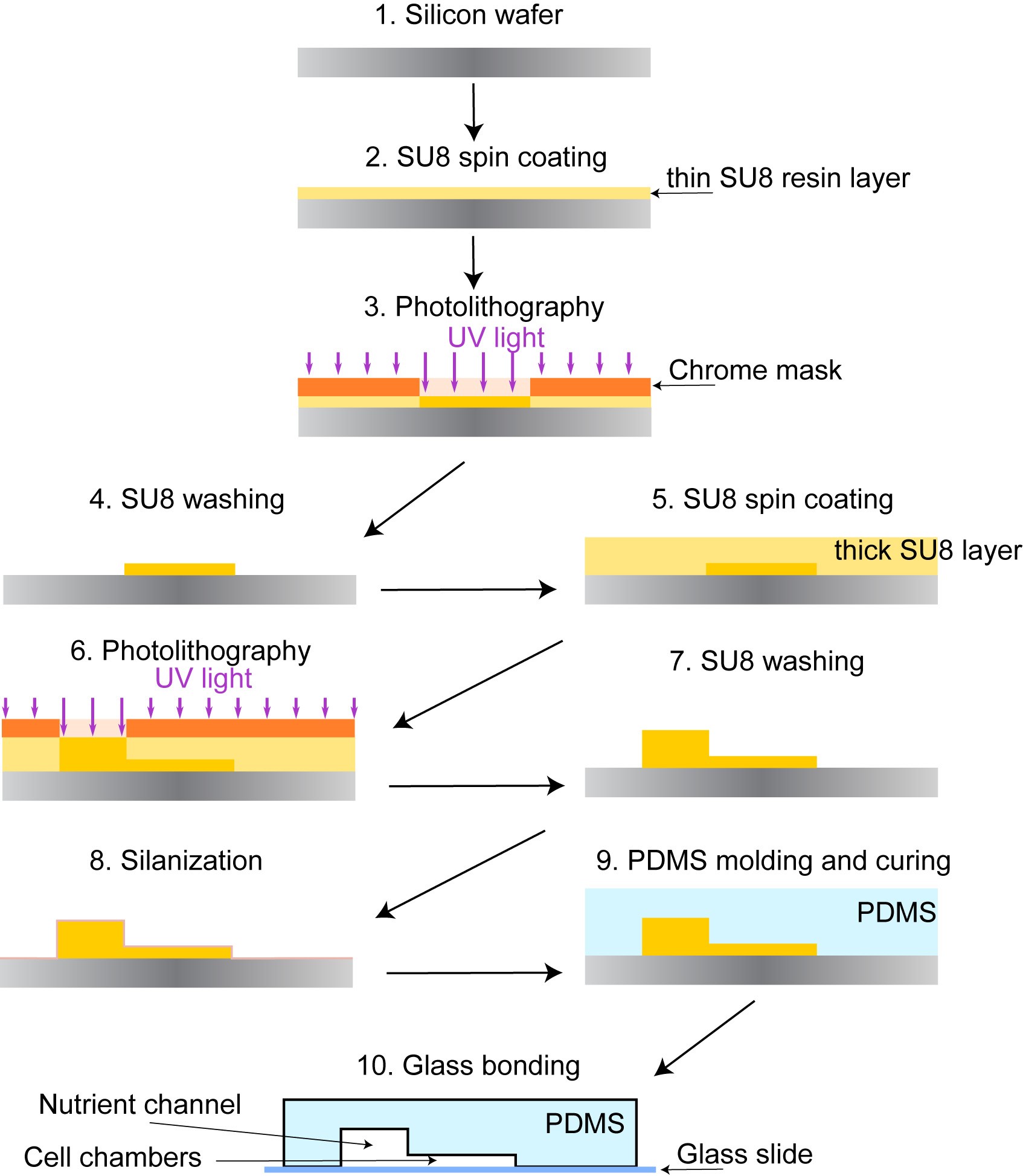
Figure 2. Microfluidic device fabrication. Epoxy-based resin is spread on a silicon wafer and illuminated with the UV light through a custom designed chrome mask. Soluble part of the resin is washed off and a new cycle of resin deposition can be made depending on the number of layers of the microfluidic device. The master wafer is silanized and used as a re-usable negative mold to produce microfluidic chips. Polydimethylsiloxane (PDMS) is then poured on the master wafer and cured until it is polymerized. The PDMS chip is bonded with a glass coverslip by plasma bonding. The final microfluidic chip is used to grow cells and deliver nutrients. The thinner parts are the cell chambers, and the thicker part is the main nutrient channel. This figure is modified from the original article (Marinkovic et al., 2019).
- Casting the “yeast machine” in polydimethylsiloxane (PDMS)
Now that the desired pattern on the master wafer is made, it can be used as a negative for replica molding of the “yeast machine” chip with the help of PDMS. PDMS is a transparent biocompatible silicon polymer permeable to gases. It is a viscous fluid when it is monomeric and it is elastic once it homopolymerizes in a reaction catalyzed by a curing agent.- Mix PDMS base and curing agent in a plastic weighing dish at a mass ratio of 10:1. When casting the whole PDMS mold for the first time use 30 g of PDMS base and 3 g of curing agent. Once only the “yeast machine” part is cut out, it is sufficient to re-cast only 3 g of PDMS base and 0.3 g of curing agent.
- Mix well with a pipet tip or a similar device.
- Degas the PDMS in a vacuum desiccator until all bubbles disappear, approximately 30 min.
- Pour degassed PDMS over the master wafer in a Petri dish. If some bubbles appear again, degas the whole wafer until the bubbles are gone.
- Cure PDMS at 65 °C for 2 h minimum or overnight in a laboratory oven.
- Assembling the “yeast machine”
For each experiment always use a newly assembled microfluidic chip. “Yeast machine” cannot be recycled for additional experiments.- Gently cut the PDMS in rectangular shape around the patterns. Be careful not to crack the master wafer by putting too much pressure on the scalpel. It is also advisable to let the cured PDMS cool down to room temperature to avoid damage to the master wafer.
- Carefully peel off the PDMS from the master wafer.
- Inspect the PDMS and its patterns under the microscope in order to detect possible defects. If defects come from a damaged master wafer, make new master wafer.
- Place the cut PDMS on a cutting mat with pattern side up.
- Punch out carefully the holes at each end of the nutrient channel with the biopsy punch. Always use a sharp biopsy punch and check under a microscope that there are no cracks in the hole, otherwise the media might leak.
- Clean the surface of the PDMS and glass cover slips using tape.
- Place the PDMS pattern side up and glass cover slip in plasma cleaner.
- Turn on the vacuum pump and close the plasma cleaner.
- Set the power to high to ignite the plasma.
- After pink glowing plasma appears, wait for 1 min and turn off the power.
- Open the valve of the plasma cleaner gently to allow air to flow in. It is safe to turn off the vacuum pump now.
- Immediately and carefully place the PDMS pattern side down onto glass cover slip side that was exposed to plasma.
- Use tweezers to touch gently the top of the PDMS to facilitate bonding. Be careful, pressing down the patterns could collapse them.
- Put a piece of tape on top of the chip to prevent dust from bonding to the chip.
- Bake the bonded device at 65 °C for 1 h or overnight to cure the bonding.
- Inject 10 µl of filter sterilized 1% Pluronic F-127 dissolved in distilled water and wait for 30 min at room temperature to passivate interior surfaces. Be careful, before injecting the liquid into the channel with a pipette, pour a drop of liquid onto the inlet to prevent bubbles entering the channel.
- Flush the channel with sterile distilled water or SC media to remove Pluronic F-127. Be careful again not to inject bubbles into the channel. Pour a drop of liquid onto the inlet first.
- The microfluidic chip is now ready to be loaded with yeast.
- Cell culturing and loading
- Preculture yeast cells overnight the day before the experiment in 5 ml of synthetic complete (SC) medium containing 2% w/vol glucose in a shaking incubator at 30 °C.
- On the day of the experiment dilute cells 50-fold into 50 ml of SC + 2% w/vol glucose.
- Culture them for 5-6 h in a shaking incubator at 30 °C until they reach and OD600 of 0.2-0.4.
- Put 1 ml of cells in 1.5 ml Eppendorf tube and centrifuge them for 1 min at RCF of 300 x g.
- Resuspend the cells in 20 µl SC media + 2% w/vol glucose.
- Load 10 µl of resuspended cells into the channel with a pipette. To make sure that no bubbles get in, pour a drop of liquid onto the inlet first and then load the cells.
- Tape the chip on a custom made 3D printed holder (Figure 3) with cell chambers facing outwards, so that the centrifugal force can push the cells from media channel into the cell chamber. The chip must be firmly pressed against the holder and taped on one side as shown in Figure 3A. After taping the chip should be completely immobilized.
- Attach the 3D printed holder onto the Laurell WS-650 spin coater and centrifuge for 2 min at 110 x g.
- Alternatively, tape the chip into a centrifuge making sure that the forces push cells into the cell chambers.
- Check under a microscope that cells have been loaded into the cell chambers. Usually, a couple of layers of cells are pushed inside towards the dead end (point furthest away from media channel) of the cell chamber.
- Flush the remaining cells out of the media channel with SC media + 2% w/vol glucose, making sure that no bubbles enter the system. To prevent bubbles from entering, pour a drop of liquid onto the inlet first and then flush the cells.

Figure 3. Cell-loading equipment. A. 3D printed chip holder for centrifugation with the “yeast machine” mounted on it (channels are colored with different dyes for better visualization). B. Spin coater that we use for centrifugation of cells into the cell chamber. This figure is modified from the original article (Marinkovic et al., 2019).
- Media perfusion and imaging
There are several options to perfuse media in microfluidic chips using hydrostatic pressure, peristaltic pumps, syringe pumps, or pressure based pumps. We opted for pressure-based pumps as they offer seamless control over flow of media through the system. The flow rate control module, assures constant media flow rate, no matter if there are clogs or bubbles in the system. These are often eliminated as the control system attempts to maintain the same flow rate in time.- Turn on the microscope and heat it up to 30 °C.
- Prepare 50 ml of SC media with appropriate glucose concentration in 50 ml Falcon tube.
- Assemble the pressure-based microfluidic flow control system (MFCS, Fluigent) coupled with a flow rate platform (Fluigent) and a flow rate control module (Fluigent) according to the instructions provided with the system. See Figure 4 for details.
- Connect input and output tubing to the chip. Make sure that the media is flowing very slowly out of the tubing prior to and while connecting it to the chip to prevent bubbles entering the system.
- Set the flow in All-in-One (A-i-O, Fluigent) software to maximum to flush out all the remaining cells from the nutrient channel and then set the flow to constant 5 µl/min.
- Keep the output at a constant pressure of 100 mbar above atmospheric pressure to minimize formation of air bubbles inside the nutrient channel.
- Inspect the chip for potential leaks before mounting it to the microscope.
- Place the chip onto the microscope stage and fix it in place with tape.
- Set up the image acquisition parameters relevant to your experiments. Ideally, image as many positions as possible at the lowest intensity and exposure as possible and at shortest time intervals as possible. If you want to observe a set of several complete cell chambers in one frame, use 10x objective. If you want to observe details at the single cell level, use 60x or 100x oil-immersed objective. Duration of a typical experiment is 24-72 h. However, we did manage to run experiments for up to one week without major disruptions.
- Check the first few intervals of imaging to make sure that the focus is stable and that the field is not drifting.
- Check occasionally that no clogs or leaks are impeding the ongoing experiment. Clogs mainly occur if bubbles are formed in the system and leaks mainly occur if inlet or outlet is cracked.

Figure 4. Microfluidic device setup. A. Media reservoirs are pressurized with Fluigent MFCS pressure pump resulting in flow through the flow sensors, into the chip and then to waste. Flow sensors and pressure pump are connected to the flow-rate control module, which maintains a constant flow through the system. Nutrient supply and media conditions can be changed in real time. B. Microfluidic chip mounted on a microscope. This figure is modified from the original article (Marinkovic et al., 2019).
Data analysis
Information about data processing and analyses, Matlab code, statistics, criteria for data inclusion/exclusion, details of number of replicates, exact data sets for each figure, and notes about reproducibility and variability can be found here (Marinkovic et al., 2019).
In order to optimize acquisition time and image analysis, microscopic images were most often taken with a 10x objective. This allows an entire set of cell chambers to fit within a single image without the need to stich multiple images. Open-source µManager microscopy software (Edelstein et al., 2014) was used to control the microscope’s components and to set up image acquisition. A stack of time-lapse images acquired through a particular optical filter was saved into a single .tif (Tagged Image File Format) file and analyzed using an open-source ImageJ (1.51p) software (Schneider et al., 2012).
- Front and Local velocity
Image analysis for calculating the front and local velocity is explained in the methods part of the original article under “image analysis” (Marinkovic et al., 2019). - Gene expression
During the image analysis, each cell chamber was tracked using Region of Interest (ROI) manager (ImageJ package) that covered 50 µm width and 800 µm length of a single cell chamber. All the values of obtained pixel intensities were normalized with the values of “yeast machine” background (i.e., the background noise) for each position along the 800 µm long chamber. By using Multi Plot function within ROI manager, the mean pixel intensity of the stacked images at a timepoint of interest was computed for each longitudinal position. To export position and intensity data from ImageJ to Excel, the Save function in the Profiles window was used. Data in Excel sheet was then imported into Matlab for further processing. This way we obtained information about average cell fluorescence for each region of the chamber and processed it with custom made Matlab scripts available in the original article (Marinkovic et al., 2019).
Notes
- Representative results can be seen in figures and a movie in the original article (Marinkovic et al., 2019).
- A comprehensive video of typical results obtained with the “yeast machine” can be seen at the following link https://doi.org/10.7554/eLife.47951.005.
- Production of master wafer from part A of “Procedure” can be outsourced to commercial microfabrication companies (e.g., https://www.micruxfluidic.com).
Recipes
- Synthetic complete (SC) medium + 2% w/vol glucose (1 L)
6.7 g Yeast nitrogen base without amino acids
0.79 g Complete supplement mixture (CSM) of amino acids
20 g Glucose
Distilled autoclaved water to 1 L
Keep each component as filter sterilized 10x stock solution in distilled water
Acknowledgments
This work was supported by the Agence Nationale de la Recherche (ICEBERG-ANR-10-BINF-06-01; ANR-16-CE12-0025-01), the interdisciplinary program of the University Sorbonne Paris Cité, the Who am I? Laboratory of Excellence (ANR-11-LABX-0071 and ANR-11-IDEX-0005-01) and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No 724813). This protocol was adapted from Marinkovic et al. (2019).
Competing interests
The authors declare no competing interests.
References
- Antwis, R. E., Griffiths, S. M., Harrison, X. A., Aranega-Bou, P., Arce, A., Bettridge, A. S., Brailsford, F. L., de Menezes, A., Devaynes, A., Forbes, K. M., Fry, E. L., Goodhead, I., Haskell, E., Heys, C., James, C., Johnston, S. R., Lewis, G. R., Lewis, Z., Macey, M. C., McCarthy, A., McDonald, J. E., Mejia-Florez, N. L., O'Brien, D., Orland, C., Pautasso, M., Reid, W. D. K., Robinson, H. A., Wilson, K. and Sutherland, W. J. (2017). Fifty important research questions in microbial ecology. FEMS Microbiol Ecol 93(5).
- Broach, J. R. (2012). Nutritional control of growth and development in yeast. Genetics 192(1): 73-105.
- Bryers, J. D. (2008). Medical biofilms. Biotechnol Bioeng 100(1): 1-18.
- Cap, M., Stepanek, L., Harant, K., Vachova, L. and Palkova, Z. (2012). Cell differentiation within a yeast colony: metabolic and regulatory parallels with a tumor-affected organism. Mol Cell 46(4): 436-448.
- Carmona-Fontaine, C., Bucci, V., Akkari, L., Deforet, M., Joyce, J. A. and Xavier, J. B. (2013). Emergence of spatial structure in the tumor microenvironment due to the Warburg effect. Proc Natl Acad Sci U S A 110(48): 19402-19407.
- Carmona-Fontaine, C., Deforet, M., Akkari, L., Thompson, C. B., Joyce, J. A. and Xavier, J. B. (2017). Metabolic origins of spatial organization in the tumor microenvironment. Proc Natl Acad Sci U S A 114(11): 2934-2939.
- Conrad, M., Schothorst, J., Kankipati, H. N., Van Zeebroeck, G., Rubio-Texeira, M. and Thevelein, J. M. (2014). Nutrient sensing and signaling in the yeast Saccharomyces cerevisiae. FEMS Microbiol Rev 38(2): 254-299.
- Cooper, R. M., Tsimring, L. and Hasty, J. (2017). Inter-species population dynamics enhance microbial horizontal gene transfer and spread of antibiotic resistance. Elife 6: 25950.
- Dal Co, A., van Vliet, S. and Ackermann, M. (2019). Emergent microscale gradients give rise to metabolic cross-feeding and antibiotic tolerance in clonal bacterial populations. Philos Trans R Soc Lond B Biol Sci 374(1786): 20190080.
- Delarue, M., Montel, F., Vignjevic, D., Prost, J., Joanny, J. F. and Cappello, G. (2014). Compressive stress inhibits proliferation in tumor spheroids through a volume limitation. Biophys J 107(8): 1821-1828.
- Edelstein, A. D., Tsuchida, M. A., Amodaj, N., Pinkard, H., Vale, R. D. and Stuurman, N. (2014). Advanced methods of microscope control using muManager software. J Biol Methods 1(2).
- Estrela, S. and Brown, S. P. (2018). Community interactions and spatial structure shape selection on antibiotic resistant lineages. PLoS Comput Biol 14(6): e1006179.
- Gilbert, J. A., Blaser, M. J., Caporaso, J. G., Jansson, J. K., Lynch, S. V. and Knight, R. (2018). Current understanding of the human microbiome. Nat Med 24(4): 392-400.
- Gonzalez, A., King, A., Robeson, M. S., 2nd, Song, S., Shade, A., Metcalf, J. L. and Knight, R. (2012). Characterizing microbial communities through space and time. Curr Opin Biotechnol 23(3): 431-436.
- Koschwanez, J. H., Foster, K. R. and Murray, A. W. (2011). Sucrose utilization in budding yeast as a model for the origin of undifferentiated multicellularity. PLoS Biol 9(8): e1001122.
- Horák, J. (2013). Regulations of sugar transporters: insights from yeast. Curr Genet 59(1-2): 1-31.
- Hornung, R., Grunberger, A., Westerwalbesloh, C., Kohlheyer, D., Gompper, G. and Elgeti, J. (2018). Quantitative modelling of nutrient-limited growth of bacterial colonies in microfluidic cultivation. J R Soc Interface 15(139).
- Kim, H. J., Boedicker, J. Q., Choi, J. W. and Ismagilov, R. F. (2008). Defined spatial structure stabilizes a synthetic multispecies bacterial community. Proc Natl Acad Sci U S A 105(47): 18188-18193.
- Liu, J., Prindle, A., Humphries, J., Gabalda-Sagarra, M., Asally, M., Lee, D. Y., Ly, S., Garcia-Ojalvo, J. and Süel, G. M. (2015). Metabolic co-dependence gives rise to collective oscillations within biofilms. Nature 523(7562): 550-554.
- Marinkovic, Z. S., Vulin, C., Acman, M., Song, X., Di Meglio, J. M., Lindner, A. B. and Hersen, P. (2019). A microfluidic device for inferring metabolic landscapes in yeast monolayer colonies. eLife 8: 47951.
- Nadell, C. D., Drescher, K. and Foster, K. R. (2016). Spatial structure, cooperation and competition in biofilms. Nat Rev Microbiol 14(9): 589-600.
- Nadell, C. D., Foster, K. R. and Xavier, J. B. (2010). Emergence of spatial structure in cell groups and the evolution of cooperation. PLoS Comput Biol 6(3): e1000716.
- Palková, Z. and Váchová, L. (2006). Life within a community: benefit to yeast long-term survival. FEMS Microbiol Rev 30(5): 806-824.
- Ratcliff, W. C., Denison, R. F., Borrello, M. and Travisano, M. (2012). Experimental evolution of multicellularity. Proc Natl Acad Sci U S A 109(5): 1595-1600.
- Schneider, C. A., Rasband, W. S. and Eliceiri, K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nature Methods 9(7): 671-675.
- Shapiro, J. A. (1998). Thinking about bacterial populations as multicellular organisms. Annu Rev Microbiol 52: 81-104.
- Shou, W., Ram, S. and Vilar, J. M. (2007). Synthetic cooperation in engineered yeast populations. Proc Natl Acad Sci U S A 104(6): 1877-1882.
- Vulin, C., Di Meglio, J. M., Lindner, A. B., Daerr, A., Murray, A. and Hersen, P. (2014). Growing yeast into cylindrical colonies. Biophys J 106(10): 2214-2221.
- Widder, S., Allen, R. J., Pfeiffer, T., Curtis, T. P., Wiuf, C., Sloan, W. T., Cordero, O. X., Brown, S. P., Momeni, B., Shou, W., Kettle, H., Flint, H. J., Haas, A. F., Laroche, B., Kreft, J. U., Rainey, P. B., Freilich, S., Schuster, S., Milferstedt, K., van der Meer, J. R., Grobetakopf, T., Huisman, J., Free, A., Picioreanu, C., Quince, C., Klapper, I., Labarthe, S., Smets, B. F., Wang, H., Isaac Newton Institute, F. and Soyer, O. S. (2016). Challenges in microbial ecology: building predictive understanding of community function and dynamics. ISME J 10(11): 2557-2568.
- Xavier, J. B. and Foster, K. R. (2007). Cooperation and conflict in microbial biofilms. Proc Natl Acad Sci U S A 104(3): 876-881.
Article Information
Copyright
![]() Marinkovic et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Marinkovic et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Marinkovic, Z. S., Vulin, C., Acman, M., Song, X., Di Meglio, J. M., Lindner, A. B. and Hersen, P. (2020). Observing Nutrient Gradients, Gene Expression and Growth Variation Using the "Yeast Machine" Microfluidic Device. Bio-protocol 10(13): e3668. DOI: 10.21769/BioProtoc.3668.
- Marinkovic, Z. S., Vulin, C., Acman, M., Song, X., Di Meglio, J. M., Lindner, A. B. and Hersen, P. (2019). A microfluidic device for inferring metabolic landscapes in yeast monolayer colonies. eLife 8: 47951.
Category
Microbiology > Community analysis > Spatial interaction
Cell Biology > Cell metabolism > Carbohydrate
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.